")
Back to Journals » Journal of Blood Medicine » Volume 13
Impact of Abnormal Leukocyte Count in the Pathophysiology of Sickle Cell Anemia
Authors Yousif TYE
Received 19 June 2022
Accepted for publication 10 November 2022
Published 16 November 2022 Volume 2022:13 Pages 673—679
DOI https://doi.org/10.2147/JBM.S378133
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Tagwa Yousif Elsayed Yousif
Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, Jazan University, Gizan, 45142, Saudi Arabia
Correspondence: Tagwa Yousif Elsayed Yousif, Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, Jazan University, Gizan, 45142, Saudi Arabia, Email [email protected]
Background: Sickle cell disease (SCD) is a hereditary disease that causes deoxygenated erythrocytes to stiffen, resulting in vaso-occlusive crises, endothelial damage, organ failure and systemic consequences. An abnormal leukocyte count is associated with a worse clinical picture in SCD.
Methods: I studied the link between an abnormal leukocyte count and sickle cell disease. I used the PRISMA guidelines to conduct a systematic review of 5 different clinical literatures that discussed the link. Use of databases including Web of Science, Scopus, Cochrane Library, MEDLINE, ScienceDirect, EMBASE, and JSTOR to make our findings.
Findings: SCD is a red cell disorder, but an elevated leukocyte count plays a significant role in its pathophysiology. Sickled red cells adhere better to leukocytes when compared to normal red cells, which explains the worse clinical picture seen in sickle cell patients. Hydroxyurea therapy inhibits the recruitment and invasion of neutrophils, which lowers vaso-occlusion.
Interpretations: An elevated leukocyte count is responsible for worse clinical manifestations in SCD, eg, clinical stroke, acute chest syndrome, end organ damage, and infarction pain. Leukocytes attach to erythrocytes and vascular endothelium to cause vaso-occlusion, responsible for these effects. They also elaborate inflammatory mediators to aid the process. The higher the leukocyte count, the worse the effects. Hydroxyurea is very helpful in disrupting several pathways of leukocyte contribution to SCD pathophysiology. Therefore, more research on other pharmacologic agents that interfere with this disease process is necessary. Lastly, there is a need for further research into the effects of a reduced leukocyte count in SCD pathophysiology.
Keywords: SCD pathophysiology, leukocyte count, infraction, effects, sickle cell
Introduction
In Sickle Cell Anemia (SCA), valine is abnormally switched out for glutamine at position 6 of the hemoglobin chain (SCA). Any person who expresses this mutation homozygously has the genotype associated with Sickle Cell Disease (SS).1 The aberrant sickle shape that the erythrocytes adopt is due to a genetic abnormality, which also causes the endothelial damage, vas blockage, hemolysis, ischemia, and inflammation that appear around 5 to 6 months of age after the loss of fetal hemoglobin (HbF).2 The disease’s clinical manifestation differs from person to person and is more prevalent in the black race while having a clearly characterized monogenic inheritance pattern.3
Although there is no doubt that SCD is caused by a red cell hemoglobin mutation, SCD patients frequently have an abnormal white blood cell count.
When the WBC count is inadequate, infections are more likely to spread and are more difficult to treat. Her white blood cell count will often be normal or higher if the child has sickle cell disease. The WBC count, however, might briefly increase or decrease as a result of disease and some medications. White cell control-focused treatments may be advantageous for patients. Approximately half of the 290 SCD patients in a 2016–2017 study by Ahmed et al5 in Saudi Arabia exhibited abnormally increased or decreased white cell counts of 116 or 15, respectively. Other studies have revealed that an elevated white cell count is the most common observation in SCD patients with abnormal white cell counts.6–8 By adhering to vascular endothelium and obstructing the lumen, leukocytes participate in the SCD process. They achieve this by stimulating the production of vascular endothelial ligands for the blood cell adhesion molecules in order to be successful. That results in tissue damage, inflammation, and ultimately vaso-occlusion.9
This study will review the pertinent literature in order to comprehend how an unusually high or low white cell count contributes to sickle cell anemia at the tissue level.
Method
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses standards were followed for this study’s systematic literature review.10
Information Sources
I searched these databases for relevant literature: Google Scholar, PubMed, Web of Science, Scopus, Cochrane Library, MEDLINE, ScienceDirect, EMBASE, and JSTOR.
Search Strategy
The terms “sickle cell anemia”, “sickle cell illness”, “white blood cell”, and “sickle cell disease” were used in the search.
Eligibility Criteria
The inclusion criteria for selection include studies that attempt to establish a link between abnormal leukocyte count and sickle cell anemia between January, 2002 and May, 2022; literature published in English; and those with an observational, qualitative, or systematic review design.
Selection Process
Studies unrelated to the link between abnormal leukocyte count and sickle cell anemia, duplicated ones, and abstract-only papers were excluded.
Results
A total of 20 studies that satisfied my inclusion criteria were identified, of which I selected 4. The study selection process goes as follows: After identifying 20 studies, 15 passed the screening stage, while the other 5 were duplicates. Of these 15 studies, only six were eligible, as the remaining nine were abstract-only studies. I arrived at the inclusion of 2 studies and 3 articles6,9,11 following a careful review of all options. The characteristics of the samples, types of intervention, and outcomes of the 2 studies are shown in Table 1.
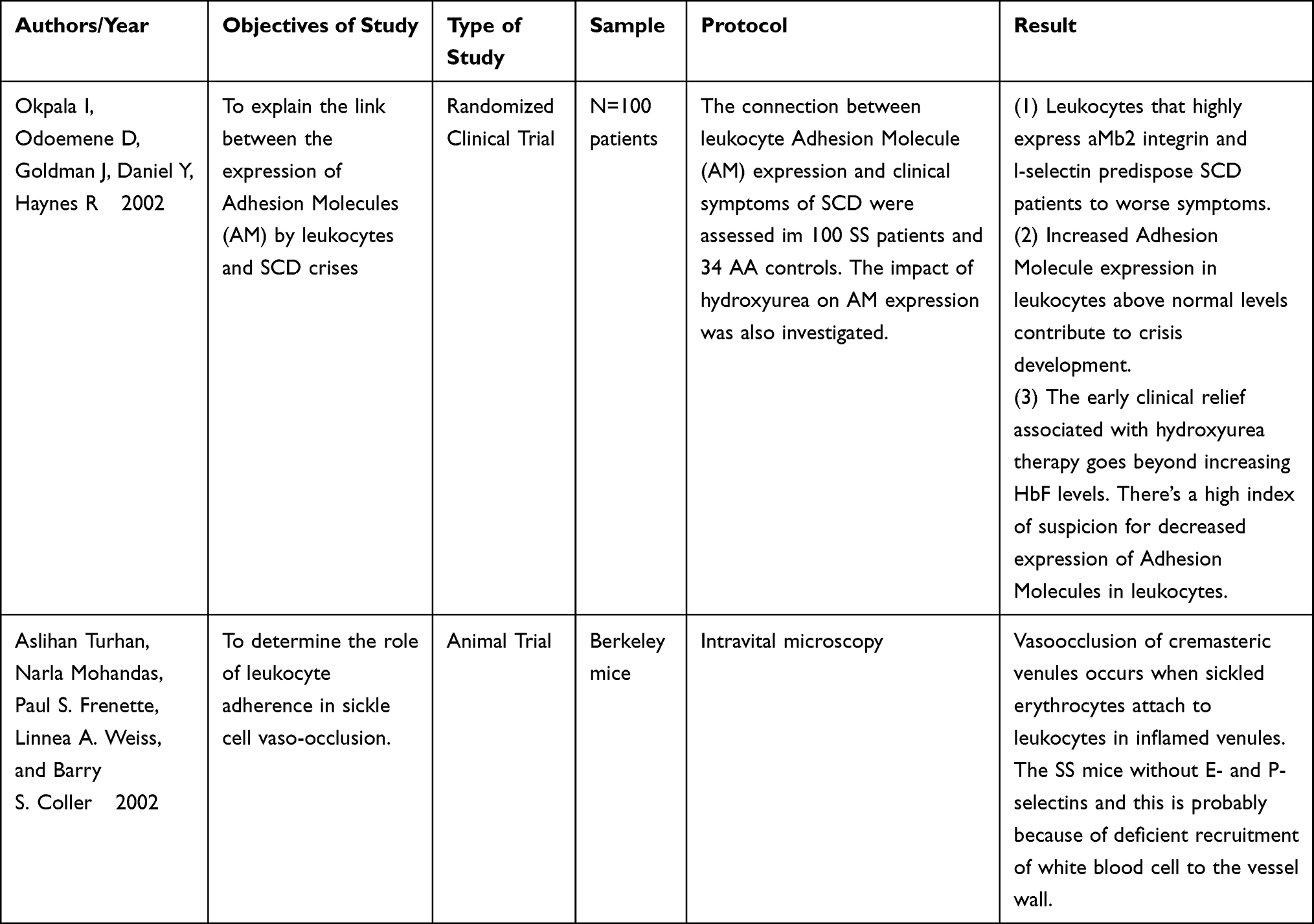 |
Table 1 Characteristics of the Samples, Types of Intervention, and Outcome of the 2 Studies |
Okpala I,9 in his article titled “The Intriguing Contribution of White Blood Cells to Sickle Cell Disease – a Red Cell Disorder”, reviews relevant literature, totaling 67, in an attempt to establish a direct link between leukocytes and the disease process in SCDA. Wun T.11 takes it a step further and also explores the role of inflammatory cytokines alongside leukocytes in “The Role of Inflammation and Leukocytes in the Pathogenesis of Sickle Cell Disease.” Zhang D.6 decides to focus on a particular class of leukocytes in his article” Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology.”
Results of Individual Studies
After a thorough search of the listed databases, three experienced reviewers selected the relevant studies for this review based on the title and abstract. They passed on the selected studies to two other reviewers, who read the full text of the selected studies and approved the most suitable options.
As Okpala I9 demonstrates, there’s ample evidence to suggest that white blood cells contribute to the clinical manifestations seen in SCD. Despite the diverse phenotypes seen in a monogenic disorder such as SCD, the most consistent finding directly related to clinical severity remains an elevated leukocyte count, particularly neutrophils.14–17 Furthermore, a drop in leukocyte count reduces the severity of the hemoglobinopathy, consistent with reports indicating SCD is exacerbated by increased leukocyte levels. The therapeutic impact of hydroxyurea in SCA is associated with a decrease in the polymorphonuclear (neutrophil) count even in people with stable hemoglobin F levels.18 SCD vaso-occlusion is thought to be caused by a complex interaction involving white blood cells, red blood cells, platelets, plasma proteins, and blood vessel walls, which explains why a high leukocyte count has such a negative influence on clinical severity. The attachment of leukocytes to the endothelium of postcapillary venules is thought to diminish the diameter of the vessels, which is worsened by the attachment of erythrocytes, both sickled and un-sickled, as well as platelets.19
The neutrophils are the most notorious leukocytes, well known for attaching to the vessels’ walls. It is the body’s most common leukocyte but it has a short circulation time, totaling about 6 hours. The huge number of white blood cells in the diapedesis process reveals one key fact: the lumen of postcapillary venules is constantly shrinking. The more adherent cells inside the vessel, the higher the leukocyte count. As a result, HbSS patients with greater leukocyte counts have a worse prognosis. White blood cells do not only attach to the walls of blood vessels. They also attach to one another and to other blood cells like red blood cells and platelets. In this way, they form cell aggregates that reduce the diameter of small arteries more effectively. Moreover, there are higher chances for entanglement of sickled red blood cells in this matrix, leading to vascular occlusion. The stability of these blood cell aggregates rests on the CD36–TSP, CD31–CD31, and CD62L–CD162 connections.20–23
Leukocytes may indirectly precipitate vascular occlusion in SCD by activating the endothelial layer of blood vessels. The inflammatory mediators TNF-a and interleukin-1b, for example, are secreted by monocytes and activate the vascular endothelium.24 The inflammatory response is heightened, and the danger of tissue damage is increased. The endothelium of blood vessels is stimulated during inflammation, and leukocytes and other blood cells express more adhesion molecule ligands. In turn, that increases the risk of lumen blockage, ischemia, organ damage, dysfunction, and infarction pain associated with SCD.
Wun T.11 remarks that raised white cell counts in SCD patients with no signs of infection, pain, or inflammation are documented.25 He goes further to list reasons for this, such as functional hyposplenism, asplenia, and bone marrow stimulation for red cell production due to the hemolysis in SCD. Furthermore, an elevated leukocyte count in SCD indicates a worse clinical picture, with an increased risk of stroke26 and acute chest syndrome.27 According to the article, what role do leukocytes play in SCD pathogenesis? Activated and stiff PMN can initiate vascular occlusion by impeding blood flow in the small calibers arterioles and increasing the time needed for the sickled erythrocytes to transit. All these happen in a bid to increase the chances of HbS polymerization. Furthermore, SS-RBC adhered to neutrophil monolayers better than control red cells (AA-RBC). In the case of SSRBC, autologous plasma-enhanced adhesion, but not in the case of AA-RBC. This revealed that SSRBC attached more easily to PMN, and that the plasma contained chemicals that aided this attachment. Surprisingly, the dense fraction had the most adhesive SSRBC, but the thin fraction had the most adherent SSRBC to active endothelium. Activated PMNs release free oxygen radicals, which increase the expression of AMs on the vascular endothelium, causing vascular injury.28
Monocytes are not left out. In sickle cell disease, monocytosis is prevalent. They attach to active platelets and SS-RBC. This most likely activates them and has been linked to the etiology of vascular disease.29–31 In vitro investigations show that monocytes from SCD patients produce greater superoxide anion in response to agonists than controls, leading to vascular injury. Finally, in sickle cell disease patients, the action of procoagulants is enhanced, with practically all of the procoagulant activity found in the mononuclear component of the blood.
According to Zhang D,6 the identification of absolute contraindications for myeloid growth factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) in SCD patients suggests a role for neutrophils in SCD pathogenesis. Early reports indicate that SCD patients given either growth factors to cure leg ulcers, mobilize hematopoietic stem cells, or correct diminished neutrophil counts experienced severe or fatal crises.32–34 A patient with SCD who had severe congenital neutropenia was recently found to have significantly improved clinical symptoms when compared to his brothers and sister. However, things changed when he was given G-CSF for neutropenia, as his condition quickly worsened.35 The significance of hydroxyurea in SCD therapy is also explained in further detail in the article. Sickle RBC adherence to the endothelium is reduced by hydroxyurea treatment, as are the levels of soluble vascular cell adhesion molecule (VCAM)-1 in patient plasma.36 Furthermore, current research reveals that hydroxyurea treatment increases NO species, which is probably related to HbF induction.37,38 Furthermore, things remain the same in patients who do not have a detectable increase in HbF as hydroxyurea medication is still helpful. On the other hand, patients who have a positive clinical response to hydroxyurea therapy have lower neutrophil counts.39 Furthermore, the correction of neutrophil activation indicators reveals that hydroxyurea treatment inhibits neutrophil activation.40 Additional data suggests that hydroxyurea treatment lowers vascular occlusion in sickle cells by limiting the recruitment and activation of PMNs via the NO-cGMP pathway.41 The discovery that sickled erythrocytes bind to PMN in-vitro adds to the growing body of evidence indicating neutrophils may have a direct impact on SCD pathogenesis. In vivo studies in SCD mice have discovered that intravital imaging can be used to study the pattern of blood cell flow in the cremasteric microvasculature. Sickled erythrocytes primarily interact with attached leukocytes in postcapillary venules in this model. Surgical trauma induces these interactions, which are increased and extended by the administration of tumor necrosis factor (TNF), resulting in a fatal vaso-occlusive crisis (VOC). In the test subjects lacking E-and P-selectins, which usually block leukocyte migration to the vascular endothelial layer, VOC is not seen.39 First, inflammatory stimuli from within and outside the body activate endothelial cells, allowing circulating neutrophils to be attracted to postcapillary venules. The attached PMNs then connect with circulating sickled erythrocytes via activated Mac-1, restricting venular blood flow for a short or long time. Increased RBC transit time, ischemia, erythrocyte sickling, the recruitment of neutrophils, and interaction within this blood cell recruitment all result in vascular occlusions, which can lead to life-threatening crises.
The role of monocytes is not ignored by Zhang D. Monocytes with SCD have a functional characteristic that encourages inflammation—they can activate the nuclear factor B (NF-B) pathway in the cells of the endothelium. By enhancing the production of leukocyte AMs like E-selectin, ICAM-1 (intercellular adhesion molecule), and VCAM-1, the NF-B pathway increases mononuclear leukocyte attachment to endothelial cells. Endothelial cells are stimulated by monocytes via proinflammatory cytokines produced by monocytes rather than cell-cell contact. When tumor necrosis factor-alpha and IL-1 from monocytes are blocked by using neutralizing antibodies, there is a decrease in the expression of E-selectin by the endothelial cells.42,43
The in-vivo study of Turhan et al13 to demonstrate that sickled erythrocytes attach to adhering leukocytes in inflamed venules has already been covered briefly in previous study reviews. They used intravital microscopy to view the microvasculature of genetically engineered mice that exclusively express human hemoglobin S (HbS). These mice have irreversibly sickled circulating cells and exhibit several symptoms of SCD, such as anemia, increased reticulocyte counts, and end-organ damage. Using two separate animal models of SCD, the researchers discovered that sickled erythrocytes interact with white blood cells attached to the cremasteric postcapillary and collect venules. These interactions were sustained or increased when TNF-alpha was given shortly after the surgical alteration (30 minutes). Although most interactions between the sickled erythrocytes and white blood cells were short-lived, some lasted several seconds, withstood blood shear, and resulted in vascular occlusion. These findings show that the attachment of leukocytes to the walls of the blood vessels has a direct impact on the arterial occlusions seen in SCD.
Okpala et al12 looked at 100 HbSS patients, 43 males and 57 females between the ages of 18 and 65. There were 34 HbAA controls in all, 11 men and 23 females between 19 and 60. The connection between leukocyte adhesion molecules and SCD clinical symptoms was investigated using flow cytometry. HbSS patients with any condition other than SCD, HbF levels greater than 10%, and controls with any known medical diagnosis were all excluded from the study. SCD and control participants were both excluded if they had been pregnant or had given birth in the previous three months. Significant levels of aMb integrin expression in neutrophils and l-selectin expression in lymphocytes and neutrophils were seen in patients with SCD (P < 0.03).
The neutrophils of sickle nephropathy patients had a high level of CD18 expression (P = 0.018), whereas the lymphocytes of stroke patients had a high level of l-selectin (P = 0.03). Monocyte l-selectin increased (P = 0.04) in the sickle cell crisis compared to steady-state. Within one month of starting hydroxyurea therapy, there was a decrease in aLb2 integrin expression by leukocytes-neutrophils, monocytes, and lymphocytes (P = 0.05), with clinical improvement in the patients with a 3.3% increase in HbF level. They also discovered that the steady-state expression of neutrophil aMb2 integrin and l-selectin was much higher in patients with complications than in people without problems. Because adhesion molecules mediate the attachment of white blood cells to the vascular endothelial layer, this discovery is very likely. As the number of neutrophils in the blood increases, SCD becomes more severe, and leukocytosis increases the chances of the worst complication-death. The first contact of PMNs and leukocytes with the blood vessel wall is controlled by selectin (CD62L).44 CD11b and CD18 together make up the aMb2 integrin molecule, mediating leukocyte adhesion to vascular endothelium.45 It should come as no surprise that HbSS patients with elevated levels of leukocyte selectin and aMb2 integrin are more likely to develop SCD complications.46
Based on their data, they came to the following conclusions: (1): Leukocytes with high aMb2 integrin and l-selectin steady-state expression predispose patients to severe symptoms. (2) AM expression in leukocytes increased above steady-state levels and may have contributed to the beginning of the crisis. (3). Other processes, such as decreased AM expression in leukocytes, may be involved in the early clinical improvement associated with hydroxyurea therapy. (4) Other therapies that reduce leukocyte AM expression may benefit patients.
Discussion
An abnormal leukocyte count and the SCD disease process have only sometimes been directly linked in research. Due to our review of the articles, we have presented quite a few. First, there is a ton of evidence that leukocytes play a part in the development of SCD. Adults with SCD and elevated white cell counts were more likely to visit the Emergency Department (ED) as a result of sickle cell crises, according to Curtis et al.4 Leukocytes, like erythrocytes, may aid in SCD vaso-occlusion by sticking to the endothelium. The severity of SCD is highly correlated with the number of neutrophils in the blood. Unsurprisingly, among all leukocytes, neutrophils have the greatest effect on the SCD disease process. This location is closely followed by monocytes. Other leukocytes, such as basophils and eosinophils, are also present. Blood cell-vessel wall contacts and intercellular interactions are potentiators of the process.
Leukocytosis has been associated with an elevated mortality rate from illness. In some SCD patients, the advantages of hydroxyurea (HU) therapy are accompanied with a decreased neutrophil count without an increase in hemoglobin F levels.
Leukocytes undoubtedly contribute to SCD vaso-occlusion, which results in serious clinical conditions, as shown by the aforementioned study as well as several other ones that have already been mentioned.
Limitations
This study could not link the effect of reduced leukocyte count in the disease process of SCD. Furthermore, the impossibility of in-vivo human studies limits us to the use of studies in mice, which may not necessarily be an exact representation of the pathophysiology in humans.
Conclusion
The involvement of abnormal leukocyte counts in the etiology of SCD is not only supported by evidence. There is also a lot of proof that therapies that focus on these pathways enhance the clinical results of SCD patients. But there is little proof to prove that a low leukocyte count contributes to the pathophysiology of SCD.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Stamatoyannopoulos G. The molecular basis of hemoglobin disease. Annu Rev Genet. 2003;6:47–70. doi:10.1146/annurev.ge06120172000403
2. Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: an update on management. Sci World J. 2012;2012:55. PMC3415156.
3. Solovieff N, Hartley SW, Baldwin CT, et al. Ancestry of African Americans with sickle cell disease. Blood Cells Mol Dis. 2011;47(1):41. PMC3116635. doi:10.1016/j.bcmd.2011.04.002
4. Curtis SA, Danda N, Etzion Z, Cohen HW, Billett HH, Lam W. Elevated steady state WBC and platelet counts are associated with frequent emergency room use in adults with sickle cell anemia. PLoS One. 2015;10(8):e0133116. doi:10.1371/journal.pone.0133116
5. Ahmed AE, Ali YZ, Al-Suliman AM, et al. The prevalence of abnormal leukocyte count, and its predisposing factors, in patients with sickle cell disease in Saudi Arabia. J Blood Med. 2017;8(185):185–191. PMC5661844. doi:10.2147/JBM.S148463
6. Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801–809. doi:10.1182/blood-2015-09-618538
7. Akinbami A, Dosunmu A, Adediran A, Oshinaike O, Adebola P, Arogundade O. Haematological values in homozygous sickle cell disease in steady state and haemoglobin phenotypes AA controls in Lagos, Nigeria. BMC Res Notes. 2012;5(1):1–6. doi:10.1186/1756-0500-5-396
8. Buchanan GR, Glader BE. Leukocyte counts in children with sickle cell disease: comparative values in the steady state, vaso-occlusive crisis, and bacterial infection. Am J Dis Child. 1978;132(4):396–398. doi:10.1001/archpedi.1978.02120290068013
9. Okpala I. The intriguing contribution of white blood cells to sickle cell disease – a red cell disorder. Blood Rev. 2004;18(1):65–73. doi:10.1016/S0268-960X(03)00037-7
10. Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264–269. doi:10.7326/0003-4819-151-4-200908180-00135
11. Wun T. The role of inflammation and leukocytes in the pathogenesis of sickle cell disease. Hematology. 2000;5(5):403–412. doi:10.1080/10245332.2000.11746536
12. Okpala I, Daniel Y, Haynes R, Odoemene D, Goldman J. Relationship between the clinical manifestations of sickle cell disease and the expression of adhesion molecules on white blood cells. Eur J Haematol. 2002;69(3):135–144. doi:10.1034/j.1600-0609.2002.02775.x
13. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99(5):3047–3051. doi:10.1073/pnas.052522799
14. Hedo C, Aken’ova Y, Okpala I, Salimonu L. Serum immunoglobulin (G, A and M) classes and IgG subclasses in sickle cell anaemia. APMIS. 1993;101(5):353–357. doi:10.1111/j.1699-0463.1993.tb00121.x
15. Anyaegbu CC, Okpala IE, Aken’ova AY, Salimonu LS. Complement haemolytic activity, circulating immune complexes and the morbidity of sickle cell anaemia. APMIS. 1999;107(7):699–702. doi:10.1111/j.1699-0463.1999.tb01463.x
16. Hedo CC, Aken’ova YA, Okpala IE, Durojaiye AO, Salimonu LS. Acute phase reactants and severity of homozygous sickle cell disease. J Intern Med. 1993;233(6):467–470. doi:10.1111/j.1365-2796.1993.tb01000.x
17. Anyaegbu CC, Okpala IE, Akren’Ova A, Salimonu S. Peripheral blood neutrophil count and candidacidal activity correlate with the clinical severity of sickle cell anaemia (SCA). Eur J Haematol. 1998;60(4):267–268. doi:10.1111/j.1600-0609.1998.tb01036.x
18. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995;332(20):1317–1322. doi:10.1056/NEJM199505183322001
19. Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9(2):101–106. doi:10.1097/00062752-200203000-00003
20. Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost. 1999;81(01):1–7. doi:10.1055/s-0037-1614407
21. Silverstein RL, Asch AS, Nachman RL. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J Clin Invest. 1989;84(2):546–552. doi:10.1172/JCI114197
22. Walcheck B, Moore KL, McEver RP, Kishimoto TK. Neutrophil-neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation of P-selectin in vitro. J Clin Invest. 1996;98(5):1081–1087. doi:10.1172/JCI118888
23. Tan P, Luscinskas FW, Homer-Vanniasinkam S. Cellular and molecular mechanisms of inflammation and thrombosis. Eur J Vasc Endovasc Surg. 1999;17(5):373–389. doi:10.1053/ejvs.1998.0759
24. Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96(7):2451–2459. doi:10.1182/blood.V96.7.2451
25. Stewart West M, Wethers D, Smith J, Steinberg M. The cooperative study of sickle cell disease. Laboratory profile of sickle cell disease: a cross-sectional analysis. The cooperative study of sickle cell disease. J Clin Epidemiol. 1992;45(8):893–909. doi:10.1016/0895-4356(92)90073-V
26. Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342(2):83–89. doi:10.1056/NEJM200001133420203
27. Platt OS. The acute chest syndrome of sickle cell disease. N Engl J Med. 2000;342(25):1904–1907. doi:10.1056/NEJM200006223422510
28. Hofstra TC, Kalra VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996;87(10):4440–4447. doi:10.1182/blood.V87.10.4440.bloodjournal87104440
29. Mendoza E, Gutgsell N, Temple JD, Issitt P. Monocyte phagocytic activity in sickle cell disease. Acta Haematol. 1991;85(4):199–201. doi:10.1159/000204892
30. Gawaz M, Fateh‐moghadam S, Pilz G, Gurland HJ, Werdan K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest. 1995;25(11):843–851. doi:10.1111/j.1365-2362.1995.tb01694.x
31. Weyrich AS, Elstad MR, McEver RP, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97(6):1525–1534. doi:10.1172/JCI118575
32. Dias-Da-Motta PM, Arruda VR, Muscará MN, et al. The release of nitric oxide and superoxide anion by neutrophils and mononuclear cells from patients with sickle cell anaemia. Br J Haematol. 1996;93(2):333–340. doi:10.1046/j.1365-2141.1996.4951036.x
33. Adler BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VVB, Prchal JT. Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood. 2001;97(10):3313–3314. doi:10.1182/blood.V97.10.3313
34. Abboud M, Laver J, Blau CA. Granulocytosis causing sickle-cell crisis. Lancet. 1998;351(9107):959. doi:10.1016/S0140-6736(05)60614-9
35. Wei A, Grigg A. Granulocyte colony-stimulating factor-induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta+ thalassemia. Blood. 2001;97(12):3998–3999. doi:10.1182/blood.V97.12.3998
36. Wali Y, Beshlawi I, Fawaz N, et al. Coexistence of sickle cell disease and severe congenital neutropenia: first impressions can be deceiving. Eur J Haematol. 2012;89(3):245–249. doi:10.1111/j.1600-0609.2012.01827.x
37. Saleh AW, Hillen HFP, Duits AJ. Levels of endothelial, neutrophil, and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102(1):31–37. doi:10.1159/000040964
38. Almeida CB, Scheiermann C, Jang JE, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood. 2012;120(14):2879–2888. doi:10.1182/blood-2012-02-409524
39. Barazia A, Li J, Kim K, Shabrani N, Cho J. Hydroxyurea with AKT2 inhibition decreases vaso-occlusive events in sickle cell disease mice. Blood. 2015;126(22):2511–2517. doi:10.1182/blood-2015-02-626234
40. Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The multicenter study of hydroxyurea in sickle cell anemia. Medicine. 1996;75(6):300–326. doi:10.1097/00005792-199611000-00002
41. Benkerrou M, Delarche C, Brahimi L, et al. Hydroxyurea corrects the dysregulated L-selectin expression and increased H(2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood. 2002;99(7):2297–2303. doi:10.1182/blood.V99.7.2297
42. Safaya S, Steinberg MH, Klings ES. Monocytes from sickle cell disease patients induce differential pulmonary endothelial gene expression via activation of the NF-κB signaling pathway. Mol Immunol. 2012;50(1–2):117–123. doi:10.1016/j.molimm.2011.12.012
43. Keegan PM, Surapaneni S, Platt MO. Sickle cell disease activates peripheral blood mononuclear cells to induce cathepsins k and v activity in endothelial cells. Anemia. 2012;2012:1–7. doi:10.1155/2012/201781
44. Ivetic A, Green HLH, Hart SJ. L-selectin: a major regulator of leukocyte adhesion, migration and signaling. Front Immunol. 2019;10(May). PMC6527602. doi:10.3389/fimmu.2019.01068
45. Panés J, Perry M, Granger DN. Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention. Br J Pharmacol. 1999;126(3):537. PMC1565837, doi:10.1038/sj.bjp.0702328
46. Johnson C, Telen MJ. Adhesion molecules and hydroxyurea in the pathophysiology of sickle cell disease. Haematologica. 2008;93(4):481–485. doi:10.3324/haematol.12734
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.