Back to Journals » Clinical and Experimental Gastroenterology » Volume 9
Improving outcomes of refractory celiac disease – current and emerging treatment strategies
Authors Woodward J
Received 4 March 2016
Accepted for publication 24 May 2016
Published 3 August 2016 Volume 2016:9 Pages 225—236
DOI https://doi.org/10.2147/CEG.S87200
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Andreas M. Kaiser
Jeremy Woodward
Department of Gastroenterology and Clinical Nutrition, Addenbrooke’s Hospital, Cambridge, UK
Abstract: Intestinal inflammation and symptoms of celiac disease (CD) usually respond well to gluten withdrawal, but rare cases are refractory to diet. Two types of refractory CD are discriminated on the basis of the presence or absence of an atypical population of mucosal lymphocytes that may progress to enteropathy-associated T-cell lymphoma. Challenges remain in the secure diagnosis of both types of refractory disease, and evidence on which to base treatment recommendations is flawed by the small numbers of reported patients and the use of different diagnostic strategies. Recent advances in our understanding of the mechanisms of the condition in conjunction with the development of immunomodulatory agents for managing other inflammatory diseases are helping to shape future approaches to targeted therapy. Progression will depend on collaboration and recruitment to trials. In the meantime, there is evidence to suggest that earlier diagnosis and better follow-up and management of CD may prevent the development of refractoriness.
Keywords: celiac disease, gluten, small intestine, lymphoma, lymphocytes
Overview
Celiac disease (CD) is a reversible, proximal immune enteropathy resulting from the interaction of dietary gluten with the intestinal immune system.1 Both innate and adaptive immune mechanisms are implicated in the response2 – the latter predominating as evidenced by the dependence on the presence of specific human leukocyte antigen (HLA) class II molecules for gliadin-derived antigen presentation. In keeping with this, dietary exclusion of gluten leads to resolution of the chronic inflammation, restoration of the intestinal mucosal architecture, and resolution of downstream effects of malabsorption.3–5 In a small proportion of cases and for reasons that are poorly understood, intestinal mucosal damage persists despite the removal of the environmental antigenic trigger – hence “celiac disease refractory to gluten withdrawal” or simply “refractory celiac disease” (RCD).6,7 This condition is now recognized as a premalignant overture to enteropathy-associated T-cell lymphoma (EATL), which carries a grave prognosis.8,9
Defining RCD
Understanding any condition relies fundamentally on stringent definition, without which data on prevalence are unreliable, clinical trials of treatment are impossible or uninterpretable, and it becomes difficult to correlate data from different institutions. Unfortunately the application of existing diagnostic criteria in RCD permits significant potential for variable interpretation.
RCD is currently defined as “the recurrence or persistence of malabsorptive symptoms and signs with villous atrophy despite a strict gluten-free diet for >12 months”.1
A clinician will face a number of significant difficulties in applying this definition in practice:
- Only a minority of patients experience the “classical” symptoms of weight loss and diarrhea, and the majority present with a wide array of different symptoms, including fatigue or headache, coincidental findings of various deficiencies on blood tests (such as iron, folic acid, vitamin D, and vitamin B12), and low bone density, or frequently with no reported symptoms at all (for instance, those identified through case finding in first-degree relatives).10,11 It may seem incongruous that the diagnosis of RCD should require the presence of malabsorptive signs and symptoms that are unlikely to have been present in the initial active (gluten-induced) lesion – it is possible (and common) to have severe villous atrophy in the absence of symptoms.
- Malabsorptive symptoms and signs remain uncharacterized in this definition that requires both for the diagnosis of RCD. A common presentation of CD with bloating and irritable bowel syndrome-type symptoms is undoubtedly caused by proximal malabsorption and distal substrate fermentation.12 Whether this would be a sufficient attribution to define RCD is unclear; however, the coexistence of responsive CD and ongoing irritable bowel syndrome as a cause of persisting symptoms is overwhelmingly more likely. Similarly, diarrhea persisting despite gluten withdrawal may be due to causes that include pancreatic exocrine deficiency or microscopic colitis.13–15 Some malabsorptive symptoms and signs are likely to be multifactorial – for instance, associated with iron deficiency – and their presence as a defining feature of RCD may lead to overdiagnosis in a patient with ongoing villous atrophy on intestinal biopsy.
- A strict gluten-free diet is exceptionally difficult to follow due to the environmental ubiquity of gluten. Furthermore, there are no definitive surrogate markers of ongoing gluten ingestion – we and others have recently shown that persisting IgA antitissue transglutaminase titers are not a reliable indicator of ongoing dietary gluten ingestion,16 and this has now been adopted in the UK national guidance.17 Interview of patients by experienced dietitians and the use of food diaries are equally likely to miss sources of gluten ingestion due to recall bias. Patients who are most likely to experience inadvertent gluten exposure are those who are least likely to prepare their own food and are therefore unable to relate the ingredients or precautions taken to prevent contamination during preparation.18 It is estimated that up to 45% of patients with ongoing symptoms after starting a gluten-free diet have inadvertent gluten exposure in the diet.19
- Sensitivity to dietary gluten may also vary significantly between individuals with CD as a large number of different immunomodulatory genes have been implicated to date in its pathogenesis.20,21 It is likely that many patients with ongoing symptoms and villous atrophy 1 year after the initiation of treatment will still have gluten in the diet at a level that leads to toxic effects in that individual rather than a diagnosis of RCD. The reported prevalence of RCD is therefore related to the inverse function of the reliability of the individual or health professionals to be able to exclude sufficient gluten from the diet. It should also be borne in mind that the enforcement of national legislation, involvement of patient support groups, socioeconomic status, and cultural and ethnic dietary factors will impact on successful dietary gluten exclusion and therefore on the persistence of symptoms and villous atrophy at 1 year. This may account for the majority of the large difference between the reported prevalences of “RCD” from different cultures.
- A degree of villous atrophy is present in ∼40% of patients who are rebiopsied at 1 year despite good dietary compliance.16,22 As there is an inevitable learning curve with the gluten-free diet, it is not possible to state for certain whether this is due to delayed initiation of adequate gluten exclusion or a fixed length of time required to heal the mucosal lesion. Most studies to date that purport to indicate the length of time required for mucosal healing were not designed to do so.22–25 However, while we have found no difference in the healing rates between patients rebiopsied at 1 year and after 1 year,16 a significant recent study has demonstrated that the likelihood of ongoing villous atrophy at 1 year is related to the severity of the initial mucosal lesion, and this is the first reliable evidence that supports a time to mucosal healing after gluten exclusion of >1 year in certain cases.26 There is a significant observer variability between histopathologists in the reporting of duodenal biopsies,27 and therefore, minor degrees of ongoing intestinal inflammation need to be treated with caution and not necessarily considered to represent refractory disease.
- Many patients with severe malabsorption due to (primarily unresponsive) RCD become clearly apparent long before the 12 months required by this definition due to the severity of the symptoms and the mucosal lesion despite rigorous exclusion of gluten with artificial enteral or parenteral feeds.
- There is a need to exclude pathologies that may masquerade as RCD, such as olmesartan enteropathy28,29 or chronic norovirus infection associated with immunodeficiency30 – this is particularly relevant as many RCD patients are seronegative for antitissue transglutaminase or endomysial antibodies. While the presence of an appropriate HLA is a necessary prerequisite, positivity does not prove the case due to the high background prevalence of HLA-DQ2 or HLA-DQ8 in Caucasian populations.
Therefore, as a result of this working definition, RCD is most likely to be overdiagnosed due to the likelihood of ongoing gluten exposure in the majority of cases and the presence of villous atrophy in a high proportion of normally responsive cases at 1 year. However, the requirement for the presence of unspecified symptoms in the definition (which is not necessary for the diagnosis of CD itself) is likely to underestimate those asymptomatic or paucisymptomatic cases with severe refractory mucosal inflammation (histologically refractory) that may be at risk of late complication.31
Classification of RCD
Clarification of the diagnosis of RCD came with the finding of an aberrant population of intestinal intraepithelial lymphocytes (IELs) in some patients with RCD that lack the usual expression of CD3 and CD8 on the cell surface.32 These cells often share a clonal T-cell receptor (TCR) rearrangement and show immune-phenotypic similarity to the malignant cells in EATL,33 lending credence to a continuum between RCD and EATL.34 Patients with an excess of such IELs are described as having type 2 RCD in contrast to type 1 where they are absent (Figure 1 and Table 1).35
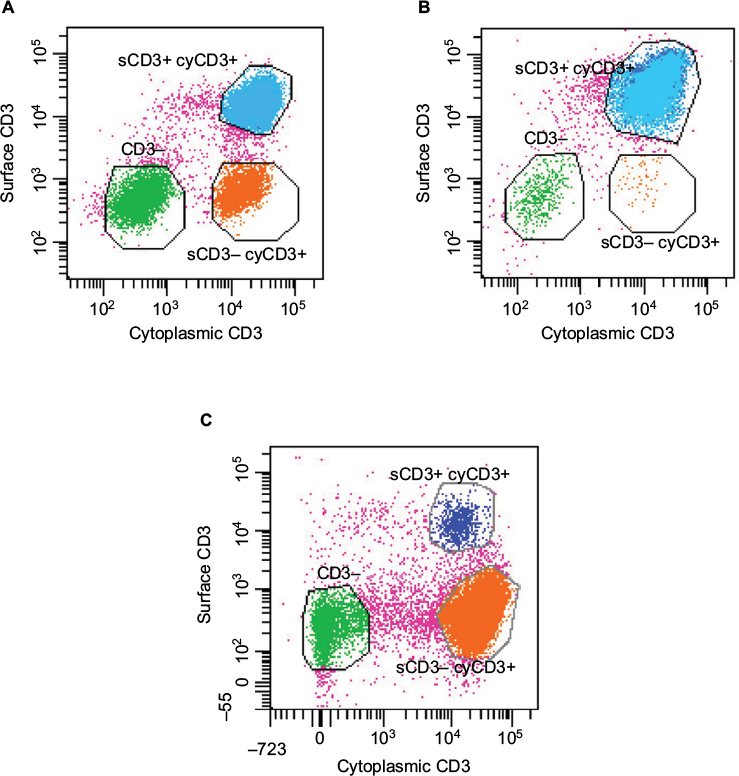 | Figure 1 Characteristic flow cytometry of intraepithelial lymphocytes isolated from intestinal biopsies incubated with anti-CD3 antibodies prior to and after permeabilization in order to identify cell sCD3 (y axis) and cyCD3 (x axis) expression. Notes: (A) Normal individual (not celiac): 26% of cells lack surface and intracellular CD3 expression, 13% demonstrate intracytoplasmic but not cell sCD3 expression, and 56% are sCD3 and intracytoplasmic CD3+ in keeping with mature T-cells. Of these, the majority (96%) express the ab TCR. (B) Patient with RCD1 (note that this is indistinguishable from the patient with active CD): 2% of cells lack sCD3 and intracellular CD3 expression, 0.3% demonstrate intracytoplasmic CD3 but lack sCD3 expression, and 95% are sCD3 and intracytoplasmic CD3+, of which roughly equal proportions express the ab and gd TCR. (C) Patient with RCD2: 12% of cells lack intracytoplasmic and sCD3, 76% express the “aberrant phenotype” of intracytoplasmic CD3 without surface expression, and only 4% are mature T-cells expressing both sCD3 and cyCD3. Abbreviations: sCD3, surface CD3; cyCD3, cytoplasmic CD3; TCR, T-cell receptor; RCD, Refractory Celiac Disease. |
 | Table 1 Characteristics, outcomes, and possible future discriminatory tests in CD and RCD Note: The requirement for symptoms in the definition of RCD results in the entity of “histologically RCD” without symptoms or signs of malabsorption that may go undetected unless routine follow-up biopsies are performed after initiating diet. Abbreviations: CD, celiac disease; RCD, refractory celiac disease; TCR, T-cell receptor; IEL, intraepithelial lymphocyte; EATL, enteropathy-associated T-cell lymphoma. |
RCD2 carries a far greater risk of transformation to EATL (Figure 2) than RCD1 and a correspondingly poor prognosis and would therefore warrant a more aggressive therapeutic approach if it were shown that this could reduce the risk of malignant transformation or resolution of symptoms.36–39 However, such an approach mandates reliable discrimination of type 1 from type 2 RCD, but unfortunately, despite a clearly identifiable cellular phenotype, the diagnosis of RCD2 is not always clear. The presence of clonal TCR rearrangements is problematic as the background IEL population is oligoclonal under normal circumstances, and a clonal rearrangement can predominate in patients who are newly diagnosed or continue to ingest gluten. The presence of clonality can be transient and is not therefore definitive evidence of RCD2.39–41
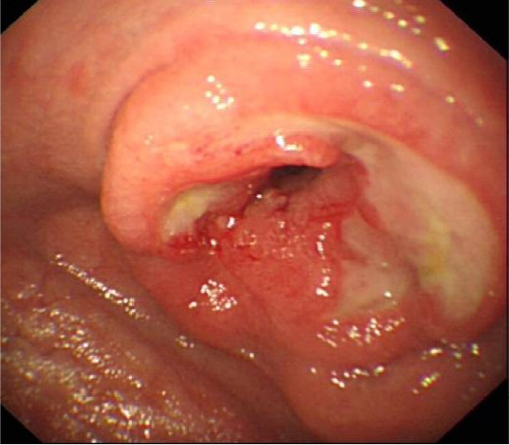 | Figure 2 An endoscopic image of T-cell lymphoma (EATL). |
The presence of immunophenotypically aberrant IELs, regardless of whether or not they are clonal, should indicate RCD as type 2. However, even the immunophenotype of the aberrant IELs is not unique to RCD2 and can occur in normal individuals (Figure 1A) – it is the relative proportion of the surface CD3-CD8- cells that is key to the diagnosis. How these cells are detected is also critical – immunohistochemistry using dual color immunostaining for CD3 and CD8 helps localization,39 but is most often carried out singly, leading to very rough approximation of the proportion of CD8-CD3+ cells (immunohistochemistry cannot differentiate between surface and intracellular CD3).42 Flow cytometry has additional benefits of multiple marker identification on cells but is affected by variable cell yields, the requirement for fresh biopsies, and the lack of localization of the cells isolated (CD4+ lymphocytes predominate in the lamina propria compared to CD8+ cells in the epithelium).43,44 Different cutoffs of the proportion of aberrant IELs are used to define RCD type 2 depending on the techniques used to quantify them. Therefore, given the lack of a clear defining test, continual monitoring of aberrant IEL numbers over time in sequential biopsies may be preferable.39
Epidemiology of RCD
A further prerequisite for optimizing the management of any condition is to understand its natural history. The main groups reporting RCD have collected cases over many years or even decades, and this reflects the rarity of the condition. However, its reported prevalence ranges from 0.3% to 10% of CD cases – an extraordinary 30-fold discrepancy. This span most likely reflects the loose definition and problems in ruling out gluten exposure in the diagnosis of type 1 RCD as the ratio of RCD1 to RCD2 is higher in those with the highest prevalence of RCD (11:1) compared to those with the lowest prevalence of RCD (3.4:1). However, even when allowing for this, there is still a sevenfold difference in the reported prevalence of RCD2.45–47 This may be due to ascertainment bias within tertiary centers; however, it is likely that the use of different diagnostic tests – and in particular the overreliance on TCR clonality rather than flow cytometry – contributes significantly to the variability that inevitably impacts on the reported outcomes of interventions.
Younger patients have been described,48 but the peak age of onset of RCD is in the sixth decade with a predominance in females – in contrast to EATL that is more frequently identified in males.
The prognosis of RCD2 is poor with a 5-year survival of only 44%–58%, largely accounted for by progression to EATL in 33%–67% of cases. In contrast, while RCD1 can progress to EATL, this is rare and the corresponding 5-year survival is >90%.36–38 Transformation to EATL carries a very poor prognosis with the majority dying within 8 months of diagnosis and a 5-year survival of only 11%.8,9,38
Whether RCD1 progresses to RCD2 is contentious – only one study has retrospectively analyzed sequential intestinal biopsies taken prior to the diagnosis of RCD and demonstrated a progressive increase in aberrant IELs averaging 1.8% per month to above the diagnostic threshold for RCD2 in tandem with the development of TCR clonality.39
Pathogenesis of RCD
The age at presentation and the finding of de novo RCD or even EATL (without a prior diagnosis of CD) in a significant number of cases implicate chronic antigen exposure, driving the transformation from inflammation to neoplasia. RCD1 is clinically and histologically indistinguishable from active CD and could represent an increased sensitivity to background gluten or a delayed response to gluten withdrawal. This would be in keeping with the findings of resolution of RCD1 over time with the use of strict antigen exclusion (elemental diet49,50), careful and sustained dietary follow-up, or immunosuppressive agents. No genetic markers of unresponsiveness have been described although homozygosity for HLA-DQ2 appears to predispose to a higher degree of gluten sensitivity.51 A recent study has demonstrated a different cytokine profile in RCD1 compared to responsive (but active) CD with the expression of tumor necrosis factor-α (TNF-α) in RCD1.52
In contrast, the aberrant IEL phenotype in RCD2 appears to be expanded in vitro and in vivo by the mucosal expression of interleukin (IL)-15, which is stimulated by innate immune pathways, including interferon-a, a nonimmunogenic gliadin epitope, and viral RNA pattern recognition via TLR3. The aberrant IELs demonstrate direct cell-mediated cytotoxicity against enterocytes, which is activated by the interaction of a stress-derived major histocompatibility complex class I-like molecule (MICA) on the surface of enterocytes with NKG2D expressed on the IELs. This direct epithelial cytotoxicity may account for the severity of the mucosal lesion in RCD2.53,54 A proportion of the aberrant IELs in RCD express the IL-15 receptor b (CD122), whereas a further subset is characterized by the IL-7 receptor CD127 and lower level expression of CD122.55 Intriguingly, CD127-IELs taken from healthy individuals or those with CD can be induced to mature into functional CD56+ conventional natural killer cells or CD3+ T-cells (respectively) in the presence of IL-15.56 However, the CD127-aberrant IELs from patients with RCD2 differ in remaining phenotypically undifferentiated.
Cytogenetic alterations have been identified in RCD2 IELs similar to those in EATL, including partial trisomy of 1q, which is common in other forms of peripheral T-cell lymphoma.57 An interesting cytogenetic finding in EATL that has not to date been replicated in RCD2 IELs is a chromosomal gain at 9q33–34, a region that encompasses both Notch (required for T-cell differentiation of CD127-IELs in the abovementioned study) and the oncogene ABL.58
Prevention of RCD
Despite our incomplete understanding of the etiology of RCD, it is likely that chronic antigenic exposure is the driving force. In most western populations, there is a seroprevalence of ∼1%, but only a small proportion of these patients with CD are diagnosed.59 Indeed, EATL or osteoporosis often arises in patients previously not known to have the condition. It is possible that early detection and good subsequent management of CD could lead to a reduction in the later development of such complications, including RCD. Such data could be gleaned from comparative studies of different health care economies where case finding and management vary significantly – for instance in Finland, there is a high detection rate of CD and a very low reported incidence of RCD46,60 in comparison to the USA with a high reported incidence of RCD and a low detection rate of CD.47,61 Quality of adherence to a gluten-free diet in preventing the development of RCD is further supported by the increase in lymphoproliferative disease in patients with incomplete mucosal healing on follow-up biopsies.62
Treatment of the mucosal lesion in RCD
Following establishment of the diagnosis, treatment of the patient with RCD requires a careful search for evidence of underlying EATL using cross-sectional imaging,18 fluorodeoxyglucose-positron emission tomography, enteroscopy, and video capsule studies.63–65 Nutritional deficiencies usually require parenteral replacement. Associated osteoporosis should not be overlooked. Treatment is aimed at the mucosal lesion for two reasons: to improve absorption and symptoms and to reduce the risk of progression to EATL.
Steroids and immunosuppressant agents
Corticosteroids are used as the initial treatment for both types of RCD. Subjective (symptomatic) improvement is common, but the extent of mucosal response is unclear. Interestingly, corticosteroids were used in the treatment of CD prior to Dicke’s thesis on the association with cereals, and brought about a significant and rapid histological and symptomatic response.66 The extent of the response in RCD is less clear. An observational study from Paris38 demonstrated good clinical response (defined as a 50% reduction in symptoms and/or a 50% regain of lost weight prior to treatment); however, histological response was only 40% in RCD1 and 33% in RCD2. Only two of eleven patients treated with steroids alone achieved histological recovery in the Mayo clinic report,36 and a small study in Argentina reported no benefit from oral steroids.67
There is a substantial risk of steroid dependence when used in this setting, in patients already at risk of complications such as osteoporosis, and two strategies have been employed to reduce this risk – topical steroids (modified release budesonide) and purine analog antimetabolites as “steroid-sparing agents”.
While mostly inactivated by first-pass metabolism, modified release budesonide has a systemic bioavailability of 12%,68 but this may be higher with mucosal disease.69 Only 30% of the delivered dose is released in the proximal small intestine. Higher doses may therefore work by the systemic route, and it is possible that in CD, where the systemic bioavailability is unknown, the patient may be exposed to a similar magnitude of long-term risks as with oral prednisolone therapy. Subjective clinical measures have improved in both RCD1 and RCD2 with modified release budesonide but without any sign of histological recovery.70,71
Azathioprine is initially combined with steroids due to the delay in onset in effect (when used in inflammatory bowel disease). Early studies showed promising subjective and histological responses in both RCD1 and RCD2,72,73 but a worrying trend to increasing progression to EATL in RCD2.73 Good responses have been reported in RCD1 but not subsequently in RCD2.38 Limiting its use to RCD1, the azathioprine metabolite, thioguanine (tioguanine), achieved an 83% clinical response and 78% histological response, but there was still a considerable degree of corticosteroid dependency.74
One center reporting exceptionally good responses to azathioprine in RCD2 reported no incidence of EATL in their patients >15 years.75 This discrepancy demonstrates the difficulty in diagnosing RCD2 by over-reliance on T-cell clonality as these patients were most likely RCD1 and the responses to Azathioprine therefore in keeping with previous reports.38,74
An alternative approach in RCD2 has been to use the adenosine nucleoside analog cladribine. Promising results have been demonstrated with an 81% response and histological recovery in 47% patients – some with complete mucosal recovery.76,77 However, the proportion of aberrant IELs, while reduced by the treatment, still remained within the diagnostic criteria for RCD2; hence, pending longer term follow-up for these patients, they cannot be considered to have been “cured”.
A variety of other immunosuppressant agents are reported to have been used in small numbers, including infliximab,78–82 campath (anti-CD52),83,84 methotrexate,38cyclosporin A,85 and a trial of immunomodulation with recombinant IL-10.86 Patient numbers are too small and outcomes too variable to consider the routine use of these agents. However, the number of case reports of benefit with infliximab coinciding with the recent findings of TNF-α upregulation in RCD might support a reevaluation of the use of anti-TNF-α monoclonal therapy in RCD1 following failure of steroids and azathioprine.
Hemopoeitic stem cell transplantation
Given that a high proportion of patients with RCD2 progress to EATL (32%–67%),36–38 which in turn carries a dire survival of ∼15% at 2 years,8,9 it is worth considering the possibility of more radical therapies. Autologous hemopoeitic stem cell transplantation (aHSCT) has been used successfully in this setting.87 In the largest report to date, 13 patients received autologous marrow precursors following melphalan and fludarabine conditioning.88 Three died subsequently – one as a result of EATL and the other two most likely as a result of the procedure itself, giving a procedural mortality rate of 15%. Of the 18 patients initially selected on the basis of nonresponse to cladribine, two failed stem cell harvest, possibly as a result of the prior use of cladribine itself and three developed EATL prior to the possibility of transplantation. Overall, excluding those who developed EATL prior to commencement, the intention to treat survival was ten out of 15 (66%), but this is based on a minimum follow-up of 10 months (median >2 years). All the survivors at the time of reporting had an excellent World Health Organization performance status. The possibility of a lead-time bias should be borne in mind as two of the patients who underwent successful transplantation had effectively normal intestinal biopsies at the time (Marsh grades 0 and 1 effectively), which makes it unclear when or whether they would have ever progressed to EATL. Against the apparent benefits of aHSCT must be balanced the subsequent risk of secondary malignancy (including acute myeloid leukemia), which may be as high as 5% at 5 years and plateau at 10% after 10 years, and the risk of solid tumors, which appears to continue beyond 20 years.89 This is clearly dependent on the type of conditioning regimen used, and with milder treatments (that then risk failure of engrafting), there is a lower risk of complications.
Unfortunately, none of the patients undergoing autologous transplantation obliterated their aberrant IEL population and thus remain at risk of further EATL after transplantation. It is well known that IELs are very resistant to chemotherapy and radiotherapy, and therefore, depletion of these cells will require much more intensive conditioning therapy that will significantly increase the risks in the patient. Patients with RCD2 therefore face an uncertain 50% 5-year survival rate with standard therapies, including cladribine, or an equally uncertain 5-year survival rate of <66% following aHSCT, but still with ongoing risks of EATL and additional complications of toxicity of the procedure, including secondary malignancies.
Potential new therapeutic approaches
With greater understanding of the molecular mechanisms of the development of RCD, it is likely that newer immunomodulatory treatments being developed for other inflammatory conditions will become available for use in RCD. However, given the small number of patients with this condition, trials will have to be focused on the most appropriate agent in order to avoid incomplete evidence based only on case reports. There is experimental evidence for the use of anti-IL-15 monoclonal antibody treatment,90 and data from the use of this agent in patients are awaited. However, recent evidence suggesting that certain aberrant IELs in RCD differ with regard to IL-15 receptor expression might also suggest that any effect would be limited.91 Therefore, it is likely that downstream regulation of intracellular signaling pathways in the aberrant IELs will be subjected to suppression in an attempt to change the natural history of the condition. One such agent already in use for rheumatoid arthritis and ulcerative colitis is the JAK3 inhibitor tofacitinib92 that inhibits downstream signaling from the common g-chain receptor component of IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, and it may find value in the setting of RCD2 in combination with other agents.93
Current and emerging strategies in the management of RCD1
There is currently no way of knowing whether patients with RCD1 have ongoing gluten ingestion, a high immunological sensitivity to gluten, or slow response to withdrawal or have already entered a different biological phase of refractoriness. Differential cytokine expression in CD and RCD52 and different epitope recognition patterns by anticalreticulin antibodies in CD and RCD94 hint, however, at a true biological entity, and novel diagnostic tests may become available as a result. Newer methods of detecting gluten ingestion – in stool95 or urine96 – may help to discriminate those with ongoing gluten ingestion.
In keeping with other conditions where chronic inflammation and antigen exposure lead to the development of lymphoproliferative disease,97 all patients with RCD must ensure strict dietary gluten exclusion, even beyond normal measures. Given the current lack of defining characteristics for RCD1, patients should undergo rigorous reappraisal of the original diagnosis and a search for alternative causes of symptoms or histological damage.
Management of RCD1 is largely symptom driven with an emphasis on surveillance in case of development of characteristics defining features of RCD2. Treatment should be initiated with oral prednisolone at 0.5–1 mg/kg/d with bone disease prophylaxis as necessary. Following an initial symptomatic response with steroids, azathioprine can be initiated at 2–2.5 mg/kg/d. Intestinal rebiopsy should be performed after 3 months on azathioprine. Failure to respond symptomatically should once again prompt a reevaluation of the diagnosis of RCD1, and failure to respond to azathioprine warrants measurement of 6-thioguanine and shunt pathway metabolites in order to optimize the dosage. If the diagnosis is not in doubt and the patient remains symptomatic despite the abovementioned measures, a trial of anti-TNF-α monoclonal therapy could be considered.
Patients who respond to treatment should undergo annual endoscopy and biopsy with quantification of aberrant IELs by flow cytometry or detailed immunohistochemistry and TCR clonality studies. Clinicians could consider withdrawing azathioprine after 2–3 years of complete response in order to confirm the diagnosis of RCD1 rather than a slow response to gluten withdrawal (Figure 3).
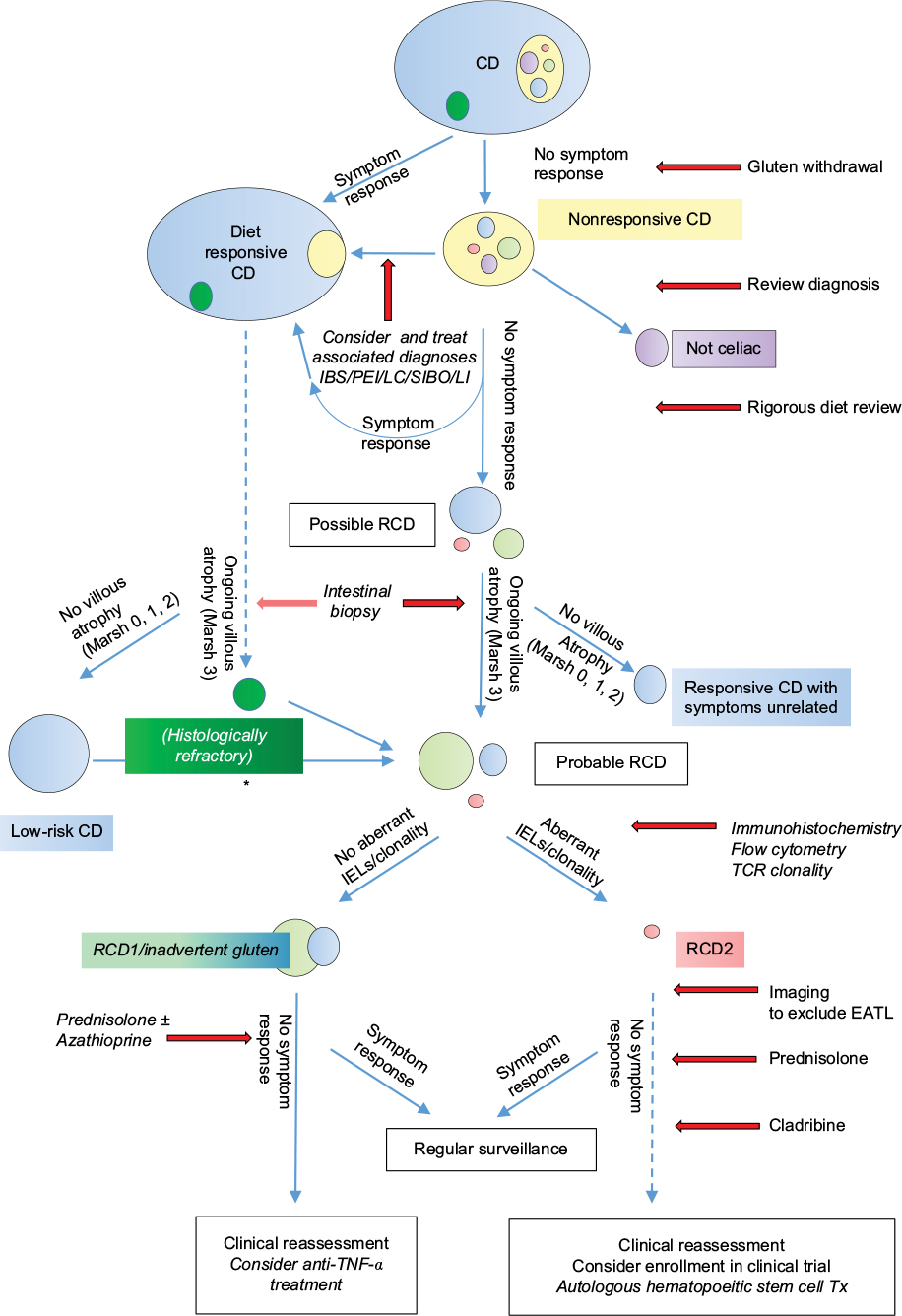 | Figure 3 A flow chart showing suggested pathways for diagnosis and management of RCD. Notes: Dotted lines and italics show pathways that remain unclear or controversial. Arrow marked with * indicates that RCD can develop secondarily after initial response to gluten withdrawal. Abbreviations: RCD, refractory celiac disease; CD, celiac disease; IBS, irritable bowel syndrome; PEI, pancreatic exocrine insufficiency; LC, lymphocytic colitis; SIBO, small intestinal bacterial overgrowth; LI, lactose intolerance; IEL, intraepithelial lymphocyte; TCR, T-cell receptor; EATL, enteropathy-associated T-cell lymphoma; TNF-α, tumor necrosis factor-α; Tx, transplantation. |
Current and emerging strategies in the management of RCD2
Identification of an aberrant IEL phenotype is fundamental to the diagnosis of RCD2. However, the mere presence of TCR clonality or an aberrant IEL phenotype is insufficient by itself and requires clinician integration of evidence from symptoms, histology, flow cytometry, and molecular studies of clonality. Patients mislabeled as RCD2 are liable to considerable harm as a result of Internet search-induced anxiety or toxic overtreatment.
Management of RCD2 is aimed at improving symptoms and removing the aberrant IEL clone – a goal that is at present difficult to achieve. The hope is that directed treatment at an early stage – when the patient is still well – could reduce the devastating risk of subsequent EATL. If this were proven, then potentially toxic therapies would be warranted; however, there is as yet no convincing evidence that the development of EATL can be prevented, and current data suggest that at best it may be delayed.
Following rigorous evaluation for the presence of EATL, initial treatment should be steroid based with subsequent biopsy reevaluation of the diagnosis regardless of the response. Reconfirmation of RCD2 – often with an increase in the number of aberrant IELs – warrants treatment intensification. Given the risk of enhancing progression to EATL with azathioprine, this agent should be avoided. Although based on limited experience, intravenous cladribine could be used at this stage at 0.1 mg/kg/d for 5 days.
Patients who continue to experience a symptomatic decline following cladribine therapy should undergo a reevaluation of their diagnosis, and at this stage, there are limited options available. The evidence in support of aHSCT is limited but remains the only available option for patients who are declining rapidly – patients who reach this stage of their disease should be managed in conjunction with a center collating data and preferably as part of a clinical trial. Better understanding of the nature of RCD2 as a diffuse EATL may lead to resetting thresholds for the use of high-dose chemotherapy in patients without a solid tumor, in such cases with aHSCT as salvage therapy. At present, there is no role for aHSCT in attempting to eradicate the aberrant IEL population in patients with RCD2 who are subjectively well (Figure 3).
Summary
The current state of the art in treating RCD depends on reliable diagnosis, and outcomes in this condition will only be improved when there is consensus and the development of better diagnostic tools that can adequately predict prognosis. Given the rarity of the condition, the development of national centers or databases and international collaborations will be required. Better case finding and management of CD (including follow-up biopsies on diet) may lead in the future to even lower rates of RCD.
Acknowledgment
The author would like to acknowledge Miss Jennifer Stevens for IEL flow cytometry.
Disclosure
The author reports no conflicts of interest in this work.
References
Ludviggson JF, Leffler DA, Bai JA, et al. The Oslo definitions for coeliac disease and related terms. Gut. 2013;62(1):43–52. | ||
Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to glaidin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362(9377):30–37. | ||
Green PH, Cellier C. Celiac disease. N Eng J Med. 2007;357(17):1731–1743. | ||
Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137(6):1912–1933. | ||
Sollid LM, Lundin KE. Diagnosis and treatment of celiac disease. Mucosal Immunol. 2009;2(1):3–7. | ||
Woodward JM. The management of refractory coeliac disease. Ther Adv Chronic Dis. 2013;4(2):77–90. | ||
Walker MM, Woodward J. A clinicopathological approach to the diagnosis of Coeliac disease. Diag Histopath. 2012;18(10):402–410. | ||
Gale J, Simmonds PD, Mead GM, Sweetenham JW, Wright DH. Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol. 2000;18(4):795–803. | ||
Nijeboer B, de Baaij LR, Visser O, et al. Treatment response in enteropathy associated T cell lymphoma – survival in a large multicentre cohort. Am J Hematol. 2015;90(6):493–498. | ||
Lo W, Sano K, Lebwohl B, Diamond B, Green PH. Changing presentation of adult celiac disease. Dig Dis Sci. 2003;48(2):395–398. | ||
Reilly NR, Fasano A, Green PH. Presentation of celiac disease. Gastrointest Endosc Clin N Am. 2012;22(4):613–621. | ||
Sanders DS, Carter MJ, Hurlstone DP, et al. Association of adult coeliac disease with irritable bowel syndrome: a case-control study in patients fulfilling ROME II criteria referred to secondary care. Lancet. 2001;358(9292):1504–1508. | ||
Leeds JS, Hopper AD, Hurlstone DP, et al. Is exocrine pancreatic insufficiency in adult coeliac disease a cause of persisting symptoms? Aliment Pharmacol Ther. 2007;25(3):265–271. | ||
Leffler DA, Dennis M, Hyett B, Kelly E, Schuppan D, Kelly CP. Etiologies and predictors of diagnosis in nonresponsive celiac disease. Clin Gastroenterol Hepatol. 2007;5(4):445–450. | ||
Mooney PD, Evans KE, Singh S, Sanders DS. Treatment failure in coeliac disease: a practical guide to investigation and treatment of non-responsive and refractory coeliac disease. J Gastrointestin Liver Dis. 2012;21(2):197–203. | ||
Sharkey L, Corbett G, Currie E, Lee J, Sweeney N, Woodward J. Optimising delivery of care in coeliac disease: comparison of the benefits of repeat biopsy and serological follow up. Aliment Pharmacol Ther. 2013;38(10):1278–1291. | ||
NICE Guidelines – NG 20 [webpage on the Internet]. Coeliac Disease: Recognition Assessment and Management. 2015. Available from: http://www.nice.org.uk/guidance/ng20. Accessed June 10, 2016. | ||
Abdulkarim AS, Burgart LJ, See J, Murray JA. Etiology of non-responsive coeliac disease: results of a systematic approach. Am J Gastroenterol. 2002;97:2016–2021. | ||
Dewar DH, Donnelly SC, McLaughlin SD, Johnson MW, Ellis HJ, Ciclitira PJ. Celiac disease: management of persistent symptoms in patients on a gluten-free diet. World J Gastroenterol. 2012;18(12):1348–1356. | ||
Trynka G, Hunt KA, Bockett NA, et al. Dense genotyping identifies and localises multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43(12):1193–1201. | ||
Tjon JM, van Bergen J, Koning F. Celiac disease: how complicated can it get? Immunogenetics. 2010;62(10):641–651. | ||
Lebwohl B, Granath A, Ekbom A, et al. Mucosal healing and mortality in coeliac disease. Aliment Pharmacol Ther. 2009;29(12):1299–1308. | ||
Hutchinson JM, West NP, Robins GG, Howdle PD. Long term histological follow-up of people with coeliac disease in a UK teaching hospital. QJM. 2010;103(7):511–517. | ||
Rubio-Tapia A, Rahim MW, See JA, Lahr BD, Wu TT, Murray JA. Mucosal recovery and mortality in adults with celiac disease after treatment with a gluten-free diet. Am J Gastroenterol. 2010;105(6):1412–1420. | ||
Wahab PJ, Meijer JW, Mulder GJ. Histologic follow up of people with celiac disease on a gluten free diet: slow and incomplete recovery. Am J Clin Pathol. 2002;118(3):459–463. | ||
Pekki H, Kurppa K, Mäki M, et al. Predictors and significance of incomplete mucosal recovery in celiac disease after 1 year on a gluten-free diet. Am J Gastroenterol. 2015;110(7):1078–1085. | ||
Picarelli A, Borghini R, Donato G, et al. Weaknesses of histological analysis in celiac disease diagnosis: new possible scenarios. Scand J Gastroenterol. 2014;49(11):1318–1324. | ||
Rubio-Tapia A, Herman ML, Ludvigsson JF, et al. Severe sprue-like enteropathy associated with Olmesartan. Mayo Clin Proc. 2012;87(8):732–738. | ||
Basson M, Mezzarobba M, Weill A, et al. Severe intestinal malabsorption associated with olmesartan: a French nationwide observational cohort study. Gut. Epub 2015 Aug 6. | ||
Woodward JM, Gkrania-Klotsas E, Cordero-Ng AY, et al. The role of chronic norovirus in the enteropathy associated with common variable immunodeficiency. Am J Gastroenterol. 2015;110(2):320–327. | ||
Kaukinen K, Peräaho M, Lindfors K, et al. Persistent small bowel mucosal villous atrophy without symptoms in coeliac disease. Aliment Pharmacol Ther. 2007;25(10):1237–1245. | ||
Cellier C, Patey N, Mauvieux L, et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology. 1998;114(3):471–481. | ||
Badgi E, Diss T, Munson P, Isaacson PG. Mucosal intraepithelial lymphocytes in enteropathy-associated T-cell lymphoma, ulcerative jejunitis and refractory celiac sprue constitute a neoplastic population. Blood. 1999;94(1):260–264. | ||
Alfsen GC, Beiske K, Bell H, Marton PF. Low-grade intestinal lymphoma of intraepithelial T lymphocytes with concomitant enteropathy-associated T cell lymphoma: a case report suggesting a possible histogenetic relationship. Hum Pathol. 1989;20(9):909–913. | ||
Rubio-Tapia A, Murray JA. Classification and management of refractory ceoliac disease. Gut. 2010;59(4):547–557. | ||
Rubio-Tapia A, Kelly DG, Lahr BD, Dogan A, Wu TT, Murray JA. Clinical staging and survival in refractory celiac disease; a single center experience. Gastroenterology. 2009;136(1):99–107. | ||
Al Toma A, Verbeek MH, Hadithi M, von Blomberg BME, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single centre experience. Gut. 2007;56(10):1373–1378. | ||
Malamut G, Afchain P, Verkarre V, et al. Presentation and long term follow up of refractory coeliac disease: comparison of type I with type II. Gastroenterology. 2009;136(1):81–90. | ||
Liu H, Brais R, Lavergne-Slove A, et al. Continual monitoring of intraepithelial lymphocyte immunophentype and clonality is more important than snapshot analysis in the surveillance of refractory celiac disease. Gut. 2010;59(4):452–460. | ||
Prisco A, Troncone R, Mazzarella G, et al. Identical T-cell receptor beta chain rearrangements are present in T cells infiltrating the jejunal mucosa of untreated celiac patients. Hum Immunol. 1997;55(1):22–33. | ||
Ubiali A, Villanacci V, Facchetti F, et al. Is TCR gamma clonality assay useful to detect early celiac disease? J Clin Gastroenterol. 2007;41(3):275–279. | ||
Patey-Mariaud de Serre N, Cellier C, Jabri B, et al. Distinction between coeliac disease and refractory sprue: a simple immunohistochemical method. Histopathology. 2000;37(1):70–77. | ||
Verbeek WH, Goerres MS, von Blumberg BM, et al. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T cell clonality analysis in refractory coeliac disease. Clin Immunol. 2008;126:48–56. | ||
Leon F. Flow cytometry of intestinal intraepithelial lymphocytes in celiac disease. J Immunol Methods. 2011;363(2):177–186. | ||
Malamut G, Cellier C. Is refractory celiac disease more severe in old Europe? Am J Gastroenterol. 2011;106(5):929–932. | ||
Woodward JM. Refractory coeliac disease – rigorous management – revealing or resulting in rarity? Aliment Pharmacol Ther. 2014;39(6):638. | ||
Roshan B, Leffler DA, Jamma S, et al. The incidence and clinical spectrum of refractory celiac disease in a north American referral center. Am J Gastroenterol. 2011;106(5):923–928. | ||
Mubarak A, Oudshoorn JH, Kneepkens CM, Butler JC, Schreurs MW, Mulder CJ. A child with refractory coeliac disease. J Pediatr Gastroenterol Nutr. 2011;53(2):216–218. | ||
Mandal A, Mayberry J. Elemental diet in the treatment of refractory coeliac disease. Eur J Gastroenterol Hepatol. 2001;13(1):79–80. | ||
Olaussen RW, Lovik A, Tollefsen S, et al. Effect of elemental diet on mucosal immunopathology and clinical symptoms in type I refractory Celiac disease. Clin Gastroenterol Hepatol. 2005;3(9):875–885. | ||
Al Toma A, Goerres MS, Meijer JW, Pena AS, Cruius JB, Mulder CJ. Human Leucocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. 2006;4(3):315–319. | ||
Caruso R, Marafini I, Sedda S, et al. Analysis of the cytokine profile in the duodenal mucosa of refractory coeliac disease patients. Clin Sci (Lond). 2014;126(6):451–458. | ||
Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009;2(1):8–23. | ||
Malamut G, El Machhour R, Montcuquet N, et al. IL-15 triggers an anti-apoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. 2010;120(6):2131–2143. | ||
Schmitz F, Kooy-WInkelaar Y, Wiekmeijer AS, et al. The composition and differentiation potential of the duodenal innate lymphocyte compartment is altered in coeliac disease. Gut. Epub 2015 May 12. | ||
Schmitz F, Tjon JM, Lai Y, et al. Identification of a potential physiological precursor of aberrant cells in refractory coelic disease type II. Gut. 2013;62(4):509–519. | ||
Verkarre V, Romana S-P, Cellier C, et al. Recurrent partial trisomy 1g22-q44 in clonal intraepithelial lymphocytes in refractory sprue. Gastroenterology. 2003;125(1):40–46. | ||
Zettl A, Ott G, Makulik A, et al. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am J Pathol. 2002;161(5):1635–1645. | ||
West J, Logan RFA, Hill PG, et al. Seroprevalence, correlates and characteristics of undetected coeliac disease in England. Gut. 2003;52(7):960–965. | ||
Ilus T, Kaukinen K, Virta LJ, et al. Refractory coeliac disease in a country with a high prevalence of clinically-diagnosed coeliac disease. Aliment Pharmacol Ther. 2014;39(4):418–425. | ||
Rubio-Tapia A, Ludvigsson JF, Brantner TL, et al. The prevalence of coeliac disease in the United States. Am J Gastroenterol. 2012;107(10):1538–1544. | ||
Lebwohl B, Granath F, Ekbom A, et al. Mucosal healing and risk for lymphoproliferative malignancy in celiac disease: a population-based cohort study. J Gastroenterol. 2013;48:1324–1331. | ||
Collins P, Rondonotti E, Lundin KE, et al. Video capsule endoscopy in celiac disease: current clinical practice. J Dig Dis. 2012;13(2):94–99. | ||
Hadithi M, Mallant M, Oudejans J, van Waesberghe JH, Mulder CJ, Comans EF. 18F-FDG PET versus CT for the detection of enteropathy-associated T-cell lymphoma in refractory celiac disease. J Nucl Med. 2006;47(10):1622–1627. | ||
Van Weyenberg SJ, Meijerink MR, Jacobs MA, van Kuijk C, Mulder CJ, van Waesberghe JH. MR enteroclysis in refractory celiac disease: proposal and validation of a severity scoring system. Radiology. 2011;259(1):151–161. | ||
Adlersberg D, Colcher H, Drachman SR. Studies on the effects of cortisone and pituitary adrenocorticotropic hormone (ACTH) in the sprue syndrome. Gastroenterology. 1951;19(4):674–697. | ||
Mauriño E, Niveoloni S, Cherñavsky AC, et al. Clinical characteristics and long-term outcome of patients with refractory sprue diagnosed at a single institution. Act Gastroenterol Latinoam. 2006;36(1):10–22. | ||
Edsbäcker S, Larsson P, Wollmer P. Gut delivery of Budesonide, a locally active corticosteroid, from plain and controlled-release capsules. Eur J Gastroenterol Hepatol. 2002;14(12):1357–1362. | ||
Edsbacker S, Bengtsson B, Larsson P, et al. A pharcoscintigraphic evaluation of oral budesonide given as controlled release capsules (Entocort). Aliment Phamacol Ther. 2003;17(4):525–536. | ||
Brar P, Lee S, Lewis S, Egbuna I, Bhagat G, Green PHR. Budesonide in the treatment of refractory celiac disease. Am J Gastroenterol. 2007;102(10):2265–2269. | ||
Daum S, Ipczynski R, Heine B, Schulzke J-D, Zeitz M, Ullrich R. Therapy with Budesonide in patients with refractory sprue. Digestion. 2006;73(1):60–68. | ||
Maurino E, Niveoloni S, Chernavsky A, et al. Azathioprine in refractory sprue: results from a prospective, open label study. Am J Gastroenterol. 2002;97(10):2595–2602. | ||
Goerres MS, Meijer JWR, Wahab PJ, et al. Azathioprine and prednisone combination therapy in refractory coeliac disease. Aliment Pharmacol Ther. 2003;18(5):487–494. | ||
Tack GJ, van Asseldonk DP, van Wanrooij RLJ, van Bodegraven AA, Mulder CJ. Tioguanine in the treatment of refractory celiac disease – a single centre experience. Aliment Pharmacol Ther. 2012;36(3):274–281. | ||
Nasr I, Nasr I, Beyers C, Chang F, Donnelly S, Ciclitira PJ. Recognising and managing refractory coeliac disease: a tertiary centre experience. Nutrients. 2015;7(12):9896–9907. | ||
Al Toma A, Goerres MS, Meijer JW, et al. Cladribine therapy in refractory coeliac disease with aberrant T cells. Clin Gastroenterol Hepatol. 2006;4(11):1322–1327. | ||
Tack GJ, Verbeek WH, Al-Toma A, et al. Evaluation of Cladribine treatment in refractory coeliac disease type II. World J Gastroenterol. 2011;17(4):506–513. | ||
Gillett HR, Arnott ID, McIntyre M, et al. Successful infliximab treatment for steroid-refractory coeliac disease: a case report. Gastroenterology. 2002;122(3):800–805. | ||
Chaudhary R, Ghosh S. Infliximab in refractory celiac disease. Eur J Gastroenterol Hepatol. 2005;17(6):603–604. | ||
Constantino G, della Torre A, Lo Presti MA, Caruso R, Mazzon E, Fries W. Treatment of life-threatening type I refractory coeliac disease with long-term infliximab. Dig Liver Dis. 2008;40(1):74–77. | ||
Schmidt C, Kasim E, Schlake W, Gerken G, Giese T, Stallmach A. TNF-alpha antibody treatment in refractory collagenous sprue: a report of a case and review of the literature. Z Gastroenterol. 2009;47(6):575–578. | ||
Turner SM, Morrghen M, Probert CS. Refractory celiac disease: remission with infliximab and immunomodulators. Eur J Gastroenterol Hepatol. 2005;17(6):667–669. | ||
Lundin KE, Farstad IN, Raki M, Benestad Y, Hoie O, Tjonnfjord G. Alemtuzumab treatment of refractory coeliac disease type II. Gastroenterology. 2006;130(suppl 2):A666. | ||
Vivas S, Ruiz de Morales JM, Ramos F, Suárez-Vilela D. Alemtuzumab for refractory celiac disease in a patient at risk of enteropathy-associated T-cell lymphoma. N Engl J Med. 2006;354(23):2514–2515. | ||
Wahab PJ, Crusius JB, Meijer JW, Uil JJ, Mulder CJ. Cyclosporin in the treatment of adults with refractory coeliac disease – an open pilot study. Aliment Pharmacol Ther. 2000;14(6):767–774. | ||
Mulder CJ, Wahab PJ, Meijer JW, Metselaar E. A pilot study of recombinant human interleukin-10 in adults with refractory coeliac disease. Eur J Gastroenterol Hepatol. 2001;13(10):1183–1188. | ||
Al Toma A, Visser OJ, van Roessel HM, et al. Autologous haemopoeitic stem cell transplantation in refractory celiac disease with aberrant T cells. Blood. 2007;109(5):2243–2249. | ||
Tack GJ, Wondergem MJ, Al Toma A, et al. Auto-SCT in refractory celiac disease type II patients unresponsive to Cladribine therapy. Bone Marrow Transplant. 2011;46(6):840–846. | ||
Burns LJ. Late effects after autologous haemopoeitic cell transplantation. Biol Blood Marrow Transplant. 2009;15(1 suppl):21–24. | ||
Yokoyama S, Watanabe N, Sato N, et al. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci U S A. 2009;106(37):15849–15854. | ||
Van Gils T, Nijeboer P, van Wanrooij RL, Bouma G, Mulder CJJ. Mechanisms and management of refractory coeliac disease. Nat Rev Gastroenterol Hepatol. 2015;12(10):572–579. | ||
Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus Kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616–624. | ||
Nijeboer P, Malamut G, Bouma G, et al. Therapy in RCDII: rationale for combination strategies? Dig Dis. 2015;33(2):227–230. | ||
Sanchez D, Palova-Jelinkova L, Felsberg J, et al. Anticalreticulin immunoglobulin-A (IgA) antibodies in refractory celiac disease. Clin Exp Immunol. 2008;153(3):351–359. | ||
Comino I, Real A, Vivas S, et al. Monitoring of gluten-free diet compliance in celiac patients by assessment of gliadin 33-mer epitope equivalents in faeces. Am J Clin Nutr. 2012;95(3):670–677. | ||
Moreno ML, Cebolla A, Munoz-Suano A, et al. Detection of gluten immunogenic peptides in the urine of patients with coeliac disease reveals transgressions in the gluten-free diet and incomplete mucosal healing Gut. Epub 2015 Nov 25. | ||
Nakamura S, Matsumoto T. Helicobacter pylori and gastric mucosa-associated lymphoid tissue lymphoma. Recent progress in pathogenesis and management. World J Gastroenterol. 2013;19(45):8181–8187. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.