Back to Journals » Infection and Drug Resistance » Volume 15
Importance of Efferocytosis in COVID-19 Mortality
Authors Erol A ![]()
Received 9 November 2021
Accepted for publication 3 March 2022
Published 10 March 2022 Volume 2022:15 Pages 995—1007
DOI https://doi.org/10.2147/IDR.S348639
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Adnan Erol
Independent Researcher, Not Affiliated to Any Institution, Silivri-Istanbul, Turkey
Correspondence: Adnan Erol, Independent Researcher, Not Affiliated to Any Institution, Silivri-Istanbul, Turkey, Email [email protected]
Abstract: COVID-19 is a generally benign coronavirus disease that can spread rapidly, except for those with a group of risk factors. Since the pathogenesis responsible for the severity of the disease has not been clearly revealed, effective treatment alternatives has not been developed. The hallmark of the SARS-CoV-2-infected cells is apoptosis. Apoptotic cells are cleared through a sterile process defined as efferocytosis by professional and nonprofessional phagocytic cells. The disease would be rapidly brought under control in the organism that can achieve effective efferocytosis, which is also a kind of innate immune response. In the risk group, the efferocytic process is defective. With the addition of the apoptotic cell load associated with SARS-COV-2 infection, failure to achieve efferocytosis of dying cells can initiate secondary necrosis, which is a highly destructive process. Uncontrolled inflammation and coagulation abnormalities caused by secondary necrosis reason in various organ failures, lung in particular, which are responsible for the poor prognosis. Following the short and simplified information, this opinion paper aims to present possible treatment options that can control the severity of COVID-19 by detailing the mechanisms that can cause defective efferocytosis.
Keywords: COVID-19, SARS-CoV-2, efferocytosis: ADAM17, phosphatidylserine
Graphical Abstract:

SARS-CoV-2-induced COVID-19 pandemic has been caused a serious increase in morbidity and mortality, as well as devastating problems in the economies of countries. Unprecedented rapid and widespread vaccination has provided significant relief in the control of the pandemic in a short period, less than a year. However, the belief that vaccines can solve everything has brought with it troubles. The dominance of the delta variant, the rapid increase in the number of cases and the rate of hospitalization despite vaccination necessitate taking new measures.1 People infected with the delta variant of SARS-CoV-2 seem to be more likely to spread the virus before developing symptoms. More importantly, there was no difference in the rate of transmission and viral load between vaccinated and unvaccinated people, suggesting that a proportion of vaccinated people can transmit this variant.2,3 It is also a strong possibility that those who have been vaccinated are more contagious because they are asymptomatic or are easily accepted at social events. Moreover, SARS-CoV-2 lambda, a new variant of interest, exhibits higher infectivity and confers resistance to antiviral immunity.4 At least for these reasons, it seems difficult to control the pandemic with vaccination alone. While COVID-19 is threatening in a certain risk group, it generally shows a moderate course. Therefore, the development of treatment methods that can control the severity and lethality of the disease seems to be the most rational option since the beginning of the pandemic.
SARS-CoV-2 Infection and Efferocytosis
SARS-CoV-2 is an enveloped virus. Phosphatidylserine (PS) is expressed in the outer leaflet of the viral envelope, with a symmetrical location rather than the asymmetrical phospholipid distribution observed in the normal plasma membrane.5 As with all enveloped viruses, this facilitates cell entry of SARS-CoV-2 using the apoptotic mimicry pathway.6 PS receptors (PSRs), Axl in particular, were shown to potentiate the binding and uptake of SARS-CoV-2.6
SARS-CoV-2 spike protein (S) is divided into two subunits: S1 unit of S binds to angiotensin-converting enzyme 2 (ACE2), while S2 mediates membrane fusion. Several arginine residues present in SARS-CoV-2 S are cleaved by the host protease, furin that is not found in SARS-CoV-related coronaviruses.7 Proteolytic processing of SARS-CoV-2 S in the virus-producing host cells, rather than during entry into target cells, may facilitate subsequent binding to the ACE2 receptors, fusion, and entry into target cells.8 Accordingly, loss of the furin cleavage site has been shown to affect the pathogenesis of SARS-CoV-2, suggesting that the furin cleavage site has a critical role in the transmission of SARS-CoV-2 infection.9
SARS-CoV-2 genome contains 29 open reading frames (ORFs), which encode 29 proteins. Among these, the ORF3a protein is an accessory protein specific to SARS-CoVs. ORF3a is involved in critical steps of the viral infection cycle and determines viral replication and virulence of SARS-CoV-2.10 In cells infected with SARS-CoV-2, the virus enforces the apoptotic process through ORF3a.11 ORF3a-mediated caspase-3 activation initiates both the apoptotic process and a series of changes for the rapid elimination of the apoptotic cell by the phagocytic system:
- Phospholipase A2 activation increases the level of lysophosphatidylcholine on the outer membrane surface.12
- The opening of membrane Pannexin channels causes an increase in extracellular ATP, UTP, and spermidine levels.12
- An increase in oxidized PS and phosphatidylcholine levels occurs.13 All of these three pathways act as “find me” signals for the phagocytic cells.
- Irreversible activation of Xkr8, a membrane scramblase, causes PS to be expressed in the membrane outer leaflet, which is the essential “eat me” signal for cells of the phagocytic system.14,15
In a healthy organism, professional and non-professional phagocytic cells detect “find me” and “eat me” signals and rapidly clear apoptotic cells without damaging the environment through a process known as efferocytosis.16–18 While macrophages, neutrophils and dendritic cells originating from the bloodstream are professional; local epithelial, endothelial, and fibrotic cells are the elements of the non-professional phagocytic system.19
A large number of different PSRs that recognize PS directly and indirectly in phagocytic cells facilitate the functioning of the efferocytosis process.5,6,14 While TIM receptors (T cell immunoglobulin and mucin domain-containing molecules) provide direct binding, TAM (Tyro3, Axl, and MerTK) receptors indirectly bind to apoptotic cells via mediator molecules, such as Gas6 and protein S (ProS).5,18 Among several other PSRs, the importance of the phagocytic cell scavenger receptor CD36 in engulfing the apoptotic cell should be particularly emphasized.20
Cells exposed to apoptotic environments undergo conformational and biochemical changes in their membranes. The hydrolysis of phosphatidylcholine to lysophosphatidylcholine renders the phosphatidylcholine group accessible for the binding of C-reactive protein (CRP). CRP irreversibly marks apoptotic cells for the activation of the classical complement pathway, ultimately those opsonized cells are disposed of by phagocytosis.21,22 Apoptotic cells tagged with CRP may also interact with the scavenger CD36 receptors, which are expressed by the macrophages and thus may facilitate efferocytosis.23 In other words, CRP can promote efferocytosis by binding to the apoptotic cell surface, leading to recognition and phagocytosis by macrophages.24 Consistently, CRP apheresis has been reported to be an effective treatment option for patients severely challenged by SARS-CoV-2 infection, most likely by regulating efferocytosis.25,26
Efficient phagocytosis of apoptotic cells, efferocytosis, is one of the most essential components of maintaining tissue homeostasis. After engulfment of the apoptotic cell, phagocytic cells actively control inflammation (“tolerate me”) by releasing anti-inflammatory cytokines such as transforming growth factor-beta (TGF-β) and IL-10.14 This specialized phagocytosis of apoptotic cells also accelerates the presentation of viral antigens to lymphocytes and T cells, enhancing the activation of antigen-specific B and T cells.27,28
If the efferocytosis-mediated clearance process is disrupted, the apoptotic cells undergo secondary necrosis. This is a highly detrimental process that results in leakage of toxic intracellular antigens, tissue destruction, and intensification of the existing inflammation.14,29 In secondary necrosis, increased neutrophil pro-inflammatory activity and excessive degranulation may cause acute respiratory distress syndrome (ARDS) or multiple organ failure as a result of neutrophil-mediated systemic inflammatory response syndrome.30,31 Membrane rupture due to secondary necrosis triggers the development of a different type of autoimmune response to auto-antigen release with auto-reactive CD4+ cell activation.32 Thus, efferocytosis, which means rapid and effective non-inflammatory and non-immunogenic apoptotic cell clearance through the interrelated “find me”, “eat me”, and “tolerate me” pathways, performs a unique function in the maintenance of homeostasis at the tissue and system level.
Collectively, COVID-19 patients with a relatively healthy premorbid history recover following a mild-to-moderate course through the functioning of the following mechanisms (Figure 1).
- SARS-CoV-2 replication may lead to the pro-apoptotic changes that would trigger the exposure of normally intracellular anionic phospholipids, PS in particular, on the outer leaflet of the membrane surface of virus-infected cells. These virus-induced apoptotic cells need to be rapidly and efficiently removed otherwise they may produce secondary necrotic debris that promotes the release and hyperactivation of pro-inflammatory molecules from the immune effector cells.
- Efficient efferocytosis is an essential component of tissue homeostasis and the resolution of inflammation. SARS-CoV-2-infected apoptotic cells expose PS as an “eat me” signal, which distinguishes them from live cells, initiates efferocytosis by professional and non-professional phagocytic cells in a non-inflammatory manner. After engulfing an apoptotic cell, macrophages actively dampen inflammation by releasing anti-inflammatory cytokines, such as (TGF-β) and interleukin-10, as well as pro-resolving lipid mediators for the resolution of inflammation.
- SARS-CoV-2-infected, apoptotic cells are phagocytosed and digested together with the virus itself through the actions of degrading enzymes that exist in lysosomes of phagocytes, leading to the inhibition of viral growth. This mode of phagocytic elimination of invading pathogens may be considered as a cellular innate immune response.33 Consistently, binding and preventing PS-exposing cells from being phagocytosed by annexin V, which binds specifically to PS, has been reported to augment the rate of mortality among virus-infected mice.34
- Under homeostatic conditions, anti-inflammatory cytokines and natural regulatory T cells channel dendritic cell antigen presentation to induce regulatory T-cell differentiation from naïve CD4+ T-cell precursors and to tolerize effector CD8+ T cells.35 Furthermore, efferocytosis of infected apoptotic cells can help to mount an adaptive immune response against the virus through the process of antigen cross-presentation.36
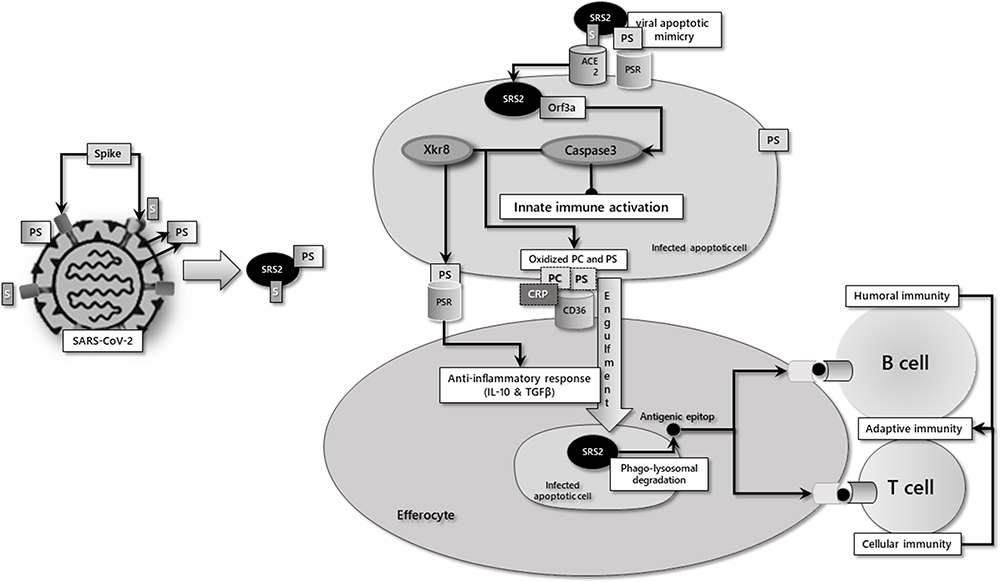 |
Figure 1 Simplified depiction of the apoptotic/efferocytic process in SARS-COV-2 infection. Notes: The phosphatidylserine (PS) in the envelope of the virus attaches to the PS receptors (PSRs) on the target cell, facilitating the binding of the viral spike protein to the host ACE2 receptor. In the cell, the viral ORF3a protein initiates the apoptotic process by the caspase-3 activation. While activated caspase-3 inhibits cellular innate immune response, caspase-3-driven permanent activation of the Xkr8 scramblase causes PS expression in the membrane outer leaflet of apoptotic cells. Caspase-3 also oxidizes PS and phosphatidylcholine (PC). PS binds to efferocytic cells via PSRs, facilitating the binding of oxidized PC-CRP and PS to CD36 and engulfment of the apoptotic cell. Successfully induced efferocytosis is a sterile process with the secretion of anti-inflammatory mediators. In addition, antigenic viral epitopes, which arise due to phago-lysosomal degradation of the virus in antigen-presenting cells, can initiate T- and B-cell-mediated adaptive immunity. Reproduced from Erol A. Defective efferocytosis as a predictor of COVID-19 mortality. OSF Preprints. 2021; September 13:1-18.119 |
Thus, while COVID-19 is highly contagious due to the multibasic furin cleavage site in the S protein,37 it follows a self-limiting course through the operation of the mechanisms briefly described. However, the disease may progress to a more severe and even fatal stage in those with chronic degenerative and cardiovascular diseases, especially in the elderly. If the underlying pathogenesis of this adverse progression could be demonstrated, it may also provide possible target(s) for the treatment.
Risk Factors for COVID-19 and Defective Efferocytosis
Despite the death of billions of cells every day, effectively functioning efferocytosis maintains normal physiology in the majority of the population; however, it becomes defective in unresolved chronic inflammatory diseases, causing apoptotic cell accumulation.38 Diseases with increased morbidity and mortality, such as aging, obesity, heart diseases, and diabetes, cause an immuno-lipidomic imbalance with lipidome disorder and chronic inflammatory pathologies.39,40 The characteristic determinants of these disorders are low-level chronic inflammation and increased apoptotic cell load.41 Altogether, defective efferocytosis is an essential pathology responsible for the development and progression of chronic inflammatory diseases, including atherosclerosis, obesity, diabetes, heart failure, chronic lung disease, and neurodegenerative disease, as well as cancer.42
The common feature of most of these degenerative chronic disorders is increased ADAM-17 (a disintegrin and metalloproteinase 17) activity in most of the cells, including bronchial epithelial cells, endothelial cells, and phagocytic cells.43,44 ADAM-17, also known as TNFα-converting enzyme (TACE), is a sheddase, capable of proteolytic cleavage and release of membrane-bound molecules.45,46 Studies revealing the relationship between ADAM-17 and a group of inflammatory degenerative diseases, including cancer, are reported with increasing frequency. Interestingly, transcriptional ADAM-17 protein expression has been observed to increase in diabetic subjects through the hyperglycemia-induced HIF-1 activity.47,48 In addition, the euglycemic effect of insulin restored ACE2 and ADAM-17 expression to the physiological levels,47 suggesting that insulin may work as an ADAM17 inhibitor.49 Furthermore, evidence is provided that ADAM-17 plays a role in metabolic syndrome-related pathologies.50,51 On the other hand, some clear data also show the possible relationship between the presence of metabolic syndrome and the severity of COVID-19.52 Accordingly, several of the features of the metabolic syndrome seem to be associated with a worse prognosis in patients with COVID-19.53
ADAM-17 is the main secretase responsible for ectodomain shedding of most of the cell surface and the transmembrane proteins. At least 90 substrates are processed by ADAM-17. Its activity towards TNFα and IL-6Rα could potentiate the proinflammatory effects of the cytokines.54 In addition, ADAM-17 activity promotes lymphocyte and neutrophil translocation to the inflammatory sites.55 Taken together, premorbid patients with increased ADAM17 and thus pro-inflammatory activity, if contracted SARS-CoV-2 infection, would have the inflammatory process more severe.56
PS translocation to the membrane surface, an exclusive marker of apoptosis, is also required for the sheddase function of ADAM-17. The externalized PS in the membrane interacts electrostatically with ADAM-17 and then enables substrate processing to take place.57 Signaling pathways destined to activate ADAM-17-sheddase function converge at the final step leading to scramblase activation with a breakdown of phospholipid asymmetry and exposure of PS at the outer leaflet.58 On the other hand, PS surface exposure is required for the regulation of cholesterol metabolism, inflammation, and hemostasis.59 Recalling that anti-inflammatory cytokines are secreted during PS-induced efferocytosis, it is possible to consider this as an effort to control the existing inflammatory pathology.60 Therefore, exposure to PS on the outer plasma membrane is increased in chronic inflammatory disorders, including atherosclerotic diseases and diseases associated with coagulation disorders,59,61 which may potentiate the activity of ADAM-17.
In principle, all-natural membranes may function as a procoagulant phospholipid surface, if sufficient PS is present in the exofacial leaflet.62 Microparticles and microvesicles refer to large extracellular vesicles, which are small membranous structures.63 Microparticles are present in the blood of healthy individuals, but their number is considerably increased in diseased states, such as inflammation, cardiovascular events, and cancer, where they may contribute to the increased risk of thrombotic events in these patients.62 Circulating microparticles are shed from the plasma membrane of a variety of cells, including platelets, erythrocytes, leukocytes, endothelial cells, and tumor cells as a result of activation and/or apoptosis.62,64 These microparticles have lost lipid asymmetry and expose PS, thus providing a procoagulant surface.62 The prothrombotic state mediated by PS-exposing microparticles is implicated in the adverse progression of chronic inflammatory diseases, including atherosclerosis.59 In addition, ADAM-17 was shown to present in microparticles released from activated cells; thus, ADAM-17 activity can be distributed to more distant cells.65 Remarkably, increased levels of microparticles in the circulation of severe COVID-19 patients that trigger the risk of thromboembolic complications were reported.66 Furthermore, the level of PS-exposing microparticles correlated more strongly with the severity of COVID-19 than indices, such as lymphopenia, IL-6, D-Dimer, fibrinogen, and other established laboratory parameters.67
ACE2, a metallopeptidase and a key component of the renin–angiotensin system (RAS), is the main receptor for the cellular entry of SARS-CoV and SARS-CoV-2. The binding of SARS viruses to ACE2 triggers the viral conformational changes, which may increase the proteolytic digestion between the S1 and S2 subunits.68,69 Indeed, ACE2 cleavage by ADAM-17 is required for efficient SARS-S-driven entry. Furthermore, ADAM-17-mediated shedding of ACE2 results in the release of the ectodomain into the circulation, which preserves its catalytic and bioactive potency, and the binding site for SARS-CoV-2.70 Recent studies demonstrated that ADAM-17-driven soluble ACE2 (sACE2) levels are elevated in COVID-19 patients. The binding of SARS-CoV-2 by sACE2 may enable cell entry of tissues where membrane-bound ACE2 (mACE2) is poorly expressed.71,72 Low expression levels of mACE2 and associated higher ADAM-17 activity have been related to cardiovascular diseases. Consequently, the activity of the sACE2 is increased in patients with cardiovascular disease and hypertension, which correlates with the severity of the disease.73 Thus, chronic diseases with increased membrane PS exposure and associated ADAM-17 activity may adversely affect the prognosis of the COVID-19 through the facilitated cellular virus entry as a result of the increased sACE2 levels.56,71
PSRs, including TIM1, TAM, and CD36, can also be proteolytically cleaved by the transmembrane protease ADAM-17.20,74 This shedding can prevent receptor functions in macrophage-mediated apoptotic cell engulfment.75 The soluble cleavage products may function as sequester ligands and decoy receptors, which can act as antagonists for those PSRs.75,76 Accordingly, the deletion of ADAM-17 has been shown to increase macrophage-mediated efferocytosis in vivo, resulting in an enhanced anti-inflammatory response.77
Programmed cell death 1 (PD-1), a surface molecule and a member of the immunoglobulin superfamily, functions as co-inhibitory receptors during immune responses against pathogens and cancer. Hence, PD-1 is important in the regulation of the magnitude and quality of T cell response.78 PD-1 interacts with the ligands PD-L1 and PD-L2. PD-L1 is expressed in all hematopoietic cells and also by many non-hematopoietic cell types, such as endothelial and epithelial cells. PD-L2 expression, on the other hand, is limited to hematopoietic cells, such as dendritic cells, B cells, and monocytes/macrophages.78,79 During the acute phase of virus infection, stimulation of the PD-1/PD-L1 signaling pathway modulates the strength and quality of cytotoxic CD8+ T cell attack, maintaining a balance between virus elimination and tissue damage.78
ADAM17 can cleave PD-L1 from the surface of vesicles and cells.80 Although circulating soluble PD-L1 (sPD-L1) has been largely known as a prognostic biomarker for cancers,81 sPD-L1 has also been detected in various pathologies, often associated with markers of inflammation.82 Of note, a recent study suggests that an increase in sPD-L1 may be a sign of poor prognosis as a result of dysregulation of the PD-1/PD-L1 axis in COVID-19 patients.83
To summarize, the following mechanisms due to the increased PS exposure and ADAM-17 activity adversely affect the prognosis of the disease in those who have comorbidities along with the COVID-19 disease (Figure 2).
- The proinflammatory burden that will determine the severity of COVID-19 is already at high levels before the onset of the disease.
- ACE2 cleavage and sACE2 levels are increased. The facilitated extracellular association between sACE2 and SARS-CoV-2 may reinforce the infection more widespread and devastating.
- Circulating microparticles have been implicated in cellular signaling processes in immune responses, inflammation, and coagulation. In addition to PS exposure on their membrane, they may contain ADAM17, which can contribute to substrate shedding on more distant cells.65
- Loss of PS receptors on phagocytic cells (efferocytes) impairs the recognition and engulfment process of apoptotic cells, thereby impairing efferocytosis.
- Lastly, decreased expression of PD-L1 on T cells may reflect the induction of hyperactivation status, which may cause an excessive immunopathology.
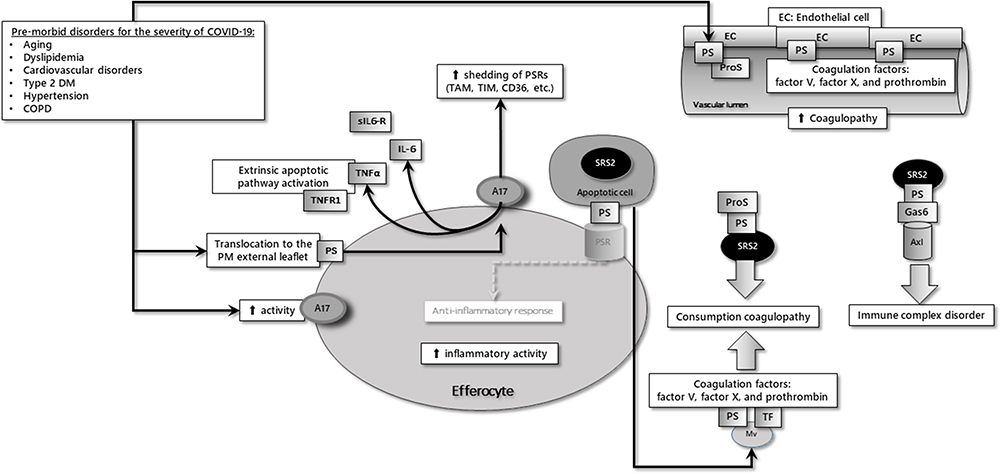 |
Figure 2 The common characteristics of co-morbid diseases responsible for the severity of COVID-19 are increased PS expression and ADAM-17 (A17) activity in cells. Notes: ADAM-17 renders efferocytosis as defective due to the shedding of the receptors required for the engulfment of apoptotic cells. PS expressed on the surface of endothelial cells, PS in the SARS-CoV-2 envelope, and microvesicles (Mv) detached from apoptotic cells reason in coagulopathy seen in COVID-19. Reproduced from Erol A. Defective efferocytosis as a predictor of COVID-19 mortality. OSF Preprints. 2021; September 13:1-18.119 |
Collectively, potentiated dysregulation of ADAM-17 activity may lead to the chronic operation of defective efferocytosis. In turn, defective efferocytosis accompanying several non-resolving, chronic inflammatory diseases may lead to the accumulation of dead cells. The dead cells can become secondarily necrotic, which can lead to tissue necrosis, autoimmunity, and pathological inflammation. Thus, defective efferocytosis is emerging as a key mechanism driving the development and progression of chronic inflammatory diseases, including atherosclerosis, obesity, diabetes, heart failure, chronic lung disease, neurodegenerative disease, and cancer.42
Defective Efferocytosis and COVID-19 Severity
Efferocytosis is a mechanism for eliminating pathogens and pathogen-infected cells in an immunologically silent manner. The quality of efferocytic clearance of virus and virus-infected apoptotic cells depends on the virus load and the operation of efferocytic machinery.17 Therefore, increased viral load and defect in efferocytic cells seem to be the crucially important factors that will determine the prognosis of the disease. Patients in the risk group who already have problems in apoptotic cell clearance due to defective efferocytosis cannot cope with the additional acute apoptotic burden that occurs with COVID-19 disease. Consequently, the intractable inflammation that increases with secondary necrosis causes destruction and inevitable organ failure. Support for this approach came from a recent study showing that dysfunctional efferocytosis is the leading cause of the increased inflammatory response and widespread tissue damage seen in COVID-19.84
Older age, obesity, diabetes, cardiovascular diseases, and active cancer have each been found to be independently associated with a higher mortality risk for COVID-19.85 Furthermore, COVID-19 does not spare young people as well. The majority of comorbidities that play a role in the prognosis of the elderly have also been shown to be risk factors that may be responsible for the severity of COVID-19 for the young adult population aged 16–28 years.86
Novel Treatment Options Targeting Defective Efferocytosis in COVID-19
The clinical trial research has occurred at an unprecedented pace during the COVID-19 pandemic. Among the many potential treatment options for COVID-19, only a few are effective and predictable for the clinical treatment of patients. Some immunomodulatory drugs such as anti-TNF, anti-IL-1, anti-IL-6 used in COVID-19 patients have been shown to support recovery, possibly by neutralizing the increased cytokine levels by the augmented ADAM17 activity.87 In a study showing the effect of glucocorticoids, another group of immune modulators, dexamethasone treatment in hospitalized COVID-19 patients provided lower mortality among those requiring respiratory support.88
Glucocorticoids are powerful anti-inflammatory agents by their ability to inhibit the recruitment of inflammatory cells and to downregulate proinflammatory cytokine expression.89 Glucocorticoids have been shown to modulate the expression of over 100 genes, including those known to be associated with efferocytosis, such as Mer tyrosine kinase (MerTK), a member of TAM family PS receptors.90 Dexamethasone was also shown to promote the expression of two PS-binding opsonins, protein S and MFG-E8, which facilitate MerTK-dependent efferocytosis of apoptotic cells.89,91 Thus, corticosterones, which provide limited support for defective efferocytosis, find wide use in clinical treatment as an effective drug that can control the severity of cases.
Peroxisome proliferator-activated receptor γ (PPARγ) agonists such as pioglitazone alter cellular metabolism of the liver, adipose tissue, and muscle cells, as well as inflammatory macrophages. In addition, they have many anti-inflammatory effects including the suppression of Th1 and Th17-induced inflammation, inhibition of NFκB, and upregulation of IL-10.92,93 Interestingly, PPARγ agonists enhance MerTK and opsonin expression by human macrophages, similar to glucocorticoids.94 Furthermore, treatment with pioglitazone, either prophylactically or during inflammation, while significantly enhancing PPARγ-mediated programming of efferocytes, reduces the accumulation of apoptotic cells.92,95 Therefore, low-dose pioglitazone alone or combination with glucocorticoids seems to be a readily available inexpensive, and effective treatment option for the correction of defective efferocytosis.93,96
While ADAM-17 plays a crucial role in the pathogenesis of defective efferocytosis, it was shown that ADAM-17 activity is also important in the development and worst prognosis of COVID-19.97 Therefore, inhibiting ADAM-17 activity seems to be a rational option in severe cases of COVID-19. Experimental evidence has been shown that the inhibition of ADAM-17 protects against lung inflammation by reducing cytokine storm and excessive neutrophil recruitment to the lung in a mouse model associated with COVID-19.98 However, ADAM-17 has a broad regulatory role in human biological processes. Consistently, dramatic disorders affecting the intestines, lungs, eyes, and hair of ADAM-17-deficient mice and humans have prompted caution regarding therapeutic interventions for ADAM-17 blockade in humans.99,100 Nevertheless, researchers are nowadays focusing on developing strategies for selective inhibition of ADAM-17. Several compounds have recently been entered into clinical trials but have been withdrawn due to emerging adverse events.97 Therefore, despite the central importance of ADAM-17 in the pathogenesis of the disease, the option of inhibiting ADAM-17 in severe cases of COVID-19 should be viewed with caution.
It has been emphasized above that the level of sACE2 in the circulation increases due to defective efferocytosis in diseases that constitute a risk group for COVID-19. However, another therapeutic approach proposes using sACE2 to trap and neutralize SARS-CoV-2. From this perspective, while mACE2 may mediate cell entry of SARS-CoV-2, a genetically modified soluble form of ACE2, called human recombinant soluble angiotensin-converting enzyme-2 (hrsACE2), may decrease cell entry of SARS-CoV-2 competing for mACE2.101 However, the plasma RAS balance in COVID-19 patients was already characterized by a strong transient increase in circulating plasma sACE2.72 Therefore, in COVID-19 patients with comorbid diseases, both Angiotensin II and virus adversely affect the prognosis by affecting the RAS balance in a way that down-regulates ACE2.102 Interestingly, a recent study reports encouraging results in the first severe COVID-19 patient treated with hrsACE2.103 However, sACE2 concentrations used in experimental studies are much higher (μg/mL) than the physiological range of sACE2 concentrations (ηg/mL). While a high range of sACE2 concentrations can compete with the SARS-CoV-2-ACE2 complex for cell entry, the physiological range of sACE2 may facilitate virus entry and thus SARS-CoV-2 infectivity. Therefore, a treatment that may alter sACE2 levels in the physiological range in patients with COVID-19 should be carefully considered.71
In severe cases of COVID-19 where defective efferocytosis is essential in the pathogenesis, the characteristic feature of infected-apoptotic cells waiting to be cleared is that they express PS on the outer surface of the membrane. Therefore, triggering antibody-dependent cellular phagocytosis (ADCP) with PS-specific antibodies can be considered as an important alternative for the treatment of such pathologies in which PS receptors are deficient. The possibility of such a specific monoclonal antibody already exists. Bavituximab (PGN401) is a monoclonal human-mouse chimeric antibody directed against the membrane phospholipid PS. Bavituximab and other antibodies that target PS were developed by Philip Thorpe’s laboratory to specifically target tumor vasculature.15,104 Bavituximab does not bind PS directly. It binds to PS-expressing membranes by crosslinking two molecules of β2-glycoprotein 1 (β2GP1) bound to PS on the membrane.14,105 Bavituximab could be expected to act by at least two mechanisms in severe COVID-19 patients: i) it can reduce the amount of virus to infect other tissues as a result of opsonization-mediated clearance of SARS-CoV2; ii) it induces ADCP of virus-infected apoptotic cells; thus, in pathologies where efferocytosis is defective, an opportunity arises for clearing apoptotic cells.106
β2GP1 is an evolutionarily conserved single-chain anionic phospholipid-binding glycoprotein. It has gained much attention since the discovery of its role in antiphospholipid syndrome (APS).107 Accordingly, β2GP1 binds to the PS expressed on apoptotic cells. When the amount of β2GP1 bound to PS reaches a certain threshold, antibodies dimerize the adjacent β2GP1 molecules. As a consequence, these high-affinity anti-β2GP1-β2GP1 complexes may activate targeted cells, causing anti-phospholipid antibodies-related manifestations.108 However, during the clinical trials, bavituximab therapy appeared safe and well tolerated, and reductions were observed in virus load in the blood. Furthermore, coagulation parameters remained within the normal range.106,109 In the light of those data and considering the FDA approval of the drug, I had planned a clinical trial to investigate the efficacy of bavituximab treatment in patients with severe COVID-19 in October 2020. Unfortunately, the opinion remained in theory, as OncXerna, the company that owns the drug, did not welcome this offer. Since the hallmark autoantibodies of APS are anti-beta2 glycoproteins, a series of fully human PS-targeting antibodies were generated by phage display technology.110 To further explore the antiviral activity of PS-targeting antibodies, my efforts to develop a specific antibody in my country, such as PGN632, that could bind directly to PS and would not need the cofactor β2GP1 protein,109 were also failed.
The majority of those who are infected with COVID-19 have a self-limiting infection and do recover. However, a minority with comorbidities proceeds to more severe disease, requiring intensive care unit admission.111 Therefore, the disease may cease to be a threat following the treatment of patients who are in the risk group or whose condition deteriorates with the monoclonal anti-PS antibody to be developed. Moreover, such a treatment would also have the potential to control the morbidity and mortality caused by other enveloped viruses such as influenza, Ebola, dengue, and even HIV.
Vaccine and therapeutic development are predominantly focused on the essential virus-encoded Spike protein. However, the variation of the S gene between SARS-like CoV viruses and SARS-CoV-2 variants may affect the efficacy and particularly for cross-protection of a vaccine. Nowadays when mutations against vaccines are confusing, other proteins of SARS-CoV-2 seem to be important targets to develop alternative treatments against potential resistance or emerging new viruses in the future.112 SARS-CoV-2 ORF3a is a putative viral ion channel implicated in autophagy inhibition, inflammasome activation, and apoptosis. As explained above, it is a specific viral protein that may be responsible for the development of the virus-induced apoptotic load and ultimately the adverse course of the disease in cases of defective efferocytosis.
SARS-CoV-2 enters the host cell by both membrane fusion and by clathrin/caveolin-mediated endocytosis after binding to the ACE2 cell-surface receptors.113 Caveolin-1 is a major component of caveolae membrane microdomains, which are involved in non-clathrin-mediated virus uptake into cells.114 Viral ORF3a protein binding to host caveolin-1 was shown to be essential for entry and endomembrane trafficking of SARS-CoV-2. In addition to viral entry, caveolin-1 was also associated with all stages of the viral life cycle.113,114 Even if ORF3a is expressed on viral particles, anti-ORF3a antibodies will not neutralize the virus by preventing attachment or fusion.115 In other words, antibodies that bind to ORF3a while disrupting the functions of the protein, the antibody-bound virus would be able to enter the cell. In turn, entry of the antibody-bound virus into the cell may present an opportunity for an unusual cytosolic antibody receptor, called TRIM21. Critically, TRIM21 is also a ubiquitin ligase and can efficiently target antibody-bound substrates for proteasomal degradation.116 Therefore, the development of an antibody or vaccine, targeting ORF3a, may provide more effective results in controlling severe cases of COVID-19 with a high risk of mortality.
Conclusion
Every virus infection and even mRNA vaccines have pro-apoptotic properties.117 Therefore, effective removal of this burden in cell death is inevitable for the maintenance of cellular homeostasis. Since defective efferocytosis is a potential threat to immune system dysregulation, a drug is urgently needed to support efferocytosis, “as it becomes more and more obvious that vaccination alone might not sufficient to curb the pandemic caused by this rapidly mutating virus”.118 Therefore, we will be awaiting further research in the hope that this work will be evaluated to respond to this urgent need.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Brown CM, Vostok J, Johnson H, et al. Outbreak of SARS-CoV-2 infections, including COVID-19 vaccine breakthrough infections, associated with large public gatherings — Barnstable County, Massachusetts, July 2021. MMWR Morb Mortal Wkly Rep. 2021;70:1059–1062. doi:10.15585/mmwr.mm7031e2
2. Riemersma KK, Grogan BE, Amanda Kita-Yarbro GE, David H, O’Connor TC, Friedrich KMG. Shedding of infectious SARS-CoV-2 despite vaccination when the delta variant is prevalent - Wisconsin, July 2021. Prepr medRxiv. 2021. doi:10.1101/2021.07.31.21261387
3. Musser JM, Christensen PA, Olsen RJ, et al. Delta variants of SARS-CoV-2 cause significantly increased vaccine breakthrough COVID-19 cases in Houston, Texas. Prepr medRxiv. 2021. doi:10.1101/2021.07.19.21260808
4. Kimura I, Kosugi Y, Wu J, et al. SARS-CoV-2 Lambda variant exhibits higher infectivity and immune resistance. Prepr bioRxiv. 2021. doi:10.1016/j.celrep.2021.110218
5. Evans JP, Liu S-L. Role of host factors in SARS-CoV-2 entry. J Biol Chem. 2021;297:100847. doi:10.1016/j.jbc.2021.100847
6. Bohan D, Van Ert H, Ruggio N, et al. Phosphatidylserine receptors enhance SARS-CoV-2 infection: AXL as a therapeutic target for COVID-19. Prepr bioRxiv. 2021. doi:10.1101/2021.06.15.448419
7. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78:779–784.e5. doi:10.1016/j.molcel.2020.04.022
8. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci. 2020;117:11727–11734. doi:10.1073/pnas.2003138117
9. Johnson BA, Xie X, Bailey AL, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021;591:293–299. doi:10.1038/s41586-021-03237-4
10. Azad GK, Khan PK. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem Biophys Rep. 2021;26:100933. doi:10.1016/j.bbrep.2021.100933
11. Ren Y, Shu T, Wu D, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:881–883. doi:10.1038/s41423-020-0485-9
12. Narahari AK, Kreutzberger AJ, Gaete PS, et al. ATP and large signaling metabolites flux through caspase-activated Pannexin 1 channels. Elife. 2021;10. doi:10.7554/eLife.64787.
13. Yamashita A, Morikawa H, Tajima N, et al. Mechanisms underlying production and externalization of oxidized phosphatidylserine in apoptosis: involvement of mitochondria. Yonago Acta Med. 2012;55:11–20.
14. Birge RB, Boeltz S, Kumar S, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:962–978. doi:10.1038/cdd.2016.11
15. Dayoub AS, Brekken RA. TIMs, TAMs, and PS- antibody targeting: implications for cancer immunotherapy. Cell Commun Signal. 2020;18:29. doi:10.1186/s12964-020-0521-5
16. Kumar S, Calianese D, Birge RB. Efferocytosis of dying cells differentially modulate immunological outcomes in tumor microenvironment. Immunol Rev. 2017;280:149–164. doi:10.1111/imr.12587
17. Karaji N, Sattentau QJ. Efferocytosis of pathogen-infected cells. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.01863/full
18. Calianese DC, Birge RB. Biology of phosphatidylserine (PS): basic physiology and implications in immunology, infectious disease, and cancer. Cell Commun Signal. 2020;18:41. doi:10.1186/s12964-020-00543-8
19. Serizier SB, McCall K. Scrambled eggs: apoptotic cell clearance by non-professional phagocytes in the drosophila ovary. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.01642/full
20. Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. 2014;468–470:565–580. doi:10.1016/j.virol.2014.09.009
21. Vogt B, Führnrohr B, Müller R, Sheriff A. CRP and the disposal of dying cells: consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity. 2007;40:295–298. doi:10.1080/08916930701358925
22. Sheriff A, Kayser S, Brunner P, Vogt B. C-reactive protein triggers cell death in ischemic cells. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.630430/full
23. Zwaka TP, Hombach V, Torzewski J. C-reactive protein–mediated low density lipoprotein uptake by macrophages. Circulation. 2001;103:1194–1197. doi:10.1161/01.CIR.103.9.1194
24. DeBerge M, Zhang S, Glinton K, et al. Efferocytosis and outside-in signaling by cardiac phagocytes. links to repair, cellular programming, and intercellular crosstalk in heart. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.01428/full
25. Torzewski J, Zimmermann O, Kayser S, et al. Successful treatment of a 39-year-old COVID-19 patient with respiratory failure by selective C-reactive protein apheresis. Am J Case Rep. 2021;22. doi:10.12659/AJCR.932964.
26. Ringel J, Ramlow A, Bock C, Sheriff A. Case report: c-reactive protein apheresis in a patient with COVID-19 and fulminant CRP increase. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.708101/full
27. Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi:10.1038/32183
28. Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: formulating future vaccines. J Clin Invest. 2016;126:799–808. doi:10.1172/JCI81083
29. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi:10.1016/j.febslet.2010.10.046
30. Laforge M, Elbim C, Frère C, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol. 2020;20:515–516. doi:10.1038/s41577-020-0407-1
31. Wang J, Li Q, Yin Y, et al. Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.02063/full
32. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. 2017;22:1189–1204. doi:10.1007/s10495-017-1413-z
33. Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol. 2007;178:2448–2457. doi:10.4049/jimmunol.178.4.2448
34. Watanabe Y, Hashimoto Y, Shiratsuchi A, Takizawa T, Nakanishi Y. Augmentation of fatality of influenza in mice by inhibition of phagocytosis. Biochem Biophys Res Commun. 2005;337:881–886. doi:10.1016/j.bbrc.2005.09.133
35. Tian L, Choi S-C, Lee H-N, et al. Enhanced efferocytosis by dendritic cells underlies memory T-cell expansion and susceptibility to autoimmune disease in CD300f-deficient mice. Cell Death Differ. 2016;23:1086–1096. doi:10.1038/cdd.2015.161
36. Cho K-J, Ishido S, Eisenlohr LC, Roche PA. Activation of dendritic cells alters the mechanism of MHC class II antigen presentation to CD4 T cells. J Immunol. 2020;204:1621–1629. doi:10.4049/jimmunol.1901234
37. Wrobel AG, Benton DJ, Xu P, et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol. 2020;27:763–767. doi:10.1038/s41594-020-0468-7
38. Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2018;4. doi:10.3389/fcvm.2017.00086/full
39. Schwarz B, Sharma L, Roberts L, et al. Cutting edge: severe SARS-CoV-2 infection in humans is defined by a shift in the serum lipidome, resulting in dysregulation of eicosanoid immune mediators. J Immunol. 2021;206:329–334. doi:10.4049/jimmunol.2001025
40. Arienti S, Barth ND, Dorward DA, Rossi AG, Dransfield I. Regulation of apoptotic cell clearance during resolution of inflammation. Front Pharmacol. 2019;10. doi:10.3389/fphar.2019.00891/full
41. Sendama W. The effect of ageing on the resolution of inflammation. Ageing Res Rev. 2020;57:101000. doi:10.1016/j.arr.2019.101000
42. Doran AC, Yurdagul A, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20:254–267. doi:10.1038/s41577-019-0240-6
43. Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi:10.3109/10409231003628015
44. Shalaby L, Thounaojam M, Tawfik A, et al. Role of endothelial ADAM17 in early vascular changes associated with diabetic retinopathy. J Clin Med. 2020;9:400. doi:10.3390/jcm9020400
45. Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. doi:10.1038/385729a0
46. Patel VB, Clarke N, Wang Z, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–176. doi:10.1016/j.yjmcc.2013.11.017
47. Salem ESB, Grobe N, Elased KM. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am J Physiol Physiol. 2014;306:F629–39. doi:10.1152/ajprenal.00516.2013
48. Li R, Uttarwar L, Gao B, et al. High glucose up-regulates ADAM17 through HIF-1α in mesangial cells. J Biol Chem. 2015;290:21603–21614. doi:10.1074/jbc.M115.651604
49. Stepanova G. Biologia futura: is ADAM 17 the reason for COVID-19 susceptibility in hyperglycemic and diabetic patients? Biol Future. 2021;72:291–297. doi:10.1007/s42977-021-00092-2
50. Menghini R, Fiorentino L, Casagrande V, Lauro R, Federici M. The role of ADAM17 in metabolic inflammation. Atherosclerosis. 2013;228:12–17. doi:10.1016/j.atherosclerosis.2013.01.024
51. Matthews J, Villescas S, Herat L, Schlaich M, Matthews V. Implications of ADAM17 activation for hyperglycaemia, obesity and type 2 diabetes. Biosci Rep. 2021;41. doi:10.1042/BSR20210029
52. le Roux CW. COVID-19 alters thinking and management in metabolic diseases. Nat Rev Endocrinol. 2021;17:71–72. doi:10.1038/s41574-020-00449-y
53. Finucane FM, Davenport C. Coronavirus and obesity: could insulin resistance mediate the severity of Covid-19 infection? Front Public Health. 2020;8. doi:10.3389/fpubh.2020.00184/full
54. Yang G, Cui M, Jiang W, Sheng J, Yang Y, Zhang X. Molecular switch in human diseases-disintegrin and metalloproteinases, ADAM17. Aging (Albany NY). 2021;13:16859–16872. doi:10.18632/aging.203200
55. Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J Immunol. 2009;182:2449–2457. doi:10.4049/jimmunol.0802770
56. Zipeto D, da Palmeira JF, Argañaraz GA, Argañaraz ER. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front Immunol. 2020;11. Available from https://www.frontiersin.org/article/10.3389/fimmu.2020.576745/full.
57. Sommer A, Bhakdi S, Reiss K. How membrane asymmetry regulates ADAM17 sheddase function. Cell Cycle. 2016;15:2995–2996. doi:10.1080/15384101.2016.1211449
58. Reiss K, Bhakdi S. The plasma membrane: penultimate regulator of ADAM sheddase function. Biochim Biophys Acta Mol Cell Res. 2017;1864:2082–2087. doi:10.1016/j.bbamcr.2017.06.006
59. Darabi M, Kontush A. Phosphatidylserine in atherosclerosis. Curr Opin Lipidol. 2016;27:414–420. doi:10.1097/MOL.0000000000000298
60. Harel-Adar T, Ben MT, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci. 2011;108:1827–1832. doi:10.1073/pnas.1015623108
61. Kawai T, Elliott KJ, Scalia R, Eguchi S. Contribution of ADAM17 and related ADAMs in cardiovascular diseases. Cell Mol Life Sci. 2021;78:4161–4187. doi:10.1007/s00018-021-03779-w
62. Bevers EM, Williamson PL. Getting to the outer leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev. 2016;96:605–645. doi:10.1152/physrev.00020.2015
63. Morel O, Jesel L, Freyssinet J-M, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011;31:15–26. doi:10.1161/ATVBAHA.109.200956
64. Owens AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. doi:10.1161/CIRCRESAHA.110.233056
65. Groth E, Pruessmeyer J, Babendreyer A, et al. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochim Biophys Acta Mol Cell Res. 2016;1863:2795–2808. doi:10.1016/j.bbamcr.2016.09.002
66. Cappellano G, Raineri D, Rolla R, et al. Circulating platelet-derived extracellular vesicles are a hallmark of Sars-Cov-2 infection. Cells. 2021;10:85. doi:10.3390/cells10010085
67. Rausch L, Lutz K, Schifferer M, et al.Binding of phosphatidylserine-positive microparticles by PBMCs classifies disease severity in COVID-19 patients. bioRxiv Prepr. 2021. doi:10.1002/jev2.12173
68. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88:1293–1307. doi:10.1128/JVI.02202-13
69. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi:10.1016/j.cell.2020.02.052
70. Hoffmann M, Hofmann-Winkler H, Pöhlmann S. Priming time: how cellular proteases arm coronavirus spike proteins. In: Act Viruses by Host Proteases. Cham: Springer International Publishing;2018:71–98. doi:10.1007/978-3-319-75474-1_4
71. Yeung ML, Teng JLL, Jia L, et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell. 2021;184:2212–2228.e12. doi:10.1016/j.cell.2021.02.053
72. Lundström A, Ziegler L, Havervall S, et al. Soluble angiotensin‐converting enzyme 2 is transiently elevated in COVID‐19 and correlates with specific inflammatory and endothelial markers. J Med Virol. 2021;93:5908–5916. doi:10.1002/jmv.27144
73. Epelman S, Tang WHW, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure. J Am Coll Cardiol. 2008;52:750–754. doi:10.1016/j.jacc.2008.02.088
74. Driscoll WS, Vaisar T, Tang J, Wilson CL, Raines EW. Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ Res. 2013;113:52–61. doi:10.1161/CIRCRESAHA.112.300683
75. Schweigert O, Dewitz C, Möller-Hackbarth K, et al. Soluble T cell immunoglobulin and mucin domain (TIM)-1 and −4 generated by A Disintegrin And Metalloprotease (ADAM)-10 and −17 bind to phosphatidylserine. Biochim Biophys Acta Mol Cell Res. 2014;1843:275–287. doi:10.1016/j.bbamcr.2013.11.014
76. de Queiroz TM, Lakkappa N, Lazartigues E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front Pharmacol. 2020;11. doi:10.3389/fphar.2020.01154
77. Tajbakhsh A, Gheibi Hayat SM, Butler AE, Sahebkar A. Effect of soluble cleavage products of important receptors/ligands on efferocytosis: their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res Rev. 2019;50:43–57. doi:10.1016/j.arr.2019.01.007
78. Schönrich G, Raftery MJ. The PD-1/PD-L1 axis and virus infections: a delicate balance. Front Cell Infect Microbiol. 2019;9. doi:10.3389/fcimb.2019.00207
79. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153–167. doi:10.1038/nri.2017.108
80. Orme JJ, Jazieh KA, Xie T, et al. ADAM10 and ADAM17 cleave PD-L1 to mediate PD-(L)1 inhibitor resistance. Oncoimmunology. 2020;9. doi:10.1080/2162402X.2020.1744980
81. Zhu X, Lang J. Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget. 2017;8:97671–97682. doi:10.18632/oncotarget.18311
82. Bailly C, Thuru X, Quesnel B. Soluble programmed death ligand-1 (sPD-L1): a pool of circulating proteins implicated in health and diseases. Cancers (Basel). 2021;13:3034. doi:10.3390/cancers13123034
83. Sabbatino F, Conti V, Franci G, et al. PD-L1 dysregulation in COVID-19 patients. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.695242.
84. dos-Santos D, Salina AC, Rodrigues TS, et al. Efferocytosis of SARS-CoV-2-infected dying cells impairs macrophage anti-inflammatory programming and continual clearance of apoptotic cells. MedRixv. 2021. doi:10.1101/2021.02.18.21251504
85. Gupta S, Hayek SS, Wang W, et al. Factors associated with death in critically ill patients with coronavirus disease 2019 in the US. JAMA Intern Med. 2020;180:1436. doi:10.1001/jamainternmed.2020.3596
86. Sandoval M, Nguyen DT, Vahidy FS, Graviss EA. Risk factors for severity of COVID-19 in hospital patients age 18–29 years. PLoS One. 2021;16:e0255544. doi:10.1371/journal.pone.0255544
87. Burrage DR, Koushesh S, Sofat N. Immunomodulatory drugs in the management of SARS-CoV-2. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.01844
88. The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2021;384:693–704. doi:10.1056/NEJMoa2021436
89. McColl A, Bournazos S, Franz S, et al. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol. 2009;183:2167–2175. doi:10.4049/jimmunol.0803503
90. Ehrchen J, Steinmüller L, Barczyk K, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109:1265–1274. doi:10.1182/blood-2006-02-001115
91. Lauber K, Keppeler H, Munoz LE, et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ. 2013;20:1230–1240. doi:10.1038/cdd.2013.82
92. Fernandez-Boyanapalli RF, Falcone EL, Zerbe CS, et al. Impaired efferocytosis in human chronic granulomatous disease is reversed by pioglitazone treatment. J Allergy Clin Immunol. 2015;136:1399–1401.e3. doi:10.1016/j.jaci.2015.07.034
93. Erol A. Role of oxidized LDL-induced “trained macrophages” in the pathogenesis of COVID-19 and benefits of pioglitazone: a hypothesis. Diabetes Metab Syndr Clin Res Rev. 2020;14:713–714. doi:10.1016/j.dsx.2020.05.007
94. Zizzo G, Cohen PL. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR-γ in human macrophage polarization. J Inflamm. 2015;12(1):36. doi:10.1186/s12950-015-0081-4.
95. Fernandez-Boyanapalli R, Frasch SC, Riches DWH, Vandivier RW, Henson PM, Bratton DL. PPARγ activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116:4512–4522. doi:10.1182/blood-2010-02-272005
96. Agrawal S, Chanley MA, Westbrook D, et al. Pioglitazone enhances the beneficial effects of glucocorticoids in experimental nephrotic syndrome. Sci Rep. 2016;6:24392. doi:10.1038/srep24392
97. Calligaris M, Cuffaro D, Bonelli S, et al. Strategies to target ADAM17 in disease: from its discovery to the iRhom revolution. Molecules. 2021;26:944. doi:10.3390/molecules26040944
98. Lartey NL, Valle-Reyes S, Hilda Vargas-Robles KE, et al. ADAM17 inhibition prevents neutrophilia and lung injury in a mouse model of Covid-19. bioRxiv Prepr. 2021. doi:10.1002/JLB.3COVA0421-195RR
99. Blaydon DC, Biancheri P, Di W-L, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–1508.
100. Bandsma RHJ, van Goor H, Yourshaw M, et al. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum Pathol. 2015;46:923–928. doi:10.1016/j.humpath.2015.02.010
101. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.e7. doi:10.1016/j.cell.2020.04.004
102. Rahman MM, Hasan M, Ahmed A. Potential detrimental role of soluble ACE2 in severe COVID‐19 comorbid patients. Rev Med Virol. 2021;
103. Zoufaly A, Poglitsch M, Aberle JH, et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med. 2020;8:1154–1158. doi:10.1016/S2213-2600(20)30418-5
104. Belzile O, Huang X, Gong J, et al. Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer. ImmunoTargets Ther. 2018;7:1–14. doi:10.2147/ITT.S134834
105. Dowall SD, Graham VA, Corbin-Lickfett K, et al. Effective binding of a phosphatidylserine-targeting antibody to Ebola virus infected cells and purified virions. J Immunol Res. 2015;2015:1–9. doi:10.1155/2015/347903
106. Soares MM, King SW, Thorpe PE. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat Med. 2008;14:1357–1362. doi:10.1038/nm.1885
107. Bevers EM, Janssen MP, Comfurius P, et al. Quantitative determination of the binding of β2-glycoprotein I and prothrombin to phosphatidylserine-exposing blood platelets. Biochem J. 2005;386:271–279. doi:10.1042/BJ20041167
108. Ho Y, Ahuja K, Körner H, Adams M. β2GP1, anti-β2GP1 antibodies and platelets: key players in the antiphospholipid syndrome. Antibodies. 2016;5:12. doi:10.3390/antib5020012
109. Thomas JM, Thorpe PE. Protective effect of anti-phosphatidylserine antibody in a guinea pig model of advanced hemorrhagic arenavirus infection. Open Microbiol J. 2017;11:303–315. doi:10.2174/1874285801711010303
110. Shao R, Xiong C, Wen X, Gelovani JG, Li C. Targeting phosphatidylserine on apoptotic cells with phages and peptides selected from a bacteriophage display library. Mol Imaging. 2007;6:
111. Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). StatPearls; 2021. Available from https://www.ncbi.nlm.nih.gov/books/NBK554776/.
112. Kern DM, Sorum B, Mali SS, et al. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat Struct Mol Biol. 2021;28:573–582. doi:10.1038/s41594-021-00619-0
113. Gupta S, Mallick D, Banerjee K, Sarkar S, Sonny TM, Partha basuchowdhuri SSJ. D155Y substitution of SARS-CoV-2 ORF3a weakens binding with caveolin-1: an in silico study. bioRxiv Prepr. 2021. doi:10.1101/2021.03.26.437194
114. Padhan K, Tanwar C, Hussain A, et al. Severe acute respiratory syndrome coronavirus Orf3a protein interacts with caveolin. J Gen Virol. 2007;88:3067–3077. doi:10.1099/vir.0.82856-0
115. Chakraborty S, Mallajosyula V, Tato CM, Tan GS, Wang TT. SARS-CoV-2 vaccines in advanced clinical trials: where do we stand? Adv Drug Deliv Rev. 2021;172:314–338. doi:10.1016/j.addr.2021.01.014
116. Caddy SL, Vaysburd M, Papa G, et al. Viral nucleoprotein antibodies activate TRIM21 and induce T cell immunity. EMBO J. 2021;40. doi:10.15252/embj.2020106228
117. Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20:817–838. doi:10.1038/s41573-021-00283-5
118. Kunzelmann K. Getting hands on a drug for Covid-19: inhaled and intranasal niclosamide. Lancet Reg Health. 2021;4:100094.
119. Defective efferocytosis as a predictor of COVID-19 mortality. OSF Preprints. 2021; September 131–18. doi:10.31219/osf.io/cfwsh
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.