Back to Journals » Drug Design, Development and Therapy » Volume 9
Improving dissolution and oral bioavailability of pranlukast hemihydrate by particle surface modification with surfactants and homogenization
Authors Ha E, Baek I, Yoo J ![]() , Jung Y, Kim M
, Jung Y, Kim M
Received 1 May 2015
Accepted for publication 28 May 2015
Published 24 June 2015 Volume 2015:9 Pages 3257—3266
DOI https://doi.org/10.2147/DDDT.S87738
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Eun-Sol Ha,1 In-hwan Baek,2 Jin-Wook Yoo,1 Yunjin Jung,1 Min-Soo Kim1
1College of Pharmacy, Pusan National University, 2College of Pharmacy, Kyungsung University, Busan, Republic of Korea
Abstract: The present study was carried out to develop an oral formulation of pranlukast hemihydrate with improved dissolution and oral bioavailability using a surface-modified microparticle. Based on solubility measurements, surface-modified pranlukast hemihydrate microparticles were manufactured using the spray-drying method with hydroxypropylmethyl cellulose, sucrose laurate, and water and without the use of an organic solvent. The hydrophilicity of the surface-modified pranlukast hemihydrate microparticle increased, leading to enhanced dissolution and oral bioavailability of pranlukast hemihydrate without a change in crystallinity. The surface-modified microparticles with an hydroxypropylmethyl cellulose/sucrose laurate ratio of 1:2 showed rapid dissolution of up to 85% within 30 minutes in dissolution medium (pH 6.8) and oral bioavailability higher than that of the commercial product, with approximately 2.5-fold and 3.9-fold increases in area under the curve (AUC0→12 h) and peak plasma concentration, respectively. Therefore, the surface-modified microparticle is an effective oral drug delivery system for the poorly water-soluble therapeutic pranlukast hemihydrate.
Keywords: solubility, wettability, sucrose laurate, cellulose
Introduction
Pranlukast hemihydrate, N-[4-oxo-2-(1H-tetrazol-5-yl)-4H-chromen-7-yl]-4-(4-phenylbutoxy)benzamide 1/2H2O, is a selective, competitive antagonist of the cysteinyl leukotriene receptors, inhibiting binding at the cysteinyl leukotriene type 1 receptor. Pranlukast hemihydrate is insoluble in water (1.03 μg/mL at 37°C) and is formulated as a hard gelatin capsule in its commercial dosage form, which is used to treat chronic bronchial asthma and to prevent exercise-induced asthma.1–3 The absolute bioavailability of pranlukast is approximately 4.3% after oral administration to rats.4 A minimum of 12.5% of an oral dose of pranlukast administered after ingestion of food is absorbed, as indicated by the ratio of urinary and fecal excretion of the drug in healthy volunteers.1 To improve the biological performance of pranlukast hemihydrate, various formulations, such as microsuspensions, nanosuspensions, and solid dispersions, have been studied.5–9
Amorphous solid dispersions are widely used in formulations of poorly water-soluble active pharmaceutical ingredients (APIs). Generally, APIs exist in an amorphous form within solid dispersions, resulting in increased oral bioavailability of APIs via increased solubility and dissolution.10–13 Solid dispersions can be manufactured by solvent evaporation of organic solutions prepared by dissolving APIs and carriers in organic solvents such as methanol, ethanol, acetone, and methylene chloride, owing to the poor water solubility of APIs. Recently, novel solid dispersion systems for APIs have been reported, including surface-attached and surface-modified solid dispersions, which do not utilize organic solvents with potential toxicity and exhibit dramatically improved solubility, dissolution, and oral bioavailability.14–17
The present study was conducted to develop an oral formulation to improve the dissolution and oral bioavailability of pranlukast hemihydrate by incorporating it into a surface-modified microparticles. The solubility of pranlukast hemihydrate in different excipient aqueous solutions was analyzed to select the best hydrophilic polymer and surfactant. In this study, the surface-modified pranlukast hemihydrate microparticle was prepared by the spray-drying method with hydroxypropylmethyl cellulose (HPMC), sucrose laurate, and water and without the use of an organic solvent. The physicochemical properties of the surface-modified pranlukast hemihydrate microparticle were characterized using scanning electron microscopy, powder X-ray diffraction, a particle size analyzer, and contact angle determination. In addition, the dissolution and oral bioavailability of the surface-modified pranlukast hemihydrate microparticle in rats was evaluated in comparison with a commercial pranlukast hemihydrate product.
Materials and methods
Pranlukast hemihydrate was kindly donated by Yuhan Pharm Co Ltd. (Yongin, Korea). Hydroxypropyl cellulose and HPMC (2910) were purchased from Nippon Soda Co (Tokyo, Japan) and Shin-Etsu Chemical Co Ltd (Tokyo, Japan), respectively. Polyvinylpyrrolidone K30, polyvinylpyrrolidone vinyl acetate VA64, poloxamer 188, poloxamer 407, and solutol HS15 were obtained from BASF Co Ltd (Ludwigshafen, Germany). d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS, Eastman Co, Kingsport, TN, USA), Gelucire 44/14 (Gattefossè, Saint-Priest, France), sucrose laurate (Ryoto sugar ester L1695, Mitsubishi-Kagaku Foods Co, Tokyo, Japan), and sodium lauryl sulfate (Sigma-Aldrich Co Ltd, St Louis, MO, USA) were used as surfactants. Montelukast was used as an internal standard (Sigma-Aldrich). The commercial pranlukast hemihydrate product containing 112.5 mg of pranlukast hemihydrate (Onon® capsule, Dong-A ST, Seoul, Korea) was purchased from a pharmacy. Acetonitrile, ethanol, and methanol were of high-performance liquid chromatography (HPLC) grade.
Measurement of pranlukast hemihydrate solubility in various excipients
To select the best hydrophilic polymer and surfactant (Table 1), the solubility of pranlukast hemihydrate was measured in various aqueous solutions containing the excipients at 1% and 5% concentrations (w/v) for the polymer and surfactant, respectively. Approximately, 50 mg of pranlukast hemihydrate was added to 15 mL of aqueous solution containing each excipient. The suspensions were sonicated for 2 hours in an ultrasonic bath (model 5800, Branson, Danbury, CT, USA). The samples were allowed to equilibrate by incubating them in a shaking water bath (BS-21, Jeiotech Co Ltd, Daejeon, Korea) at 25°C for 24 hours. Preliminary experiments showed that 24 hours was sufficient to achieve equilibrium solubility with pranlukast hemihydrate. After 24 hours, each sample was filtered using a 0.45 μm polytetrafluoroethylene syringe filter and transferred to a volumetric flask. Subsequently, the samples were diluted with the mobile phase. The concentration of pranlukast hemihydrate was assessed by HPLC analysis using a Waters HPLC system (Milford, MA, USA). The HPLC apparatus consisted of a pump (model 600), a column oven (steel column heater 600), an autosampler (model 717 plus), an ultraviolet detector (model 486 tunable absorbance detector), and a chromatographic data processor (Empower Pro). Separations were performed on a Capcell Pak C18 MG (4.6 mm ×250 mm, 5 μm) reversed-phase column (Shiseido Fine Chemicals, Tokyo, Japan) eluted under isocratic conditions with a mobile phase consisting of 20 mM aqueous potassium dihydrogen phosphate-acetonitrile-methanol (30:58:12 v/v/v). The injection volume was 20 μL. Chromatography was performed at 30°C at a flow rate of 1.0 mL per minute. An ultraviolet detector was used at a wavelength of 262 nm.
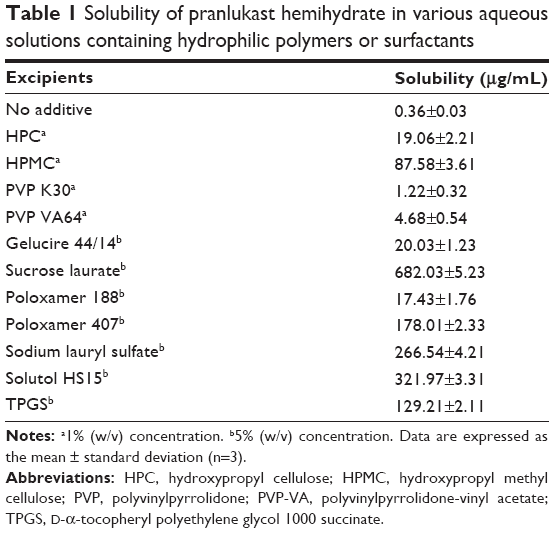 | Table 1 Solubility of pranlukast hemihydrate in various aqueous solutions containing hydrophilic polymers or surfactants |
Preparation of surface-modified pranlukast hemihydrate microparticles
Based on the results of the solubility study, HPMC and sucrose laurate were used as the hydrophilic polymer and surfactant, respectively. First, various amounts of HPMC and sucrose laurate were dissolved in water and 5 g of pranlukast hemihydrate was dispersed in 100 g of each solution using a mechanical overhead stirrer (RW16 basic, IKA, Staufen, Germany). Each suspension was further homogenized using an Ultra-Turrax® T18 (IKA) at 20,000 rpm for 10 minutes. The resulting suspension was spray-dried through a two-fluid pressure nozzle attached to a peristaltic pump using a Buchi 191 nozzle type mini-spray dryer (Buchi, Flawil, Switzerland). During processing, the spraying rate (5–10 mL per minute) and inlet air temperature (115°C–140°C) were adjusted to maintain the outlet temperature between 77°C and 84°C. The spray air pressure was 5 kPa. The flow of the feed was concurrent with the direction of the inlet drying air.
Characterization of the surface-modified pranlukast hemihydrate microparticles
Particle size analysis
The shape and morphological characteristics of the surface-modified pranlukast hemihydrate microparticles were analyzed by scanning electron microscopy (JSM-7100F, JEOL Ltd, Tokyo, Japan) at 5.0 kV. The particle size distributions of the dry powders of the surface-modified pranlukast hemihydrate microparticles were measured based on the Fraunhofer theory with a Helos laser diffraction spectrometer and a Rodos dry dispersing unit (Sympatec Ltd, Clausthal-Zellerfeld, Germany). The particle size distribution was measured using an R2 lens with a measuring range of 0.25–87.5 μm. A RODOS 4 mm disperser at 5.0 bar pressure was used. Measurements were made in triplicate.
Powder X-ray diffraction
Powder X-ray diffractograms were obtained using a D/max-IIIC X-ray powder diffractometer (Rigaku, Tokyo, Japan). All samples were exposed to Cu-Kα radiation (40 kV, 45 mA). The samples were analyzed between 5° and 60° 2θ with a step width of 0.01° at a scanning speed of 3° per minute.
Contact angle
To quantify the surface wettability of the surface-modified pranlukast hemihydrate microparticles, the water contact angle of sessile drops was measured at room temperature and in ambient air conditions using a Krüss EasyDrop goniometer (Krüss, Hamburg, Germany). For the measurements, discs of the surface-modified pranlukast hemihydrate microparticles were prepared by compressing 100 mg of powder in an hydraulic press (PerkinElmer, Waltham, MA, USA). Sessile drops comprising 3 μL distilled water were released onto the surface of the discs through a 22-gauge flat tip needle and observed through a stereo microscope and charge-coupled device camera. Contact angles were determined using Drop Shape analysis software (Krüss, Hamburg, Germany). Advancing contact angles were measured for 1 minute after addition of the droplets. Contact angles were expressed as the average of six measurements at different places.
In vitro dissolution test
The dissolution studies were conducted in a United States Pharmacopeia dissolution apparatus II (paddle method, Electrolab, Mumbai, India). The dissolution medium was maintained at 37°C±0.5°C and stirred at 50 rpm by means of an adjusted constant speed motor. Surface-modified pranlukast hemihydrate microparticles equivalent to 112.5 mg of pranlukast hemihydrate were introduced into the dissolution vessel and the time was recorded (time 0). Samples (5 mL) were withdrawn at different time intervals and replaced with fresh dissolution medium maintained at 37°C±0.5°C to maintain a constant volume. The samples were immediately filtered through a 0.45 μm polytetrafluoroethylene syringe filter and diluted with the mobile phase. The concentration of pranlukast hemihydrate was assessed by HPLC analysis. Dissolution tests were also performed for the raw material and the commercial pranlukast hemihydrate product. Dissolution media at pH 1.2 (HCl and NaCl), pH 4.0 (acetate buffer), and pH 6.8 (phosphate buffer) were used for the dissolution tests, which were performed in quadruplicate for each formulation.
Pharmacokinetic study
The animal study protocol was in compliance with the institutional guidelines for the care and use of laboratory animals and approved by the ethics committee of Kyungsung University (Busan, Korea). Ten male Sprague-Dawley rats (250±10 g, Orient Bio Inc, Seongnam, Korea) were divided into two treatment groups of five rats each. Prior to the study, the rats were fasted for 18 hours. Each group received either the surface-modified pranlukast hemihydrate microparticles or the commercial pranlukast hemihydrate product at pranlukast hemihydrate doses of 40 mg/kg by oral administration. The surface-modified pranlukast hemihydrate microparticles and the commercial pranlukast hemihydrate product were dispersed in 1 mL of water immediately prior to oral dosing. Blood samples (approximately 0.25 mL) were collected in heparinized tubes from the retro-orbital plexus of the treated rats 0.25, 0.5, 1, 1.5, 2, 4, 8, and 12 hours after dosing. The blood samples were centrifuged at 10,000 rpm for 10 minutes at 4°C. The resulting plasma was transferred to individual Eppendorf tubes and stored at −70°C. Plasma concentrations of pranlukast hemihydrate were determined by liquid chromatography with mass spectrometry according to methods previously reported by our group.5 Pranlukast hemihydrate and montelukast (internal standard) were extracted from the plasma matrix by liquid-liquid extraction using ethyl acetate and methanol (2:8, v/v). Pharmacokinetic analysis of the data was carried out with WinNonlin Standard Edition software, version 5.3 (Pharsight Corporation, St Louis, MO, USA). The area under the curve (AUC0→12 h) was calculated according to the trapezoidal method. The peak plasma concentration (Cmax) and the time to reach Cmax (Tmax) of pranlukast hemihydrate in the plasma were taken directly from the data. Statistical analysis of the dissolution data and pharmacokinetic parameters was performed using t-tests or one-way analysis of variance followed by the Student-Newman-Keuls and least squares difference tests. The analyses were performed with Statistical Package for the Social Sciences version 21.0 software (IBM Corporation, Armonk, NY, USA).
Results and discussion
In this study, surface-modified pranlukast hemihydrate microparticles were developed using the spray-drying method with a hydrophilic polymer, a surfactant, and water with the goal of improving the dissolution and oral bioavailability of pranlukast hemihydrate. First, to select the best hydrophilic polymer and surfactant for our purpose, the solubility of pranlukast hemihydrate was analyzed in different excipient aqueous solutions. As presented in Table 1, the solubility of pranlukast hemihydrate in water without an excipient was approximately 0.36 μg/mL at 25°C, indicating poor water solubility, while its water solubility significantly increased upon addition of the hydrophilic polymer and surfactant. Among the tested surfactant aqueous solutions, pranlukast hemihydrate exhibited the greatest solubility (682.03 μg/mL) in the sucrose laurate solution, which was approximately 1,900-fold greater than its solubility in water without an excipient. Among the hydrophilic polymers tested, pranlukast hemihydrate (87.58 μg/mL) exhibited the greatest solubility in the 1% (w/w) aqueous solution of HPMC. Therefore, HPMC and sucrose laurate were selected as the hydrophilic polymer and surfactant, respectively, for the development of the surface-modified pranlukast hemihydrate microparticles.
The surface-modified pranlukast hemihydrate microparticle was easily manufactured by the spray-drying method with HPMC, sucrose laurate, and water and without the use of an organic solvent. Under scanning electron microscopy (Figure 1), raw pranlukast hemihydrate powder appears as needle-shaped crystals with a smooth surface. All of the surface-modified microparticles were present as relatively rough-surfaced agglomerates with needle-like crystals and irregular particles with a similar mean particle size (6–8 μm), indicating that HPMC and sucrose laurate were attached to the surface of the pranlukast hemihydrate crystal. There were no significant differences between the surface-modified microparticles (P>0.05), as shown in Table 2. HPLC analysis showed that the drug content of each surface-modified microparticle was almost equal to the theoretical value, indicating that pranlukast hemihydrate was not degraded during the spray-drying process. As depicted in Figure 2, the crystalline state of pranlukast hemihydrate within the surface-modified microparticles was determined using a powder X-ray diffractometer. Raw pranlukast hemihydrate had characteristic diffraction patterns at 2θ values of 9.8°, 14.4°, 16.6°, and 19.9°, corresponding to a highly crystalline state. All major characteristic crystalline peaks of pranlukast hemihydrate were observed in all of the surface-modified microparticles, indicating that pranlukast hemihydrate existed in an unchanged crystalline state in the surface-modified microparticles with HPMC and sucrose laurate. Wetting is the first process in dissolution of a solid oral dosage form and the degree of wetting (wettability) is an important predictor of the dissolution of poorly water-soluble APIs.18,19 Wettability is usually determined by the contact angle.20 Theoretically, a contact angle of 0° indicated complete wetting, while a contact angle of 180° indicates no wetting. As shown in Table 2, raw pranlukast hemihydrate showed a contact angle greater than 73° due to its hydrophobic nature. However, the contact angle significantly decreased owing to surface modification with HPMC and sucrose laurate. The contact angle of the surface-modified pranlukast hemihydrate microparticles (F6 and F7) was found to be about 40°, demonstrating the improved wettability of pranlukast hemihydrate in the surface-modified microparticles. Therefore, our results suggest that pranlukast hemihydrate was present in a crystalline state in the surface-modified microparticles with enhanced wettability due to the attachment of HPMC and sucrose laurate to the surface of the poorly water-soluble pranlukast hemihydrate crystal.
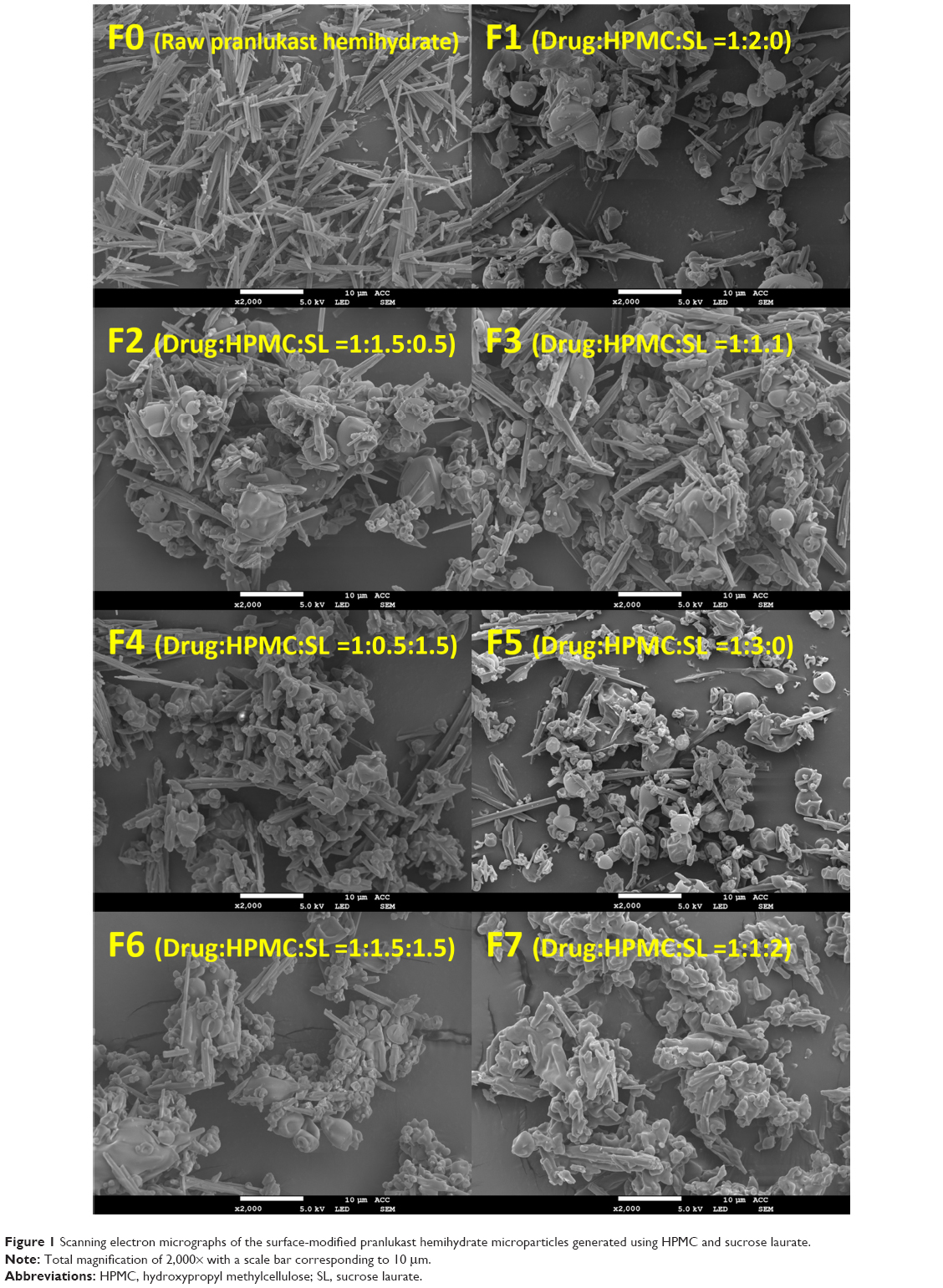 | Figure 1 Scanning electron micrographs of the surface-modified pranlukast hemihydrate microparticles generated using HPMC and sucrose laurate. |
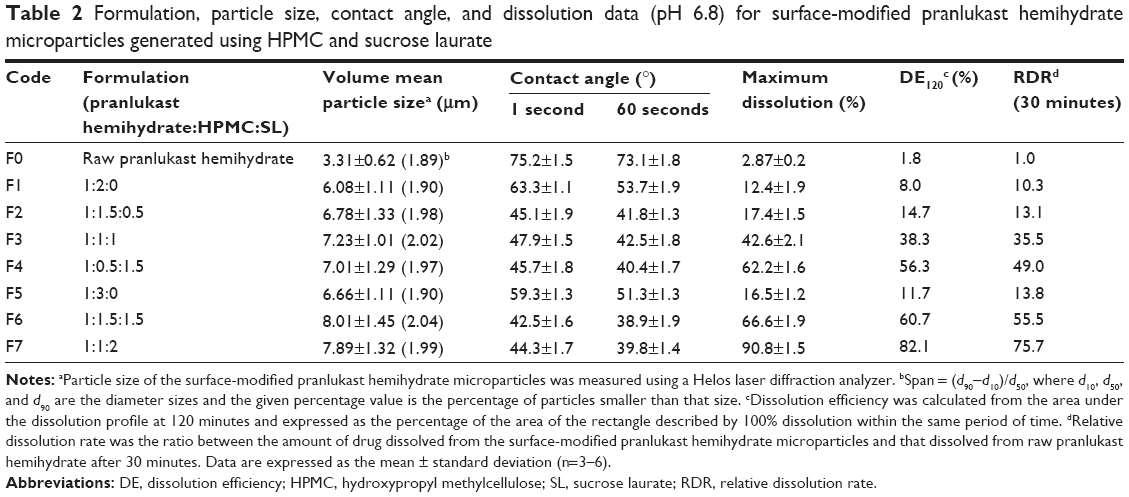 | Table 2 Formulation, particle size, contact angle, and dissolution data (pH 6.8) for surface-modified pranlukast hemihydrate microparticles generated using HPMC and sucrose laurate |
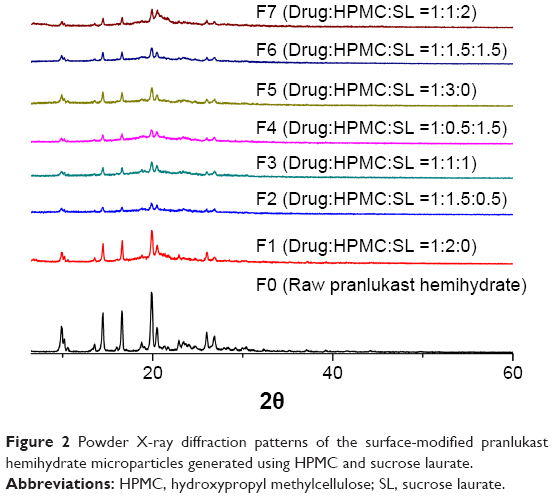 | Figure 2 Powder X-ray diffraction patterns of the surface-modified pranlukast hemihydrate microparticles generated using HPMC and sucrose laurate. |
To select an optimal formulation, the influence of the amounts of HPMC and sucrose laurate included in the formulation on the dissolution of pranlukast hemihydrate was investigated. The dissolution tests were performed by the paddle method in the pH 1.2 and pH 6.8 dissolution media under non-sink conditions. Dissolution profiles were obtained for raw pranlukast hemihydrate powder and the surface-modified pranlukast hemihydrate microparticles (Figure 3). The dissolution data obtained in the pH 6.8 dissolution medium were analyzed with respect to maximum drug dissolved (%), dissolution efficiency (DE120), and relative dissolution rate at 30 minutes (Table 2). The DE120 values for pure pranlukast hemihydrate and surface-modified pranlukast hemihydrate microparticles were calculated as defined by Khan and Rhodes.21 As shown in Figure 3, the extent of dissolution and rate of raw pranlukast hemihydrate powder were extremely low in the pH 1.2 and pH 6.8 dissolution media due to the poor water solubility and wettability of pranlukast hemihydrate. In the pH 1.2 dissolution medium, the amount of pranlukast hemihydrate dissolution from the surface-modified microparticles with HPMC and sucrose laurate was very low for all compositions, perhaps due to the acidity of pranlukast hemihydrate. Pranlukast hemihydrate contains a tetrazoleheterocylce group (estimated pKa approximately 5), which may influence its solubility in a pH-dependent manner, resulting in extremely low dissolution at acidic pH in comparison with other pH values.22 Furthermore, the solubility of pranlukast hemihydrate was less than 10 μg/mL in the pH 6.8 dissolution medium; however, the dissolution rate and extent of dissolution of the surface-modified pranlukast hemihydrate microparticles with HPMC and sucrose laurate significantly increased in the pH 6.8 dissolution medium. In particular, in terms of DE120 and relative dissolution rate at 30 minutes, the extent of dissolution of pranlukast hemihydrate from the surface-modified microparticles with an HPMC/sucrose laurate ratio of 1:2 (F7) was approximately 45.5-fold and 75.7-fold greater, respectively, than that of raw pranlukast hemihydrate powder. This result may be due to the improved wettability of the surface-modified microparticles and solubilization of HPMC and sucrose laurate. Furthermore, the rate of dissolution and extent of pranlukast hemihydrate were more dependent on the amount of sucrose laurate than on the amount of HPMC. As expected, the rate of dissolution and extent increased with the amount of sucrose laurate in terms of maximum drug dissolved (%), relative dissolution rate at 30 minutes, and DE120. The release of pranlukast hemihydrate from the surface-modified microparticles with an HPMC/sucrose laurate ratio of 1:2 (F7) had a maximum extent of dissolution of more than 85% within 30 minutes and had the greatest extent and rate of dissolution among the formulations tested. Therefore, the F7 microparticle formulation was considered the most suitable composition for surface-modified pranlukast hemihydrate microparticles synthesized with HPMC and sucrose laurate.
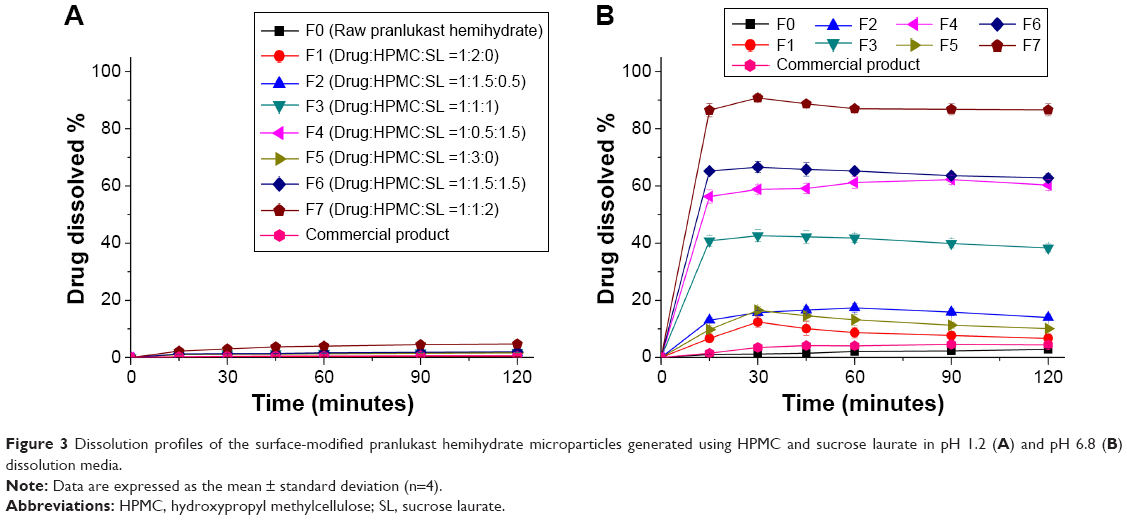 | Figure 3 Dissolution profiles of the surface-modified pranlukast hemihydrate microparticles generated using HPMC and sucrose laurate in pH 1.2 (A) and pH 6.8 (B) dissolution media. |
Dissolution of the surface-modified pranlukast hemihydrate microparticles (F7) was evaluated in three different dissolution media to allow comparison with a commercial pranlukast hemihydrate product. In the pH 1.2 and pH 4.0 dissolution media, the rate and extent of dissolution of pranlukast hemihydrate from the surface-modified microparticles (F7) was increased compared with that from the commercial product (Figure 4). In particular, dissolution of pranlukast hemihydrate dramatically increased from the surface-modified microparticles (F7) compared with that from the commercial product in the pH 6.8 dissolution medium. After 120 minutes in the pH 6.8 dissolution medium, about 90.8% of the drug in the form of the surface-modified microparticles (F7) dissolved, while only approximately 4% of the commercial product dissolved. Therefore, the surface-modified microparticle formulation generated with HPMC and sucrose laurate significantly enhanced dissolution of the poorly water-soluble drug pranlukast hemihydrate.
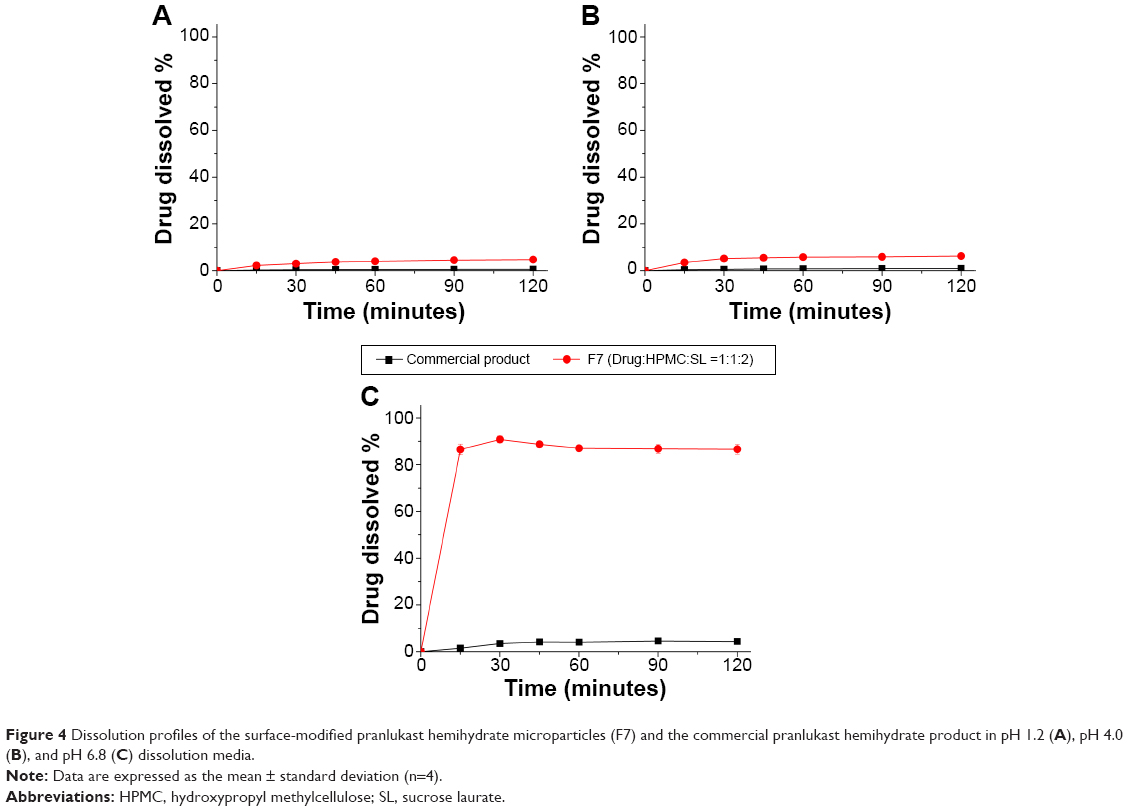 | Figure 4 Dissolution profiles of the surface-modified pranlukast hemihydrate microparticles (F7) and the commercial pranlukast hemihydrate product in pH 1.2 (A), pH 4.0 (B), and pH 6.8 (C) dissolution media. |
The enhanced dissolution of pranlukast hemihydrate from the surface-modified microparticles generated with HPMC and sucrose laurate could directly enhance the oral bioavailability of pranlukast hemihydrate. Therefore, we investigated the oral bioavailability of pranlukast hemihydrate following administration of the surface-modified pranlukast hemihydrate microparticles in Sprague-Dawley rats. Figure 5 shows the plasma concentration-time profiles for pranlukast hemihydrate after oral administration of the surface-modified microparticles (F7) or the commercial product. The pharmacokinetic parameters (AUC0→12 h, Cmax, and Tmax) of the surface-modified microparticles (F7) and the commercial product are presented in Table 3. As shown in Figure 6, the plasma concentration of the surface-modified microparticles (F7), which showed a rapid drug absorption rate, was dramatically higher than that of the commercial product. The oral absorption of the surface-modified microparticles (F7) was significantly greater than that of the commercial product, with approximately 2.5-fold and 3.9-fold increases in AUC0→12 h, and Cmax, respectively (Table 3). These results suggest that the dissolution and oral absorption of pranlukast hemihydrate significantly increased upon incorporation into the surface-modified microparticles generated with HPMC and sucrose laurate. In general, poorly water-soluble APIs exist in an amorphous form within composite particles, such as in solid dispersion systems prepared by drying organic solutions containing an API, a hydrophilic polymer, and a surfactant, dramatically decreasing the crystallinity of such APIs.23 However, in this study, we prepared surface-modified pranlukast hemihydrate microparticles by attaching HPMC and sucrose laurate to the surface of poorly water-soluble crystalline pranlukast hemihydrate, without the use of a potentially toxic organic solvent. The hydrophilicity of pranlukast hemihydrate increased upon incorporation into the surface-modified microparticles, leading to enhanced dissolution and oral bioavailability without changes in crystallinity. In addition, the pranlukast hemihydrate content within the surface-modified microparticles (F7) was 99.5%±1.7% after 1 year at 25°C±2°C and 60%±3% relative humidity, and there were no significant changes compared with the drug content of the initial sample (100.1%±1.3%, P>0.05). As shown in Figure 6, the dissolution profiles of the surface-modified microparticles (F7) were similar on day 0 and after 1 year. Therefore, the surface-modified microparticle (F7) is stable for at least 1 year under long-term condition.
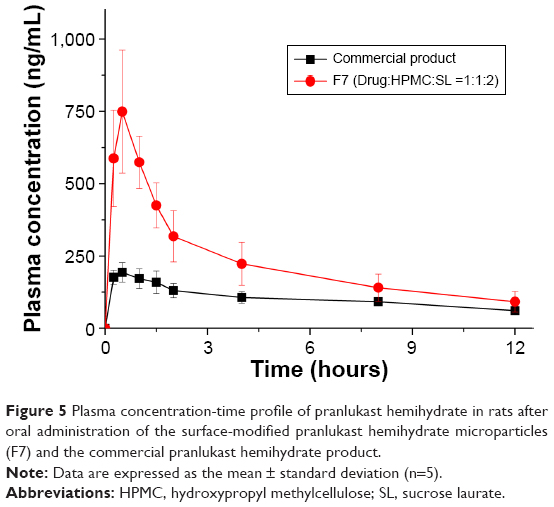 | Figure 5 Plasma concentration-time profile of pranlukast hemihydrate in rats after oral administration of the surface-modified pranlukast hemihydrate microparticles (F7) and the commercial pranlukast hemihydrate product. |
 | Table 3 Pharmacokinetic parameters for the commercial pranlukast hemihydrate product and the surface-modified pranlukast hemihydrate microparticles generated using HPMC and sucrose laurate (F7) |
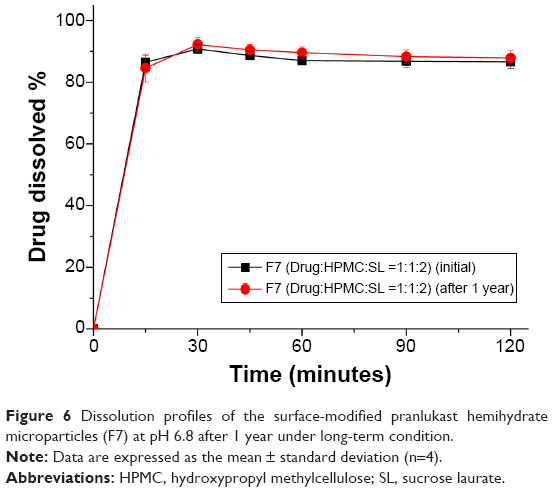 | Figure 6 Dissolution profiles of the surface-modified pranlukast hemihydrate microparticles (F7) at pH 6.8 after 1 year under long-term condition. |
Conclusion
The present study was carried out to develop an oral formulation to improve the dissolution and oral bioavailability of pranlukast hemihydrate using surface-modified microparticles. Based on solubility measurements, surface-modified pranlukast hemihydrate microparticles were developed using the spray-drying method with HPMC, sucrose laurate, and water, without the use of an organic solvent. The surface-modified microparticles with an HPMC/sucrose laurate ratio of 1:2 showed rapid dissolution of up to 85% within 30 minutes in pH 6.8 dissolution medium, as well as oral bioavailability higher than that of the commercial product, with increases of approximately 2.5-fold and 3.9-fold in AUC0→12 h and Cmax, respectively. Taken together, these results demonstrate that surface-modified microparticles generated using HPMC, sucrose laurate, and water represent an effective oral drug delivery system for the poorly water-soluble therapeutic pranlukast hemihydrate.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant funded by the Korea government (2009-0083538).
Disclosure
The authors report no conflicts of interest in this work.
References
Keam SJ, Lyseng-Williamson KA, Goa KL. Pranlukast: a review of its use in the management of asthma. Drugs. 2003;63(10):991–1019. | ||
Wang L, Hao Y, Liu N, Ma M, Yin Z, Zhang X. Enhance the dissolution rate and oral bioavailability of pranlukast by preparing nanosuspensions with high-pressure homogenizing method. Drug Dev Ind Pharm. 2012;38(11):1381–1389. | ||
Kataoka M, Sugano K, da Costa Mathews C, et al. Application of dissolution/permeation system for evaluation of formulation effect on oral absorption of poorly water-soluble drugs in drug development. Pharm Res. 2012;29(6):1485–1494. | ||
Chono S, Takeda E, Seki T, Morimoto K. Enhancement of the dissolution rate and gastrointestinal absorption of pranlukast as a model poorly water-soluble drug by grinding with gelatin. Int J Pharm. 2008;347(1–2):71–78. | ||
Baek IH, Kim JS, Ha ES, et al. Dissolution and oral absorption of pranlukast nanosuspensions stabilized by hydroxypropylmethyl cellulose. Int J Biol Macromol. 2014;67:53–57. | ||
Baek MK, Lee JH, Cho YH, Kim HH, Lee GW. Self-microemulsifying drug-delivery system for improved oral bioavailability of pranlukast hemihydrate: preparation and evaluation. Int J Nanomedicine. 2013;8:167–176. | ||
Uchiyama H, Tozuka Y, Asamoto F, Takeuchi H. α-Glucosyl hesperidin induced an improvement in the bioavailability of pranlukast hemihydrate using high-pressure homogenization. Int J Pharm. 2011;410(1–2):114–117. | ||
Wongmekiat A, Tozuka Y, Moribe K, Oguchi T, Yamamoto K. Preparation of drug nanoparticles by co-grinding with cyclodextrin: formation mechanism and factors affecting nanoparticle formation. Chem Pharm Bull. 2007;55(3):359–363. | ||
Wongmekiat A, Tozuka Y, Oguchi T, Yamamoto K. Formation of fine drug particles by cogrinding with cyclodextrins. I. The use of beta-cyclodextrin anhydrate and hydrate. Pharm Res. 2002;19(12):1867–1872. | ||
de la Torre-Iglesias PM, García-Rodriguez JJ, Torrado G, Torrado S, Torrado-Santiago S, Bolás-Fernández F. Enhanced bioavailability and anthelmintic efficacy of mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des Devel Ther. 2014;8:1467–1479. | ||
Kim MS, Kim JS, Cho W, et al. Supersaturatable formulations for the enhanced oral absorption of sirolimus. Int J Pharm. 2013;445(1–2):108–116. | ||
Ha ES, Baek IH, Cho W, Hwang SJ, Kim MS. Preparation and evaluation of solid dispersion of atorvastatin calcium with Soluplus® by spray drying technique. Chem Pharm Bull. 2014;62(6):545–551. | ||
Kim MS, Baek IH. Fabrication and evaluation of valsartan-polymer-surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine. 2014;9:5167–5176. | ||
Lee SN, Poudel BK, Tran TH, et al. A novel surface-attached carvedilol solid dispersion with enhanced solubility and dissolution. Arch Pharm Res. 2013;36(1):79–85. | ||
Tran TH, Poudel BK, Marasini N, et al. Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Int J Pharm. 2013;443(1–2):50–57. | ||
Yan YD, Sung JH, Kim KK, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422(1–2):202–210. | ||
Ha ES, Ok J, Noh J, et al. Fabrication and evaluation of celecoxib microparticle surface modified by hydrophilic cellulose and surfactant. Int J Biol Macromol. 2015;72:1473–1478. | ||
Lu Y, Tang N, Lian R, Qi J, Wu W. Understanding the relationship between wettability and dissolution of solid dispersion. Int J Pharm. 2014;465(1–2):25–31. | ||
Dahlberg C, Millqvist-Fureby A, Schuleit M. Surface composition and contact angle relationships for differently prepared solid dispersions. Eur J Pharm Biopharm. 2008;70(2):478–485. | ||
Kim MS, Kim JS, Hwang SJ. Enhancement of wettability and dissolution properties of cilostazol using the supercritical antisolvent process: effect of various additives. Chem Pharm Bull. 2010;58(2):230–233. | ||
Khan KA, Rhodes CT. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm Acta Helv. 1972;47(10):594–607. | ||
Welz R, Müller S. 5-(Benzylmercapto)-1H-tetrazole as activator for 2′-O-TBDMS phosphoramidite building blocks in RNA synthesis. Tetrahedron Lett. 2002;43(5):795–797. | ||
Choi JH, Lee K, Hong S, et al. Effect of biocompatible polymers on the physicochemical and dissolution properties of fenofibrate in nanoparticle system. J Pharm Invest. 2013;43(6):507–512. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.