Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Improving aqueous solubility and antitumor effects by nanosized gambogic acid-mPEG2000 micelles
Authors Cai L, Qiu N, Xiang M, Tong R, Yan J, He L, Shi J, Chen T, Wen J, Wang W, Chen L
Received 5 September 2013
Accepted for publication 3 November 2013
Published 27 December 2013 Volume 2014:9(1) Pages 243—255
DOI https://doi.org/10.2147/IJN.S54050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Lulu Cai,1,* Neng Qiu,2,* Mingli Xiang,3,* Rongsheng Tong,1 Junfeng Yan,1 Lin He,1 Jianyou Shi,1 Tao Chen,4 Jiaolin Wen,3 Wenwen Wang,3 Lijuan Chen3
1Department of Pharmacy, Sichuan Academy of Medical Science and Sichuan Provincial People's Hospital, 2College of Materials and Chemistry and Chemical Engineering, Chengdu University of Technology, 3State Key Laboratory of Biotherapy, West China Hospital, West China Medical School, Sichuan University, Chengdu, People's Republic of China; 4Faculty of Pharmacy, University of Montreal, Montreal, QC, Canada
*These authors contributed equally to this paper
Abstract: The clinical application of gambogic acid, a natural component with promising antitumor activity, is limited due to its extremely poor aqueous solubility, short half-life in blood, and severe systemic toxicity. To solve these problems, an amphiphilic polymer-drug conjugate was prepared by attachment of low molecular weight (ie, 2 kDa) methoxy poly(ethylene glycol) methyl ether (mPEG) to gambogic acid (GA-mPEG2000) through an ester linkage and characterized by 1H nuclear magnetic resonance. The GA-mPEG2000 conjugates self-assembled to form nanosized micelles, with mean diameters of less than 50 nm, and a very narrow particle size distribution. The properties of the GA-mPEG2000 micelles, including morphology, stability, molecular modeling, and drug release profile, were evaluated. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide) tests demonstrated that the GA-mPEG2000 micelle formulation had obvious cytotoxicity to tumor cells and human umbilical vein endothelial cells. Further, GA-mPEG2000 micelles were effective in inhibiting tumor growth and prolonged survival in subcutaneous B16-F10 and C26 tumor models. Our findings suggest that GA-mPEG2000 micelles may have promising applications in tumor therapy.
Keywords: gambogic acid, poly(ethylene glycol)-drug conjugate, micelle, antitumor, toxicity
Introduction
Gambogic acid, the major active ingredient of gamboge, a brownish to orange resin exuded from the Garcinia hanburyi tree in Southeast Asia, possesses significant anticancer activity both in vitro and in vivo.1–4 Multiple mechanisms may be involved in its potent antitumor effect, including induction of apoptosis, cell cycle arrest, telomerase inhibition, antiangiogenesis activity, and an antimetastasis effect.5–10 Unfortunately, gambogic acid has poor solubility (less than 0.5 μg/mL), a short half-life (less than 1 hour in dogs and less than 20 minutes in rats), and low bioavailability, which limits its application.11,12 Solubilizers such as Cremophor® EL (polyethoxylated castor oil, Sigma-Aldrich, St Louis, MO, USA) and L-arginine have been introduced to solve these problems.11–14 However, these agents may cause a series of side effects, such as vascular stimulation, hemolytic toxicity, hypersensitivity reactions, nephrotoxicity, neurotoxicity, and cardiotoxicity.15 Further, rapid plasma elimination of gambogic acid cannot be avoided by these formulations.12,16 These defects greatly limit the therapeutic effect of gambogic acid. Therefore, it is necessary to develop structural modifications or a new dosage form of gambogic acid.
Conjugation of hydrophilic polymers with small molecule drugs to produce polymer-drug conjugate systems has been demonstrated to be a viable formulation strategy for utilizing hydrophobic drugs in a water-soluble manner, which may offer advantages over the corresponding parent drugs, including fewer side effects, improved solubility, passive tumor targeting, an improved pharmacokinetic profile, and lower plasma concentrations. This passive tumor targeting is achieved primarily through the enhanced permeability and retention effect. In past decades, conjugation of hydrophilic polymers with hydrophobic anticancer drugs (such as paclitaxel, doxorubicin, camptothecin, and curcumin) to produce polymer-drug conjugate systems has been proven to be a viable formulation strategy.17–25 A large number of hydrophilic polymers could be chosen as water-soluble macromolecule carriers; among them, poly(ethylene glycol) (PEG) is considered to be one of the best candidates. Several chemotherapeutic agents have been conjugated to PEG with covalent bonds, including a PEG-curcumin conjugate, a PEG-paclitaxel conjugate, and a PEG-camptothecin conjugate, resulting in homogeneous water-soluble prodrugs with an extended circulatory life and altered biodistribution.26–29 Tang et al30 reported that using PEG as a water-solubilizing unit was a useful strategy for increasing the water solubility of gambogic acid, which is hydrophobic. Ding et al31 synthesized a series of gambogic acid-PEG conjugates with different amino acid and dipeptide spacers, which showed satisfactory water solubility compared with gambogic acid, and the circulatory retention time, biodistribution, and bioavailability of the conjugates were remarkably improved. However, the bioactivity and toxicity of gambogic acid-PEG conjugates in vivo have not been reported thus far.
On the basis of these studies, we developed a nanosized drug delivery system with the aims of increasing the aqueous solubility, improving the therapeutic efficacy, and reducing the toxicity of gambogic acid to normal tissues. In this study, an amphiphilic polymer-drug conjugate was prepared by condensation of low molecular weight monomethoxy-poly(ethylene glycol) (mPEG)-2000 with gambogic acid (GA-mPEG2000) through an ester linkage, which then formed micelles. The physicochemical properties of the formulations were evaluated, and their in vitro cytotoxicity, in vivo antitumor effect, and toxicity were also investigated.
Materials and methods
Materials
Gamboge resin was obtained from Chengdu Herb Market (Sichuan, People’s Republic of China). mPEG (average molecular weight 2,000) and Dulbecco’s Modified Eagle’s Medium were purchased from Sigma-Aldrich. Gambogic acid was prepared and purified in our laboratory. Acetonitrile of high-performance liquid chromatography (HPLC) grade was purchased from Fisher Scientific (Loughborough, UK). Chloroform, methanol (analytical grade), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), and N-hydroxysuccinimide were purchased from Chengdu Kelong Chemical Co, Ltd (Chengdu, People’s Republic of China). Other reagents were of analytical grade. Deionized water (>18 MU, Purelab®; Classic Components Corporation, Torrance, CA, USA) was used in all experiments.
Cells and animals
Murine colon adenocarcinoma cells (C26) and murine melanoma cells (B16-F10) were purchased from the American Type Culture Collection (Rockville, MD, USA). The cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (Sigma-Aldrich). Primary human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cord veins and grown in EBM-2 (endothelial basal medium-2) with Single Quots (Lonza, Walkersville, MD, USA) containing vascular endothelial growth factor and other growth factors. HUVECs at passages 2–4 were used for all experiments. The above cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Balb/c and C57BL/6J mice (age 6–8 weeks, weight 18–22 g) were used for the in vivo antitumor tests and toxicity studies. The animals were purchased from the Laboratory Animal Center of Sichuan University, and housed in sex-segregated cages at a controlled temperature of 20°C–22°C, a relative humidity of 50%–60%, and 12-hour light-dark cycles. The animals were provided with standard laboratory chow and tap water ad libitum. After a 1-week acclimation period, the animals were used for the experiments. All animal procedures were performed following the protocol approved by the Institutional Animal Care and Treatment Committee of Sichuan University (Chengdu, People’s Republic of China). All mice were treated humanely throughout the experimental period.
Measurements
1H nuclear magnetic resonance (NMR) spectra were determined on an Avance 400 (Bruker Corporation, Ettlingen, Germany) spectrometer (400 mHz) using deuterated chloroform as the solvent. Ultraviolet-visible spectra were recorded on a UV-2401 PC spectrophotometer (Shimadzu, Tokyo, Japan). HPLC detection was done on a 2996 detector (Waters Corporation, Milford, MA, USA). Chromatographic separations were performed on a reversed phase C18 column (4.6 mm × 150 mm, 5 μm, SunFire; Waters Corporation). The column temperature was kept at 28°C, and the detection wavelength was 292 nm. Acetonitrile/0.1% formic acid (80/20, v/v) solution was used as the mobile phase, and the flow rate was 1 mL per minute.
General synthesis method for GA-mPEG2000 conjugates
Isolation and purification of gambogic acid
Fifty grams of dry gamboge resin was suspended in 150 mL of pyridine and stirred at 80°C–90°C for 30 minutes to form a pyridine salt of gambogic acid. After filtering through Kieselguhr (Sigma-Aldrich), 20 mL of water was added to the filtrate. The mixture was cooled to 4°C overnight, and the precipitate was collected and washed with pyridine solution (70%, v/v) and water several times. After drying under vacuum, the resulting yellow powder was dissolved in 250 mL of ethyl ether and heated under reflux for 30 minutes, then filtered, concentrated, and precipitated using petroleum ether; the yellow precipitate of the pyridine salt of gambogic acid was then collected and dried. The solid obtained was dissolved in 120 mL of ethyl ether and washed with aqueous HCl (1 M) and water. The ether solution was then dried over sodium sulfate and evaporated to yield an orange powder of 3.8 g gambogic acid (purity 98.6%, w/w, analyzed by HPLC).
Synthesis of GA-mPEG2000
Gambogic acid (345 mg, 0.55 mmol), EDC (191.7 mg, 1 mmol), and 4-dimethylaminopyridine (61 mg, 0.5 mmol) were dissolved in 10 mL of CH2Cl2 in an ice water bath, and mPEG2000 (1 g, 0.5 mmol) was then added. The resulting mixture was stirred overnight at room temperature. The solution was washed in 1 M HCl (3 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL), dried over Mg2SO4, and concentrated in vacuo. The precipitate was suspended in 10 mL of ethyl ether under stirring for 30 minutes to remove the free gambogic acid. The solution was filtered and the yellow precipitate was obtained. The crude product was further purified by silica gel column chromatography using a step gradient of methanol (2%–5%) in CH2Cl2 to remove the free mPEG2000, giving rise to GA-mPEG2000 (747 mg, 57%) as a buff-colored solid. A single spot was visualized with iodine vapor by thin liquid chromatography analysis (dichloromethane to methanol, 10:1). The 1H NMR spectrum of GA-mPEG2000 was studied using an Avance 400 spectrometer (400 mHz), and CH3Cl was used as the solvent.
Preparation and characterization of GA-mPEG2000 micelles
GA-mPEG2000 micelles were prepared using a direct dissolution method assisted by ultrasonication. GA-mPEG2000 (84 mg) was dissolved in 10 mL of distilled water and sonicated at 25°C for 1 minute. The control sample was prepared by dissolving 22 mg of L-arginine solubilizer and 20 mg of gambogic acid (GA-L) in 10 mL of distilled water and then sonicating the sample at 25°C for 3 minutes. The final concentrations of GA-mPEG2000 in the micelles and gambogic acid in the GA-L solution were determined by HPLC assay. A direct observation method was used to evaluate the solubility of the GA-mPEG2000 conjugates, and the equivalent solubility value was calculated depending on the measured solubility.
The particle size and zeta potential of the micelles were determined by dynamic light scattering using a Zetasizer Nano ZS-90 instrument (Malvern Instruments, Malvern, UK). The sample suspension (gambogic acid concentration 2 mg/mL) was diluted ten times with deionized water before measurement. The refractive index was 1.330. The temperature was kept at 25°C during the measuring process. All tests were run in triplicate, and mean values are reported.
The morphology of the prepared micelles was observed by transmission electron microscopy (TEM) and atomic force microscopy (AFM). For TEM, the samples were diluted with distilled water and placed on a copper grid covered with nitrocellulose. The samples were negatively stained with phosphotungstic acid and dried at room temperature, after which images were taken using a TEM device (H-6009IV; Hitachi, Japan). For AFM, the prepared micelle suspension was diluted with deionized water and deposited onto freshly cleaved mica lamella. The sample was dried for 3 hours at room temperature. AFM images were taken by tapping mode in air on an AFM device (SPA400; Seiko, Tokyo, Japan).
After preparation, the micelles were kept at 4°C and 25°C, respectively, and the stability of the formulations was evaluated qualitatively by observation of aggregates and particle size measurement. A homogeneous solution implies stability of the drug system, whereas the presence of precipitation or marked changes of particle size indicates instability.
Molecular modeling study
Construction of three-dimensional GA-mPEG2000 structures
First, gambogic acid was built with MarvinSketch (http://www.chemaxon.com) and optimized at the molecular mechanical level using the MMFF94 method.32 Meanwhile, mPEG2000 was constructed using Hyperchem33 software (Hypercube Inc., Gainesville, FL, USA). The gambogic acid and mPEG2000 structures were then merged together in the Hyperchem workspace (Figure 1A). GA-mPEG2000 was then optimized at the molecular mechanical level using the orthogonal partial least squares34 method with the steepest descent algorithm. It was further optimized at the same level with the molecular mechanical plus method35 using the Fletcher-Reeves36 algorithm.
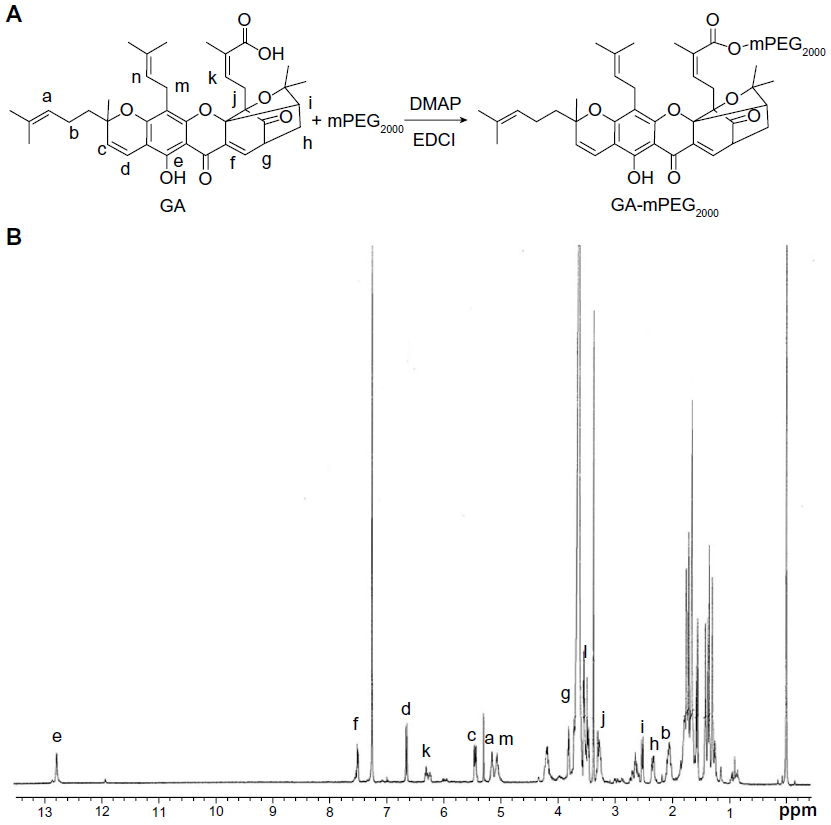 | Figure 1 Synthesis and identification of GA-mPEG2000 copolymers. (A) Chemical reaction scheme for preparing the GA-mPEG2000 conjugate. (B) 1H nuclear magnetic resonance spectrum of GA-mPEG2000 in CD3Cl. |
Next, the optimized GA-mPEG2000 was subjected to a series of molecular dynamics simulations so as to obtain a lower energy minimum. The solvent effect was implicitly considered using the CHARMM2737,38 force field in the process of molecular dynamics simulation. The molecular dynamics simulations included three stages: heating from 0 K to 400 K, simulating at 400 K, cooling from 400 K to 300 K, and running at 300 K. At each stage, the simulated time is set to 100 picoseconds.
Simulation on self-assembly of GA-mPEG2000
Two GA-mPEG2000 molecules, the structures of which had been annealed using molecular dynamics, were first merged together. Two kinds of conformations were purposefully arranged. In the first conformation, the gambogic acid moieties of the two molecules were arranged face-to-face. In the second conformation, they were arranged back-to-back. They were then simulated using Langevin dynamics. The friction coefficient and random seed were set as 0.05 per picosecond and –1,111, respectively. The solvation effect was treated as mentioned above. Simulation times for the first conformation and the second conformation were set to 100 picoseconds and 300 picoseconds, respectively.
In vitro drug release
Release of gambogic acid from the GA-L formulation and GA-mPEG2000 micelles in vitro was monitored by a dialysis method. Dialysis was carried out at 37°C using Spectra/Por dialysis membranes (Spectrum Laboratories, Inc, Rancho Dominguez, CA, USA) with a molecular weight cutoff of 1 kDa and phosphate-buffered saline (pH 7.4) as the sink solution. The molecular weight cutoff of the dialysis membrane only allows for diffusion of the free drug. The initial concentration of gambogic acid in the GA-L formulations was 20 mg/mL and the initial concentration of GA-mPEG2000 in the micelles was 84 mg/mL (gambogic acid equivalent concentration, 20 mg/mL). At scheduled intervals, 1 mL of the dialysis medium was collected and the same volume of fresh medium was added immediately. The concentration of gambogic acid in the dialysis medium was monitored by HPLC assay. The percentage release was calculated according to the following equation:
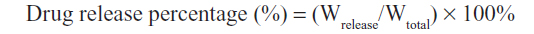
where Wtotal is the total amount of gambogic acid in the GA-L formulation or micelles and Wrelease is the amount of gambogic acid released from the GA-L formulation or GA-mPEG2000 micelles into the dialysis medium.
Cytotoxicity assay
The in vitro cytotoxicity of the conjugates was quantified by measuring the IC50 (drug concentration inhibiting 50% of cells) in a murine melanoma cell line (B16-F10), a colon carcinoma cell line (C26), and HUVECs. B16-F10 and C26 cells were cultured in Dulbecco’s Modified Eagle’s Medium containing 10% heat-inactivated fetal calf serum at a density of 1 × 105 cells per 180 μL per well in 96-well microtiter plates and allowed to proliferate at 37°C in a humidified incubator with 5% CO2 for 24 hours. HUVEC cells were cultured in Dulbecco’s Modified Eagle’s Medium containing low serum growth supplement, 100 IU/mL penicillin, and 100 μg/mL streptomycin at a density of 1 × 105 cells per 180 μL per well in 96-well microtiter plates and allowed to proliferate at 37°C in a humidified incubator with 5% CO2 for 24 hours. Next, 20 μL serial dilutions of gambogic acid and GA-mPEG2000 (final equivalent gambogic acid concentration, 0.1–3.2 mg/mL) were added to the respective wells, and all samples were prepared and measured in quintuplicate for each concentration. After 48 hours of incubation, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT, 5 mg/mL per well) was added and the plates were incubated for a further 4 hours. The supernatant was then removed, followed by addition of 150 μL of dimethylsulfoxide to each well. Fifteen minutes later, the absorbance at 570 nm was detected using a microplate reader and the IC50 values for each compound were calculated from absorbance versus dilution factor plots.
In vivo tumor model and treatment plan
The antitumor activity of GA-mPEG2000 micelles was investigated in the B16-F10 and C26 models. In the B16-F10 model, C57BL/6J mice were injected subcutaneously with 50 μL of cell suspension containing 5 × 105 B16-F10 cells on day 0. In the C26 model, Balb/c mice were injected subcutaneously into the right flank with 50 μL of C26 cell suspension containing 2 × 106 cells at day 0. Once tumors reached 150–250 mm3 in volume, the mice were randomized into three groups (ten mice per group). Tumor-bearing mice were injected intravenously every 2 days for 2 weeks with 100 mL of normal saline (control), GA-L (4 mg/kg bodyweight), or GA-mPEG2000 micelles (16.8 mg/kg body weight, gambogic acid 4 mg/kg equivalent), respectively. Tumor measurements were performed (according to the formula 0.52 × length × width2) and the mice were weighed and examined for antitumor activity every two (B16-F10 model) or three (C26 model) days. Mouse survival times were observed to document further the antitumor activity and toxicity of GA-L and GA-mPEG2000 micelles.
Statistical analysis
The data are expressed as the mean and standard deviation. Statistical differences were determined using the Student’s t-test. Differences were considered to be statistically significant at P<0.05.
Results
Synthesis and identification of GA-mPEG2000
The synthesis of GA-mPEG2000 was according to the references in the literature with some modifications.30,39 Hydroxy-terminated mPEG2000 was directly attached to gambogic acid at the 30-carboxy group. The main advantage of coupling one gambogic acid molecule at one end of mPEG2000 is the high selectivity of the only one hydroxyl in mPEG2000, which results in high yield and purity of GA-mPEG2000. The mPEG2000 was reacted with excess gambogic acid to guarantee that mPEG2000 was totally reacted. However, there was still some free mPEG2000 observed with iodine vapor by thin layer chromatography (dichloromethane to methanol, 10:1). The crude product was firstly purified in ethyl ether to remove free gambogic acid and then purified by gradient elution column chromatography.
Figure 1A shows the synthesis procedure for GA-mPEG2000. The 1H NMR spectrum of GA-mPEG2000 demonstrates successful synthesis of GA-mPEG2000. Figure 1B shows a multiplet at δ3.58–3.72 (ppm) that was attributed to the repeating units in mPEG2000, and the peaks at 3.38 ppm were assigned to the three methyl protons in mPEG2000. Protons of gambogic acid can be found at 12.78 ppm (6-H), 7.50 ppm (10-H), 6.65 ppm (4-H), 6.33–6.23 ppm (27-H), 5.48–5.42 ppm (3-H), 5.16–5.07 ppm (32-H, 37-H), 3.82 ppm (11-H), 3.58–3.54 ppm (31a-H), 3.50–3.46 ppm (31b-H), 3.30–3.26 ppm (26-H), 2.52 ppm (22-H), 2.32 ppm (21a-H), and 2.09–2.02 ppm (36-H), and all of the other multiple signals at 1.78–1.30 came from the protons in gambogic acid. These results demonstrate further that the conjugate was successfully prepared.
Characterization of GA-mPEG2000 micelles
Solubility of GA-mPEG2000 conjugates
The appearance of the prepared GA-mPEG2000 micelles is shown in Figure 2. Gambogic acid formed a turbid yellow suspension in water, indicating that gambogic acid could not be dissolved in aqueous solution (Figure 2A), whereas a clear and transparent solution of GA-mPEG2000 micelles could be observed (Figure 2B), indicating its good solubility in water. Compared with the extremely poor solubility of gambogic acid (0.5 μg/mL), GA-mPEG2000 exhibited satisfactory aqueous solubility, being 2.7 × 105-fold (135.8 mg/mL) that of gambogic acid.
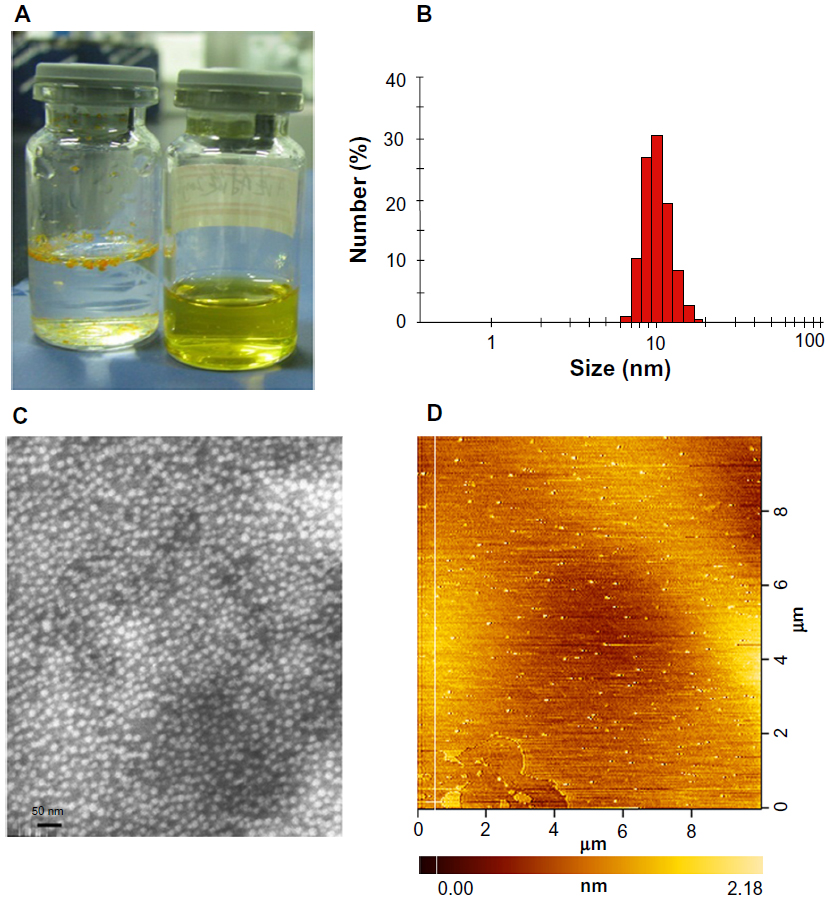 | Figure 2 Preparation and characterization of GA-mPEG2000 micelles. (A) Appearance of gambogic acid in various dosage forms (left) mixture of gambogic acid and water, (right) GA-mPEG2000 micelles. (B) Size distribution of GA-mPEG2000 micelles. (C) Transmission electron microscopic images of typical GA-mPEG2000 micelles. (D) Atomic force microscopic images of typical GA-mPEG2000 micelles. |
Particle size and morphology of GA-mPEG2000 micelles
The GA-mPEG2000 micelles were suitable for an injectable formulation with a small particle size. The mean (± standard deviation) vesicle diameter of the micelles was 9.41 ± 2.91 nm (n=3) as determined by dynamic light scattering, and the polydispersity index (PDI) was 0.193 ± 0.072 (Figure 2B). Further, AFM and TEM analyses confirmed that the micelles were spheroids with a regular shape and a size distribution of <50 nm (Figure 2C–D). The diameter of the micelles observed by AFM and TEM agreed with the results of the particle size analysis. These results demonstrate that the GA-mPEG2000 micelles were uniformly dispersed and had a very narrow particle size distribution.
Stability of GA-L and GA-mPEG2000 micelles
From observation of the aggregates, we found that the GA-L solution became turbid after 3 days and yellow precipitation could be observed after 5 days, indicating that the GA-L solution was not a stable formulation, whereas the GA-mPEG2000 micelle solution remained clear and transparent for 30 days. From particle size measurement of the GA-mPEG2000 micelles, we found that the average size and PDI of the micelles did not change significantly over 30 days (Figure 3). These results indicate that the GA-mPEG2000 micelle formulation was a homogeneous and stable drug system.
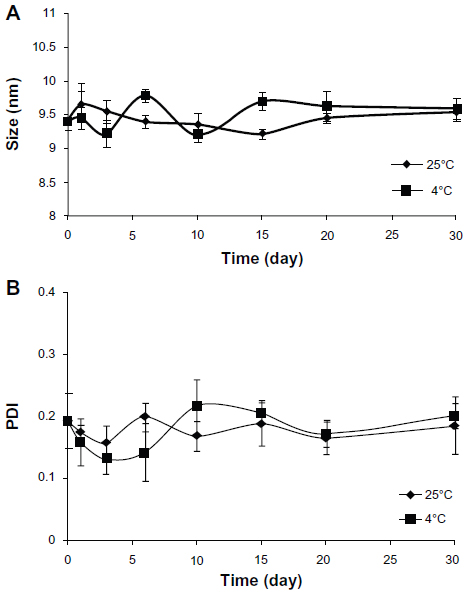 | Figure 3 Stability of GA-mPEG2000 micelles. (A) Average size of GA-mPEG2000 micelles stored at 25°C and 4°C. (B) PDI of GA-mPEG2000 micelles stored at 25°C and 4°C. |
Molecular modeling study
Three-dimensional structure of GA-mPEG2000
As demonstrated in Figure 4A, the initial conformation of GA-mPEG2000 was a curve-like mPEG2000 covalently linking with gambogic acid. Kinetic simulations were done to find an energy minimum for GA-mPEG2000. At the heating stage, GA-mPEG2000 was heated from 0 K to 400 K with a temperature step of 30 K. It can be seen from Figure 4A that the mPEG2000 moiety of GA-mPEG2000 changes its shape first and then gradually entwines around gambogic acid.
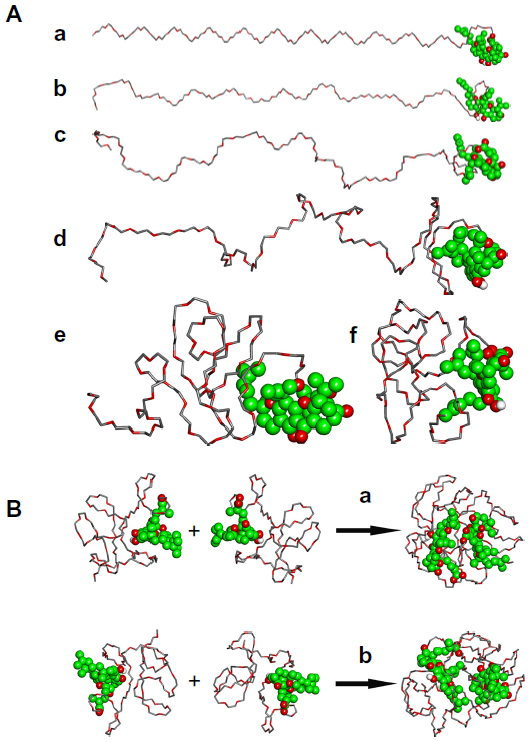 | Figure 4 Molecular modeling studies of GA-mPEG2000 micelles. (A) Conformations of GA-mPEG after being heated for 0 (a), 9.875 (b), 20.010 (c), 30.082 (d), 39.925 (e), and 100 (f) picoseconds, respectively. (B) Interaction between GA-mPEG2000 molecules resulting from face-to-face GA-GA initial conformation (a) and back-to-back GA-GA conformation (b). GA was depicted with Corey-Pauling-Koltun space filling style, its carbon atoms were colored with green, and its oxygen atoms were colored with red. |
It must be a reasonable phenomenon due to the fact that the GA-mPEG2000 is in a polar solvent environment. As mentioned above, the solvation (water) effect was considered implicitly in the process of molecular dynamics simulation, because we know that gambogic acid is hydrophobic. The GA-mPEG2000 tries to increase its interaction with the polar solvent environment by reducing the hydrophobic moiety on its exposing surface. When mPEG2000 changes its shape and entwines around gambogic acid, the exposed hydrophobic area is reduced effectively. The equilibrated structure for GA-mPEG2000 is shown in Figure 4A(f).
Simulation on the self-assembly of GA-mPEG2000
In order to understand in detail the mechanism of interaction between GA-mPEG2000 molecules, Langevin dynamics simulations were conducted on clusters composed of two GA-mPEG2000 molecules. When two GA-mPEG2000 molecules were arranged so that the gambogic acid parts of the molecules were face-to-face (as shown in Figure 4B[a]), the gambogic acid moieties approached each other after 100 picoseconds of Langevin dynamics simulation. When two GA-mPEG2000 molecules were arranged so that the gambogic acid moieties were back-to-back (as shown in Figure 4B[b]), the gambogic acid parts of the two molecules were finally close to each other after 300 picoseconds of Langevin dynamics simulation. These results suggest that, in a polar solvent environment, the gambogic acid moieties of GA-mPEG2000 tend to move closer and form the core of the micelle, whereas the mPEG2000 moieties tend to stay in the external area and form the shell of the micelle, agreeing with the data in Figure 2.
In vitro release profile
A modified dialysis method was used to investigate the in vitro drug release behavior of the GA-L and GA-mPEG2000 micelles. The molecular weight cutoff of the dialysis membrane only allows for diffusion of free gambogic acid. As shown in Figure 5, the GA-mPEG2000 micelles showed a much slower cumulative release rate compared with the rapid release profile for GA-L. Approximately 69% of the gambogic acid was released into the medium in the GA-L group within 6 hours, whereas only 24% of gambogic acid was released from the micelles. Over a 2-week period, the mean cumulative release rate in the GA-mPEG2000 micelle group was much lower than that in the GA-L group (58.30% ± 4.28% versus 89.37% ± 7.43%, respectively). The in vitro drug release data show that gambogic acid could be released from the micelles in a sustained manner for an extended period.
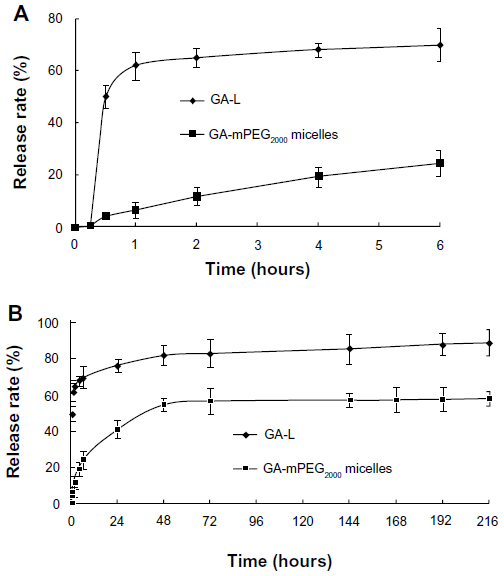 | Figure 5 Time course of GA release from the GA-L formulation (t) and GA-mPEG2000 micelles (n) at 37°C and pH 7.4 in 6 hours (A) and 216 hours (B). |
Cell cytotoxicity assay
The in vitro biological efficacy of the GA-mPEG2000 micelles was evaluated against B16-F10, C26, and HUVEC cells using the MTT method. The data in Table 1 show that both GA-L and the micelles were cytotoxic to tumor cells and HUVEC cells. Compared with GA-L, the IC50 values for the GA-mPEG2000 micelles were 2.7–3.5 times greater than those for gambogic acid.
 | Table 1 Half maximal inhibitory concentration of different types of cells after incubation with the different drugs for 48 hours |
Antitumor effects and toxicity of GA-L and GA-mPEG2000 in vivo
Mice subcutaneously injected with B16-F10 melanoma cells or C26 colon carcinoma cells were used to compare the antitumor activity of GA-L with that of GA-mPEG2000 micelles. In both models (Figures 6 and 7), tumor growth rates in mice treated with GA-L or GA-mPEG2000 micelles were markedly delayed, and GA-mPEG2000 micelles were more efficient than GA-L in suppressing growth of tumors. On day 20 after treatment, the T/C, defined as the ratio of the mean tumor volume in the treated group to that in the control group treated with normal saline, was 0.52 (P<0.05) in mice treated with GA-L and 0.39 (P<0.05) in mice treated with GA-mPEG2000 micelles (Figure 6B). Similar results were observed in C26 tumor-bearing mice (Figure 7B); on day 22 after treatment, the T/C value was 0.35 (P<0.05) in mice treated with GA-L and 0.20 (P<0.01) in mice treated with GA-mPEG2000 micelles. In addition, in both tumor models, tumor weights in the GA-mPEG2000 micelle group after the final treatment were markedly lower than in the free drug and normal saline control group (Figures 6C and Figures 7C). In the B16-F10 tumor-bearing mouse model (Figure 6C), mean tumor weights in the GA-mPEG2000 micelle group (2.07 ± 1.24 g) decreased by 20% and 55% (P<0.05) compared with the GA-L group (2.6 ± 0.24 g) and normal saline control group (4.51 ± 0.65 g), respectively. In the C26 tumor-bearing mouse model (Figure 7C), mean tumor weights in the GA-mPEG2000 micelle group (0.504 ± 0.42 g) decreased by 58% and 83% (P<0.05) compared with the GA-L group (1.2 ± 0.72 g) and normal saline control group (2.922 ± 0.83 g), respectively. These results suggest that the GA-mPEG2000 micelles enhanced the antitumor effects of gambogic acid alone, and markedly delayed tumor growth rates in mice.
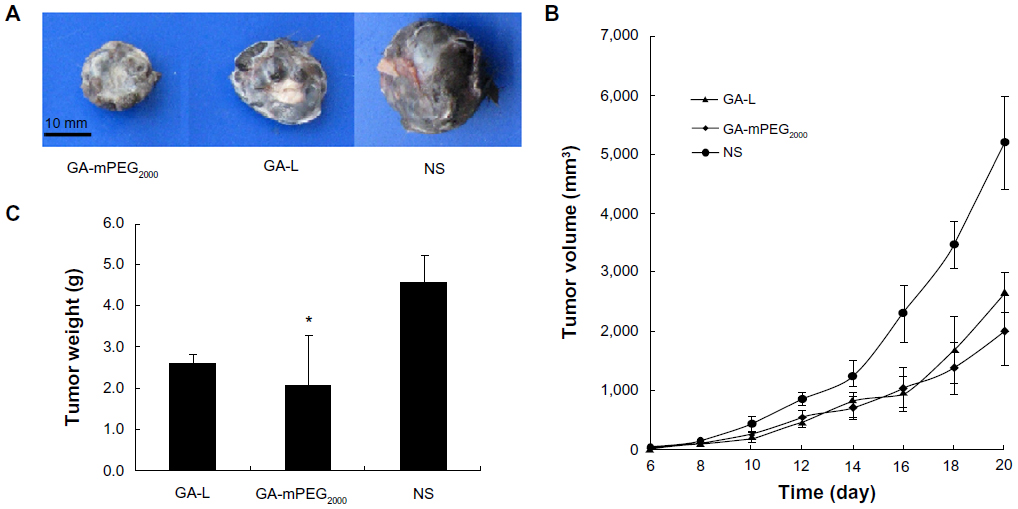 | Figure 6 Antitumor study of GA and GA-mPEG2000 micelles in B16-F10 model. (A) Representative photographs of subcutaneous tumors in the GA-L group, GA-mPEG2000 micelle group, or NS group. (B) Comparison of tumor volume after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). (C) Weight of subcutaneous tumors in each group. |
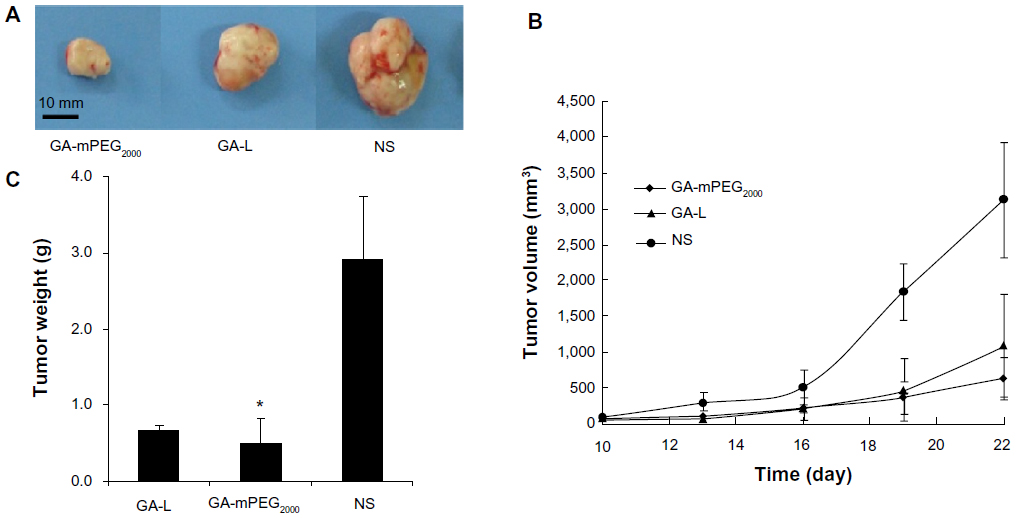 | Figure 7 Antitumor study of GA and GA-mPEG2000 micelles in C26 model. (A) Representative photographs of subcutaneous tumors in the GA-L group, GA-mPEG2000 micelle group, or NS group. (B) Comparison of tumor volume after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). (C) Weight of subcutaneous tumors in each group. |
Throughout the experiment, the mice tolerated the scheduled doses of GA-mPEG2000 micelles and did not show obvious changes in fur, behavior, or eating. However, when mice were treated with GA-L intravenously, they struggled and demonstrated piloerection every time. As shown in Figure 8A and B, weight gain was normal in mice from the normal saline group, but decreased slightly in the GA-mPEG2000 micelle group, while that in the gambogic acid group was significantly lower than that of the other two groups. During our study, we did not observe any treatment-related mortality in the GA-mPEG2000 micelle group; however, mice in the GA-L group started to die after the third (B16-F10 model) and second (C26 model) drug treatments. Moreover, for both models, we observed a substantial increase in the life span of mice in the GA-mPEG2000 micelle group. In the B16-F10 model, 50% of mice treated with GA-mPEG2000 micelles survived for more than 24 days (P<0.01, Figure 8C). In contrast, all mice treated with GA-L died within 18 days. In the C26 model, 70% of mice treated with GA-mPEG2000 micelles survived for more than 31 days (P<0.01, Figure 8D). In contrast, all mice treated with GA-L died within 28 days. These results suggest that GA-L is lethal in mice, whereas GA-mPEG2000 micelles decrease the toxicity of gambogic acid and prolong the lifespan of the animals.
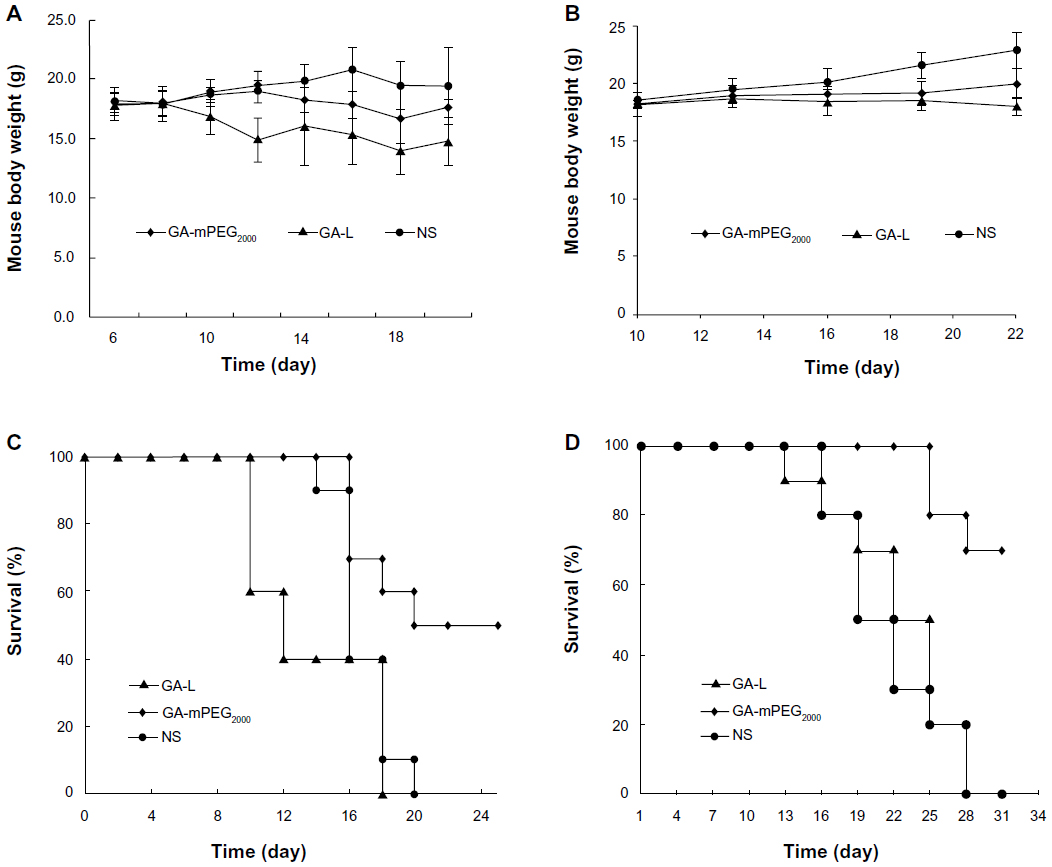 | Figure 8 Toxicity study of GA and GA-mPEG2000 micelles in B16-F10 and C26 models. (A) Curves for body weight in B16-F10 tumor-bearing mice after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). (B) Curves for body weight in C26 tumor-bearing mice after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). (C) Survival curves for B16-F10 tumor-bearing mice after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). (D) Survival curves of C26 tumor-bearing mice after treatment with GA-L (▴), GA-mPEG2000 micelles (♦), or NS (•). |
Discussion
In this study, low molecular weight PEG (ie, 2 kDa) was conjugated to the hydrophobic drug, gambogic acid (molecular weight 628.75 Da), and this amphiphilic conjugate was used to form micelles as a colloidal carrier. As shown in Figure 2, the GA-mPEG2000 conjugate formed nanosized micelles with a mean particle size of about 10 nm. These particles could be evenly dispersed in water, enabling the micelles to penetrate leaky vasculature and spontaneously accumulate in solid tumors via the enhanced permeability and retention effect.40 The maximum drug loading achieved in this formulation was 135.8 mg/mL of physically entrapped gambogic acid, indicating that the polymer-drug conjugate markedly improved the solubilization capacity of gambogic acid. Achievement of this high drug loading level highlights the influence of drug-excipient compatibility as demonstrated in previous studies.41
Over 30 days, the GA-mPEG2000 micelle solution remained clear and transparent, and the average size and PDI did not change significantly (Figure 3). Therefore, conjugating gambogic acid with biocompatible water-soluble mPEG2000 resulted in a homogeneous and stable dosage form in aqueous solution with high drug loading and a small particle size, which makes gambogic acid more suitable for intravenous administration.
From the in vitro drug release profile (Figure 5), we found that GA-mPEG2000 micelles had much slower release behavior compared with GA-L, and the gambogic acid in the GA-mPEG2000 micelles had greatly increased stability compared with GA-L. Drug release from micelles might be due to hydrolysis of the conjugate.42 This attenuated drug release suggests potential applicability of polymeric micelles as a controlled drug delivery system that could result in a more favorable pharmacokinetic profile in vivo and minimize exposure of healthy tissues, while increasing accumulation of drug at the tumor site.43–45
An in vitro cytotoxicity study showed that GA-mPEG2000 micelles retain the cytotoxicity of gambogic acid to B16-F10, C26, and HUVECs, indicating that polymeric prodrugs do not lose the anticancer and antiangiogenetic activity of gambogic acid. However, compared with free gambogic acid, the IC50 values for the GA-mPEG2000 micelles were increased by 2.7–3.0-fold. This suggests that the cytotoxicity is mainly due to the native gambogic acid released from the GA-mPEG2000 micelles, and the relatively lower cytotoxicity seen might be induced by the slower release behavior of gambogic acid from micelles over 48 hours. These results are consistent with the drug release profile mentioned earlier.
Conjugation of drug molecules with biocompatible water-soluble polymers, eg, PEG, has been shown to increase their apparent aqueous solubility, provide protection from degradation, and/or improve their pharmacokinetics and biodistribution.46–49 Conjugation to PEG as a carrier could enable passive targeting to solid tumors via the enhanced permeability and retention effect.50–52 Our in vivo antitumor study showed that GA-mPEG2000 micelles were more efficient in suppressing growth of tumors and prolonging the life span of mice compared with GA-L (Figures 6–Figures 8). The underlying mechanisms might involve three phases. First, aggregation of the GA-mPEG2000 conjugate to form micelles could protect the ester linkage from hydrolysis and protect the encapsulated drug from enzymatic degradation. Second, micelles with a small size could penetrate the leaky vasculature and accumulate in tumor tissue via the enhanced permeability and retention effect. Third, the micelles may release the drug slowly at tumor sites, resulting in higher drug concentrations in the tumor and lower drug concentrations in other tissues. Multiple mechanisms may be involved in the antitumor effect of gambogic acid, including induction of apoptosis, cell cycle arrest, telomerase inhibition, antiangiogenesis activity, and an antimetastasis effect.5–10 Our in vitro cytotoxicity study showed that gambogic acid kills tumor cells, and this also occurs in vivo. Gambogic acid can also inhibit angiogenesis, resulting in suppression of tumor growth in vivo. Therefore, the anticancer effect of GA-mPEG2000 micelles may be induced by inhibiting angiogenesis and directly killing tumor cells.
In this study, all mice showed abnormal changes in their fur and behavior during and after administration of GA-L, indicating that this formulation is irritating to mice. Interestingly, administration of the GA-mPEG2000 micelles did not induce such changes in these animals, suggesting that the nanosized assemblies of GA-mPEG2000 caused less toxicity and irritation compared with GA-L. Further, the body weight of mice in the GA-L group was significantly decreased after treatment, but not in the GA-mPEG2000 micelle group. Given that body weight loss is used as an indicator of the adverse effects of drugs and chemicals,53 our results demonstrate the decreased toxicity of GA-mPEG2000 micelles compared with GA-L. Meanwhile, we observed high mortality in the GA-L group when tumor volume was <300 mm3, suggesting that these deaths were not caused by the tumor but by drug toxicity. Encouragingly, we did not observe these phenomena in the GA-mPEG2000 micelle group, suggesting that the GA-mPEG2000 micelles did not have lethal toxicity in mice. Our results further confirm the decreased toxicity of GA-mPEG2000 micelles compared with GA-L. Two underlying mechanisms might be involved. First, conjugation with mPEG2000 may decrease the toxicity of gambogic acid.31 Second, PEGylated micelles could prolong blood residence time and reduce the risk of nonspecific accumulation of the drug in the body.54 As a result, the drug might be more distributed at the tumor site, but less accumulated in normal tissue, thus reducing drug side effects.
To our knowledge, this work is the first concerning use of GA-mPEG2000 in melanoma and colon cancer therapy in vivo. Our results suggest that intravenous administration of GA-mPEG2000 micelles may have potential application in treating melanoma and colon cancer.
Conclusion
Biodegradable mPEG2000 conjugates of the natural antitumor agent gambogic acid were prepared. These GA-mPEG2000 conjugates could self-assemble into micelles with better solubility in water as compared with gambogic acid. Moreover, the GA-mPEG2000 micelles enhanced drug stability and antitumor effects, prolonged survival time, and were less toxic to mice than gambogic acid. Our results indicate that such polymer-drug conjugate systems could solve many of the problems associated with insoluble chemicals and natural anticancer agents, and at the same time achieve excellent properties in drug delivery systems.
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (81071251) and the Open Research Fund of State Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine. We acknowledge Shichao He and Wen Yang for their help with the synthesis and antitumor studies. We also acknowledge Joseph Rohr and Xianhuo Wang of the Department of Pathology and Microbiology at the University of Nebraska Medical Center, Omaha, NE, USA, for editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Auterhoff H, Frauendorf H, Liesenklas W, Schwandt C. [The chief constituents of gamboge resins. 1. Chemistry of gamboge]. Arch Pharm. 1962;295:833–846. German. | |
Ollis WD, Ramsay MVJ, Sutherland IO, Mongkolsuk S. The constitution of gambogic acid. Tetrahedron. 1965;21:1453–1470. | |
Cai SX, Jiang S, Zhang HZ, inventors. Derivatives of gambogic acid and analogs as activators of caspases and inducers of apoptosis. World Intellectual Property Organization Patent Application PCT/US2004/042292. December 17, 2004. | |
Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull. 2004;27:998–1003. | |
Yu J, Guo QL, You QD, et al. Repression of telomerase reverse transcriptase mRNA and hTERT promoter by gambogic acid in human gastric carcinoma cells. Cancer Chemother Pharmacol. 2006;58:434–443. | |
Guo QL, Lin SS, You QD, et al. Inhibition of human telomerase reverse transcriptase gene expression by gambogic acid in human hepatoma SMMC-7721 cells. Life Sci. 2006;78:1238–1245. | |
Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. Anticancer effect and apoptosis induction of gambogic acid in human gastric cancer line BGC-823. World J Gastroenterol. 2005;11:3655–3659. | |
Qiang L, Yang Y, You QD, et al. Inhibition of glioblastoma growth and angiogenesis by gambogic acid: an in vitro and in vivo study. Biochem Pharmacol. 2008;75:1083–1092. | |
Yang Y, Yang L, You QD, et al. Differential apoptotic induction of gambogic acid, a novel anticancer natural product, on hepatoma cells and normal hepatocytes. Cancer Lett. 2007;256:259–266. | |
Sun S Y, Yue P, Kelloff GJ, et al. Identification of retinamides that are more potent than N-(4-hydroxyphenyl) retinamide in inhibiting growth and inducing apoptosis of human head and neck and lung cancer cells. Cancer Epidemiol Biomarkers. 2001;10:595–601. | |
Liu YT, Hao K, Liu XQ, Wang GJ. Metabolism and metabolic inhibition of gambogic acid in rat liver microsomes. Acta Pharmacol Sin. 2006;27:1253–1258. | |
Hao K, Liu XQ, Wang GJ, Zhao XP. Pharmacokinetics, tissue distribution and excretion of gambogic acid in rats. Eur J Drug Metab Pharmacokinet. 2007;32:63–68. | |
Dai JG, inventor. The preparation of a kind of gambogic acid injection. CN Patent No 03131511.9, Dec 5, 2003. | |
You OD, Guo QL, Ke X, et al, inventors. The preparations of gambogic acid and its compound. CN Patent No 03132386.3, July 21, 2004. | |
Qi Q, You Q, Gu H, et al. Studies on the toxicity of gambogic acid in rats. J.Ethnopharmacol. 2008;117:433–438. | |
Han J, Xiao J, Wang H, et al. Measurement and correlation of taxol solubility in methanol, ethanol and methanol-water systems. J Chem Ind Eng. 2001;52:64–67. | |
Khandare J, Minko T. Polymer-drug conjugates: progress in polymeric prodrugs. Prog Polym Sci. 2006;31:359–397. | |
Satchi-Fainaro R, Duncan R, Barnes CM. Polymer Therapeutics for Cancer: Current Status and Future Challenges. Polymer Therapeutics II. Berlin, Germany: Springer; 2006. | |
Li H, Huo M, Zhou J, et al. Enhanced oral absorption of paclitaxel in N-deoxycholic acid-N, O-hydroxyethyl chitosan micellar system. J Pharm Sci. 2010;99:4543–4553. | |
Hou L, Fan Y, Yao J, et al. Low molecular weight heparin-all-trans-retinoid acid conjugate as a drug carrier for combination cancer chemotherapy of paclitaxel and all-trans-retinoid acid. Carbohydr Polym. 2011;86:1157–1166. | |
Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Deliv Rev. 1996;21:107–116. | |
Greenwald RB, Pendri A, Conover C, Gilbert C, Yang R, Xia J. Drug delivery systems. 2. Camptothecin 20-O-poly (ethylene glycol) ester transport forms. J Med Chem. 1996;39:1938–1940. | |
Sun M, Su X, Ding B, et al. Advances in nanotechnology-based delivery systems for curcumin. Nanomedicine. 2012;7:1085–1100. | |
Tang H, Murphy CJ, Zhang B, et al. Curcumin polymers as anticancer conjugates. Biomaterials. 2010;31:7139–7149. | |
Manju S, Sreenivasan K. Conjugation of curcumin onto hyaluronic acid enhances its aqueous solubility and stability. J Colloid Interface Sci. 2011;359:318–325. | |
Murphy CJ, Tang H, Van Kirk EA, Shen Y, Murdoch WJ. Reproductive effects of a pegylated curcumin. Reprod Toxicol. 2012;34:120–124. | |
Li J, Wang Y, Yang C, et al. Polyethylene glycosylated curcumin conjugate inhibits pancreatic cancer cell growth through inactivation of Jab1. Mol Pharmacol. 2009;76:81–90. | |
Ceruti M, Crosasso P, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing water-soluble prodrugs of paclitaxel. J Control Release. 2000;63:141–153. | |
Fleming AB, Haverstick K, Saltzman WM. In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjug Chem. 2004;15:1364–1375. | |
Tang X, Zhang P, Ye H, Zhang C, Shen W, Ping Q. Water-soluble gambogic acid PEGylated prodrugs: synthesis, characterization, physicochemical properties and in vitro hydrolysis. Pharmazie. 2008;63:711–717. | |
Ding Y, Zhang P, Tang XY. PEG prodrug of gambogic acid: amino acid and dipeptide spacer effects. Polymer. 2012;53:1694–1702. | |
Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. | |
HyperChem. Professional version 8.0. Gainesville, FL, USA; Hypercube Inc; 2007. | |
Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. | |
Allinger NL. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J Am Chem Soc. 1977;99:8127–8134. | |
Fletcher R, Reeves CM. Function minimization by conjugate gradients. Comput J. 1964;7:149–154. | |
Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. | |
Brooks BR, Brooks CL, Mackerell AD, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. | |
Ping QN, Zhang C, Ye H, et al. Polyethylene glycol prodrug of gambogic acid, its preparation method, formulation and use for preparing of drug. CN Patent No 100531801 C, Aug 26, 2009. | |
Tang N, Du G, Wang N, et al. Improving penetration in tumors with nanoassemblies of phospholipids and doxorubicin. J Natl Cancer Inst. 2007;99:1004–1015. | |
Liu J, Xiao Y, Allen C. Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine. J Pharm Sci. 2004;93:132–143. | |
Liu J, Zahedi P, Zeng F, et al. Nano-sized assemblies of a PEG-docetaxel conjugate as a formulation strategy for docetaxel. J Pharm Sci. 2008;97:3274–3290. | |
Lundberg BB, Risovic V, Ramaswamy M, et al. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J Control Release. 2003;86:93–100. | |
Gabizon A, Horowitz AT, Goren D, et al. Targeting folate receptor with folate linked to extremities of poly (ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10:289–298. | |
Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233:134–144. | |
Greenwald RB. PEG drugs: an overview. J Control Release. 2001;74:159–171. | |
Minko T. Soluble polymer conjugates for drug delivery. Drug Discov Today. 2005;2:15–20. | |
Langer CJ. CT-2103: a novel macromolecular taxane with potential advantages compared with conventional taxanes. Clin Lung Cancer. 2004;6:S85–S88. | |
Vasey PA, Kaye SB, Morrison R, et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83–94. | |
Seymour LW, Ferry DR, Anderson D, et al. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–1676. | |
Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, et al. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs. 2001;12:315–323. | |
Schoemaker NE, van Kesteren C, Rosing H, et al. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Br J Cancer. 2002;87:608–614. | |
Tofovic SP, Jackson EK. Effects of long-term caffeine consumption on renal function in spontaneously hypertensive heart failure prone rats. J Cardiovasc Pharmacol. 1999;33:360–366. | |
Stoyanova E, Mitova V, Shestakova P, et al. Reversibly PEGylated nanocarrier for cisplatin delivery. J Inorg Biochem. 2013;120:54–62. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.