")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Supplement 2
Immunotoxicity of zinc oxide nanoparticles with different size and electrostatic charge
Authors Kim C, Nguyen H, Ignacio RM, Kim J, Cho H, Meang E, Kim Y, Kim M, Park B, Kim S
Received 20 November 2013
Accepted for publication 17 February 2014
Published 15 December 2014 Volume 2014:9(Supplement 2) Pages 195—205
DOI https://doi.org/10.2147/IJN.S57935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Cheol-Su Kim,1,* Hai-Duong Nguyen,1,* Rosa Mistica Ignacio,2 Jae-Hyun Kim,1 Hyeon-Cheol Cho,1 Eun Ho Maeng,3 Yu-Ri Kim,4 Meyoung-Kon Kim,4 Bae-Keun Park,5 Soo-Ki Kim1,5
1Department of Microbiology, Wonju College of Medicine, Yonsei University, Wonju-si, Gangwon-do, Republic of Korea; 2Department of Environmental Medical Biology, Wonju College of Medicine, Yonsei University, Wonju-si, Gangwon-do, Republic of Korea; 3Healthcare Laboratory, Medical Device Evaluation Team, Korea Testing and Research Institute, Gimpo-si, Gyeonggi-do, Republic of Korea; 4Department of Biochemistry and Molecular Biology, Medical School and College, Korea University, Seoul, Republic of Korea; 5Institute of Lifestyle Medicine, Wonju College of Medicine, Yonsei University, Wonju-si, Gangwon-do, Republic of Korea
*These authors contributed equally to this work
Abstract: While zinc oxide (ZnO) nanoparticles (NPs) have been recognized to have promising applications in biomedicine, their immunotoxicity has been inconsistent and even contradictory. To address this issue, we investigated whether ZnO NPs with different size (20 or 100 nm) and electrostatic charge (positive or negative) would cause immunotoxicity in vitro and in vivo, and explored their underlying molecular mechanism. Using Raw 264.7 cell line, we examined the immunotoxicity mechanism of ZnO NPs as cell viability. We found that in a cell viability assay, ZnO NPs with different size and charge could induce differential cytotoxicity to Raw 264.7 cells. Specifically, the positively charged ZnO NPs exerted higher cytotoxicity than the negatively charged ones. Next, to gauge systemic immunotoxicity, we assessed immune responses of C57BL/6 mice after oral administration of 750 mg/kg/day dose of ZnO NPs for 2 weeks. In parallel, ZnO NPs did not alter the cell-mediated immune response in mice but suppressed innate immunity such as natural killer cell activity. The CD4+/CD8+ ratio, a marker for matured T-cells was slightly reduced, which implies the alteration of immune status induced by ZnO NPs. Accordingly, nitric oxide production from splenocyte culture supernatant in ZnO NP-fed mice was lower than control. Consistently, serum levels of pro/anti-inflammatory (interleukin [IL]-1Β, tumor necrosis factor-α, and IL-10) and T helper-1 cytokines (interferon-γ and IL-12p70) in ZnO NP-fed mice were significantly suppressed. Collectively, our results indicate that different sized and charged ZnO NPs would cause in vitro and in vivo immunotoxicity, of which nature is an immunosuppression.
Keywords: immunosuppression, cytokine, ZnO, immune response, cytotoxicity, innate immunity
Introduction
Nanotechnology has enabled nanoparticles (NPs) to be designed at the molecular (nanometer) level. Thus, NPs have received the tremendous advantage of their small size, novel physicochemical properties, as well as their interactions with biological systems. In particular, inorganic NPs with metal oxides have been pre-clinically employed for diagnostic and therapeutic use in biomedicine.1–4 Of these metal oxides, zinc oxide (ZnO) NPs have received considerable attention, with a promising biological application for drug delivery and cancer therapy5–7 due to their great photo-catalytic and photo-oxidizing ability against chemical and biological species.
Despite the potential biomedical application of ZnO NPs, biohazards and toxicities of ZnO NPs remain unclear. Of these toxicities, the effects of ZnO NPs on the immune system are poorly documented. Here, immunotoxicity is defined as the adverse effects on the immune system such as hypersensitivity, chronic inflammation, immunosuppression, immunostimulation, and autoimmunity. Much evidence suggests that ZnO NPs would function as immunotoxicants.8–11 ZnO NPs have unique physicochemical properties, therefore, they could easily access several immune tissues and cells through various routines such as inhalation, ingestion, skin uptake, and injection. ZnO NP oral administration could cause severe damage to heart, lung, liver, and kidney,12 consequently leading to inflammation. In addition, in vitro exposure of ZnO leads to generation of reactive oxygen species (ROS), upregulation of genes involved in apoptosis, and cell death in human keratinocyte HaCaT cells.13,14 Likewise, profuse release of inflammatory mediators induced by ZnO NPs15,16 may result in immune stimulation or aggravation of immune diseases17 or even break down T helper (Th)-1/Th-2 balance.18
It is well known that immunotoxicity of NPs is intimately linked to oxidative stress. For instance, the toxicity of ZnO NPs to immune cells is involved in ROS generation.19–22 The high production of superoxide in mitochondria reduces the mitochondrial membrane potential,23 causing cell cycle arrest at S/G2 phase,24 and increases the ratio of Bax/Bcl-2, leading to the mitochondria-mediated pathway involved in apoptosis.25 However, the mechanism of immunotoxicity of ZnO NPs in relation to oxidative stress is unclear.
To trigger immunotoxicity, several traits or features of NPs are essential.26 Emerging evidence implies that the toxicity of ZnO NPs could be affected by size and/or electrostatic charge.27,28 For instance, an increase of ZnO NPs size might conversely decrease their toxicities.19,29 Positively charged NPs could exert higher immunotoxicity than negatively charged NPs due to effective interaction with the negative charge of acidic acid on the surface of macrophages.30 However, these studies27,28 failed to address whether immunotoxicity of ZnO NPs would be affected by the size and/or charge and whether this immunotoxicity could be categorize as immunostimulation and/or immunosuppression.
In this study, we explored the in vitro potential immunotoxicity of ZnO NPs on Raw 264.7 cells, and the systemic in vivo immunotoxicity of ZnO NPs using C57BL/6 mice. Further, we investigated the role of size and charge in ZnO NP-induced immunotoxicity.
Material and methods
Preparation of particle suspensions
ZnO (ZnO-310, Lot No 141319) was purchased from Sumitomo Osaka Cement Co, Ltd, (Tokyo, Japan). ZnOAE100 (Zn-OX-01-NP.100N, Lot No 1871511079-673) was purchased from American Elements (Los Angeles, CA, USA). The ZnO NPs used in this study were sized at 20 nm (ZnOSM20, Sumitomo Osaka Cement Co, Ltd) and 100 nm (ZnOAE100; American Elements), with approximately 99.5% purity, milky white color, and nearly spherical shape. According to the manufacturer information, the actual size and zeta potential of the ZnO NPs used in this study were as follows: 29±3 nm, −44.4±1 mV (ZnOSM20(−)); 35±5 nm, 26.3±0.5 mV (ZnOSM20(+)); 72±11 nm, −41.6±0.6 mV (ZnOAE100(−)); and 79±12 nm, 26.1±0.4 mV (ZnOAE100(+)). The surface charge (zeta potential) was modified with coating reagents, citrate to make the ZnO NPs negatively charged and L-serine to make them positively charged, as reported previously.31 In brief, 20 mg dry powder of ZnO NPs was dissolved into 100 mL of L-serine/HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 6.2, and citrate/HEPES pH 7.3 to make the NP surfaces electrostatically charged. Indicated buffers were made as follows: for the positive charge buffer, 99 mL of 20 mM HEPES pH 6, L-serine (1 g) adjusted to pH 6.2, and for the negative charge buffer, 99 mL of 20 mM HEPES pH 7, sodium citrate (1 g) adjusted to pH 7.3. Subsequently, the ZnO NP suspension was vortexed for 5 minutes at room temperature and then kept at 4°C until use. Before using, the suspension was sonicated at 4°C for 10 minutes with a sonicator (Hielscher-Ultrasound Technology, Teltow, Germany).
Cell culture
Raw 264.7, mouse macrophage cell line (American Type Culture Collection [ATCC], Manassas, VA, USA) was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Hyclone Laboratories, Inc., South Logan, UT, USA) supplemented with 10% heat-activated fetal bovine serum (FBS, Hyclone Laboratories Inc.) and 1% antibiotic–antimycotic (Gibco®, Invitrogen Corporation, Carlsbad, CA, USA) at 37°C in a 5% CO2 incubator.
In vitro cell viability study
A commercially available cell viability assay Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Rockville, MD, USA) was employed to evaluate the cytotoxic effect of ZnO NPs. Approximately 1×105 of Raw 264.7 cells was seeded into 96-well plates then incubated with various concentrations of ZnO NPs for 24 hours at 37°C in a 5% CO2 incubator. Cells not treated with ZnO NPs served as a control well in the experiment. Non-modified zinc chloride (ZnCl2) dissolved in 1X phosphate buffered saline (PBS) was used as a control, without modification of surface. Afterwards, 10 μL of CCK-8 solution was added to each well, incubated for 1 hour, and then absorbance was determined at 450 nm by a DTX-880 multimode microplate reader (Beckman Coulter, Fullerton, CA, USA). The percentage of cell viability was calculated by the following formula: cell viability (%) = (mean absorbency in test wells)/(mean absorbency in control wells) ×100. All experiments were performed in triplicate.
Maintenance of animals and ZnO NP treatment
Six-week-old inbred C57BL/6 mice (n=5) were purchased from Orient Bio Inc. (Seongnam, South Korea) and were maintained in a pathogen-free condition, and fed with a standard commercial diet. Mice were randomly assigned into five groups: normal control group, which was PBS treated, and four experimental groups treated with four types of ZnO NPs. Briefly, the ZnO NP suspension was orally administered in the mice with the dosage of 750 mg/kg every day continuously for 14 days. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Wonju College of Medicine, Yonsei University.
Induction and evaluation of delayed-type hypersensitivity (DTH)
Mice were orally administered with 750 mg/kg of different sized and charged ZnO NPs for 2 weeks. DTH assays were performed 4 days before sacrifice. To assess DTH response, mice were subcutaneously injected in the left footpad with 20 μL of saline as a control, and in the right footpad with 20 μL of ZnO NP suspension. At the indicated times after challenge (24 hours and 48 hours), footpad thickness was measured with a digital caliper (Mitutoyo Corporation, Tokyo, Japan). The level of the DTH response was determined as the difference between the left and right footpad.
Preparation of splenocytes
To isolate splenocytes, the spleen was removed aseptically from C57BL/6 mice at the endpoint treatment and was placed in a tube containing 1× PBS on ice. Splenocyte suspensions were prepared by gently pressing the spleen between the frosted ends of two sterile microscope slides into a 100 mm tissue culture grade petri dish. The slides were rinsed at regular intervals with 1× PBS. Cell suspensions were filtered by a sterile plastic strainer and then centrifuged at 1,500 rpm for 3 minutes. For optimal lysis of erythrocytes, the pellets were resuspended in 5 mL red blood cell lysis buffer and incubated on ice for 5 minutes with occasional shaking. The reaction was stopped by diluting the lysis buffer with 25 mL of 1× PBS. Thereafter, the cells were spun (1,500 rpm at 4°C for 5 minutes), and the supernatant was carefully removed. The pellet was then washed two times in 1X PBS and resuspended in Roswell Park Memorial Institute (RPMI)-1640 supplemented with 3% FBS (Hyclone Laboratories Inc.) and 1% antibiotic-antimycotic (Invitrogen Corporation). Cells were then counted. The viability of the cells used in all the experiments was higher than 95%, as measured by the trypan blue exclusion method (Sigma-Aldrich, St Louis, MO, USA).
Splenocyte proliferative responses to concanavalin A (Con A) and lipopolysaccharide (LPS)
Splenocytes were seeded at 1×105 cells per well into a 96-well, flat-bottom microtiter plate in 100 μL RPMI-1640 (Hyclone Laboratories, Inc.) supplemented with 10% heat-activated FBS (Hyclone Laboratories Inc.) and 1% antibiotic/antimycotics. Thereafter, 1.25 μg/mL Con A (Sigma-Aldrich, St Louis, MO, USA) and 500 μg/mL of LPS (Sigma-Aldrich) were used. The plates were incubated at 37°C in a humidified atmosphere under 5% CO2 for 6 hours. Cell proliferation was evaluated using a CCK-8 (Dojindo Molecular Technologies, Inc.) according to the manufacturer’s instructions. Absorbance was measured at 450 nm by a DTX-880 multimode microplate reader (Bechman Counter Inc.).
Cytotoxicity assay of natural killer (NK)-cells
NK-enriched murine splenocytes were used as effector cells, and YAC-1 (ATCC) as target cells. Briefly, various effector cell dilutions were prepared: 1×106 cells/well for 100:1, 5×105 cells/well for 50:1, and 2.5×105 cells/well for 25:1. A constant number of target cells (1×103 cells/well) were then co-cultured with the different effector cell dilutions prepared in a 96-well plate to test several effector/target cell ratios. The final combined volume was 100 μL/well. The effector/target cells were incubated 6 hours at 37°C humidified incubator with 5% CO2. The cytotoxic activity of NK-cells was assessed by a Cytotox 96 non-radioactive cytotoxicity assay (Promega Corporation, Fitchburg, WI, USA). Briefly, 50 μL supernatant from the cell culture was collected and was moved to the new 96-well round-bottom plate. Then, 50 μL of the lactate dehydrogenase (LDH) substrate mixture was added to each well and incubated for 30 minutes at room temperature, protected from light. The LDH that was released upon cell lysis was measured in the supernatant by optical density (OD) measurement at 490 nm. Target cell lysis was calculated as: (OD of sample − OD with spontaneous release of LDH from target cells − OD with spontaneous release of LDH from effector cells) × 100/(OD with maximal release of LDH from target cells − OD with spontaneous release of LDH from target cells). All experiments were performed in triplicate.
Immunophenotyping of splenocytes
Specific leukocyte subtypes of cells derived from mouse spleen were also determined by immunofluorescent antibody staining and analyzed with flow cytometry. Lymphocyte subpopulations were identified and gated using forward versus side scatter characteristics. All monoclonals were directly conjugated and were obtained from BD Biosciences (San Jose, CA, USA). Th cells (CD4+), cytotoxic T-cells (CD8+), T-cells (PE-CD4+, FITC-CD8+), B-cells (PE-CD19+, FITC-B220[CD45R+]), NK-cells (CD16+), macrophages (PE-CD11b+; FITC-CD14+), and monocytes (CD14+) were identified using the anti-mouse antibodies. Thereafter, approximately 5×105 cells were resuspended in flow cytometry buffer (2% FBS, 0.02% sodium azide in PBS) containing Fc-block to reduce nonspecific antibody binding. Cells were then incubated in the dark with the appropriate fluorochrome-conjugated antibody (10 μL of 1 μg/mL) for 30 minutes at 4°C. Afterwards, cells were washed twice with 500 μL FACS (fluorescence activated cell sorter) buffer, and flow cytometry analysis was performed on the Cytomics™ FC 500 Flow Cytometer (Beckman Coulter). Control samples were matched for each fluorochrome. Data were analyzed using Modfit LT 3.2 (Verity Software House, Topsham, ME, USA) software.
Measurement of nitric oxide (NO)
NO production in the primary splenocyte culture medium was quantified spectrophotometrically using the Griess reagent G2930 (Promega Corporation). The nitrite (NO2−) present in the supernatant of splenocytes was used as an indicator of NO. Nitrite is a stable degradation product of NO. Briefly, 50 μL of the splenocyte supernatant was mixed with an equal volume of Griess reagent in a 96-well round-bottom microtiter plate and incubated at room temperature for 15 minutes. The absorbance at 540 nm was measured, and the NO concentration was determined using a calibration curve, with sodium nitrite as a standard chemical.
Measurement of serum cytokine level
The level of the cytokines (interleukin [IL]-1β, IL-6, tumor necrosis factor [TNF]-α, interferon [IFN]-γ, IL-12p70, and IL-10) in serum was determined using Multiplex Bead Suspension Array System (Bio-Rad Laboratories, Hercules, CA, USA). The cytokine multiplex bead suspension array kit was purchased from Bio-Rad Laboratories. Briefly, each set of premixed beads coated with the target antibodies was added to a well, and incubated with the sample in a 96-well round-bottomed microtiter plate to react with specific analytes. Then, premixed detection antibodies were added to the wells followed by a fluorescently labeled reporter molecule that specifically binds the analyte. Standard curves for each cytokine were generated using the standard control concentrations provided in the kit. Each step requires specific incubation time, with shaking at room temperature and washing steps. All washes were performed using a Bio-Plex Pro™ (Bio-Rad Laboratories) wash station. Finally, the samples were then read using the Bio-Plex suspension array reader, and data acquisition and analysis were carried out with the five-parameter logistic method.
Statistical analysis
All data are presented as the mean ± standard error of the mean. The mean values among different groups were analyzed and compared using one-way analysis of variance (ANOVA) followed by subsequent multiple comparison test (Tukey) with Graph Prism (GraphPad Software, La Jolla, CA, USA) version 5.0 software packages. One-way ANOVA with repeated measurements followed by Tukey’s test was applied to test the influence of ZnO NPs on bodyweight gain. A P≤0.05 was considered to be statistically significant.
Results
Cell viability study
The influence of ZnO NP size and charge on immune cells remains unclear. To clarify this, using four types of ZnO NPs, we examined in vitro cell viability. The effect of ZnO NPs on the viability of Raw 264.7 cells was examined using CCK-8 assay, and ZnCl2 was used as a positive control group (Figure 1). ZnO NPs at the concentrations 0–5 μg/mL had minimal effect on the viability of Raw 264.7 cells, although they significantly reduced the viability at a higher concentration range (10–80 μg/mL) after 24 hours incubation. To compare the potency of each ZnO NP, the half maximal effective concentration (EC50) value was calculated according to sigmoidal dose–response regression (Table 1). The range of EC50 of ZnO NPs was 6.584–8.413 μg/mL: ZnOAE100(+) exerted the highest cytotoxicity against Raw 264.7 cells (EC50 =7.499 μg/mL), while the cytotoxicity strength of other ZnO NPs was ZnOAE100(−) > ZnOSM20(+) > ZnOSM20(−) > ZnCl2 in descending order. Of note, ZnCl2 was less cytotoxic than ZnO NPs (EC50 =14.82 μg/mL). Additionally, EC50 values of 100 nm-sized ZnO NPs were higher than those of 20 nm-sized ZnO NPs. Moreover, positively charged NPs showed higher cytotoxicity compared with their positively charged counterpart, suggesting that the size and charge of ZnO NPs could affect their cytotoxicity.
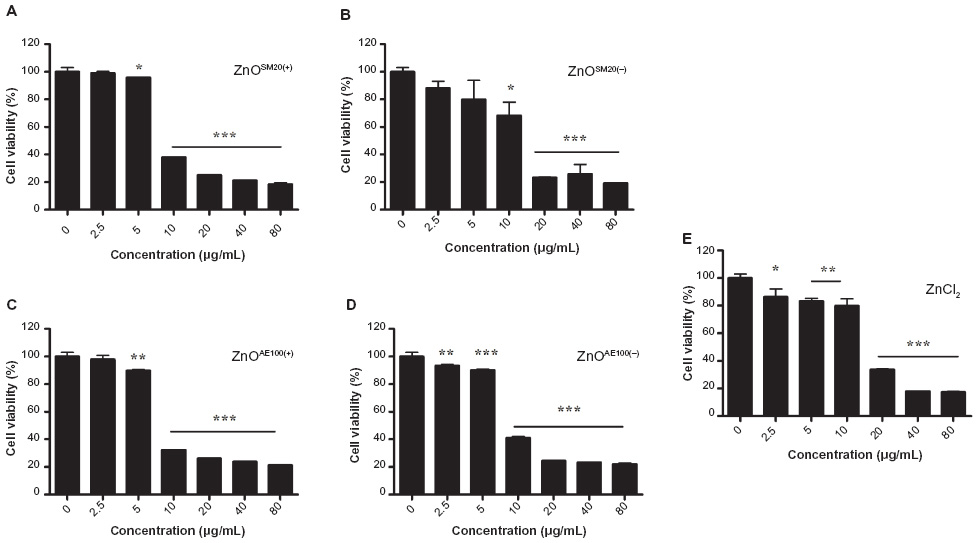 | Figure 1 Effect of different sized and electrostatically charged ZnO NPs on the viability of Raw 264.7 cells. Cells were incubated, with indicated concentrations of ZnO NPs for 24 hours: (A) ZnOSM20(+), (B) ZnOSM20(−), (C) ZnOAE100(+), (D) ZnOAE100(−), and (E) ZnCl2. DMEM media was used as a negative control. Cell viability was then determined by a CCK-8 assay. |
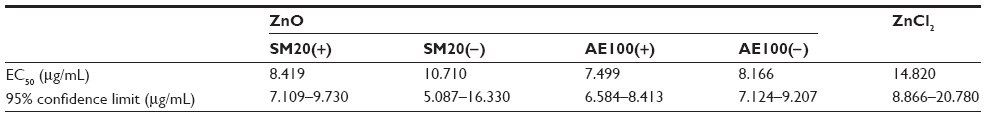 | Table 1 EC50 values (24 hours growth inhibition) of ZnO NPs on Raw 264.7 cells |
Immunophenotyping
ZnO NP treatment slightly induced the alteration in cell distribution of splenocytes (Table 2). While Th (CD4+) and cytotoxic T (CD8+) cells accounted for 16.6% and 9.1%, respectively, of the control, the distribution of Th cells and cytotoxic T-cells in splenocytes of treated groups (ZnOSM20(+/−), ZnOAE100(+/−)) was changed to 14.66%/9.18%, 15.44%/11.62%, 14.53%/10.22%, 16.80%/11.60%, respectively. Notably, the percentage of Th cells was significantly reduced when treated with ZnOAE100(+) as compared with control (Table 2). Moreover, the ratio of CD4+ T-cells to CD8+ T-cells, which are subpopulations of T-cells (Th cells and cytotoxic T-cells), significantly changed from 1.634-fold to 1.301-fold in ZnOAE100(+)-fed mice. However, little differences were noted in the proportion of B-cell, NK-cell, macrophage, and monocyte subpopulations.
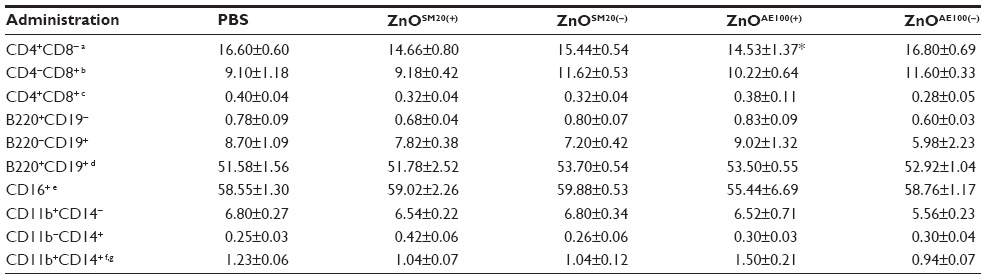 | Table 2 Immunophenotype of splenocytes in C57BL/6 mice fed with ZnO NPs for 14 days |
Innate, cell-mediated immune response (DTH and mitogenic response) against ZnO NPs
To assess the effect of ZnO NPs on regulation of innate immune response, NK-cell activity was examined using NK-sensitive YAC-1 target cells. As shown in Figure 2, a decrease of NK-cell activity was observed in the mice treated with ZnO NPs. At the ratio of 100:1, ZnO NPs significantly inhibited NK-cell activity. Next, we examined the effect of ZnO NPs on cell-mediated immunity (CMI) using DTH response on C57BL/6 mice. The swelling volume was similar after 24-hour and 48-hour challenges (Table 3). An increased DTH response to ZnO NPs was observed. However these differences were not statistically significant. Further, we analyzed the mitogen-stimulated proliferative responses of T- and B-lymphocytes (Figure 3). These responses were elevated compared with control but not significant. Taken together, ZnO NPs affected NK activity but not CMI.
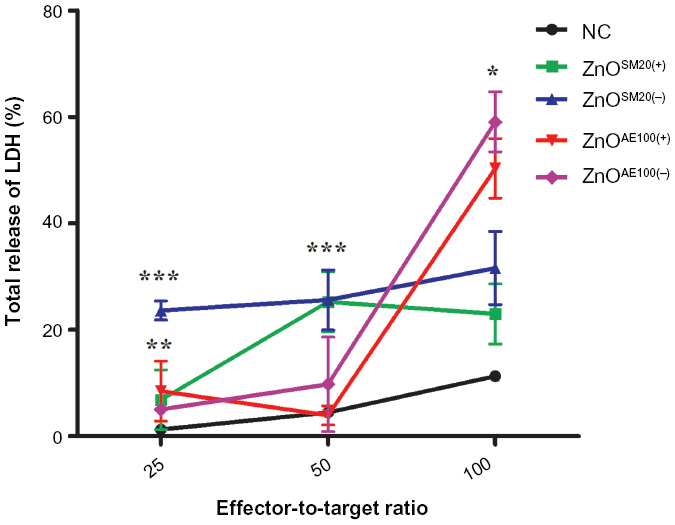 | Figure 2 Effects of ZnO NP treatment on NK-cell activity. Effector cells (NK-cells) were isolated from the spleen of mice fed with a 750 mg/kg dose of ZnO NPs for 14 days. YAC-1 cells were used as target cells. ZnO NP treatment reduced NK-cell cytotoxicity in the treated groups compared with control. |
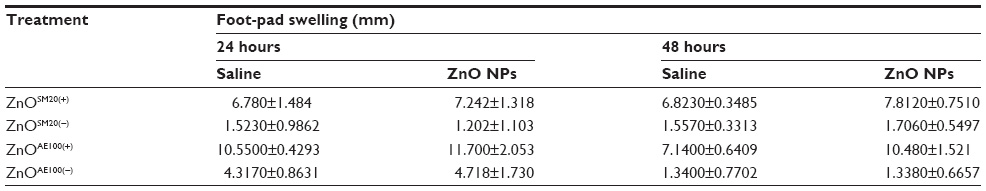 | Table 3 Delayed type hypersensitivity in ZnO NP-primed mice |
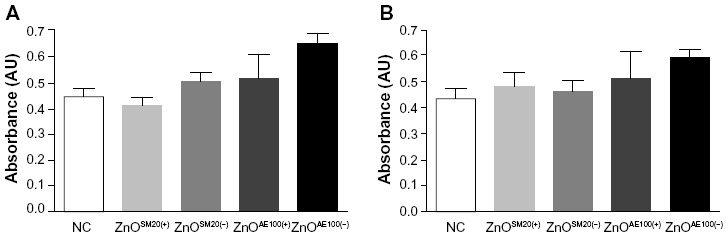 | Figure 3 Mitogenic response of mouse splenocytes. Splenocytes (1×106 cells/mL) were incubated for 24 hours with concanavalin A or lipopolysacchride to evaluate T-lymphocyte (A) and B-lymphocyte (B) proliferation, respectively. |
NO production of splenocytes
NO is a reactive form of nitrogen and is an important cellular signaling molecule involved in several biological processes, and moreover, serves as one of the key mediators of immune defense. To further explore the impact of ZnO NPs on immune defense, we measured the level of splenic NO production after 14 days treatment. As shown in Figure 4, a significant decrease in NO level occurred after administration of ZnOSM20(−) and ZnOAE100(+/−). However, there was no substantial difference of NO level in ZnOSM20(+)-fed mice as compared with the control.
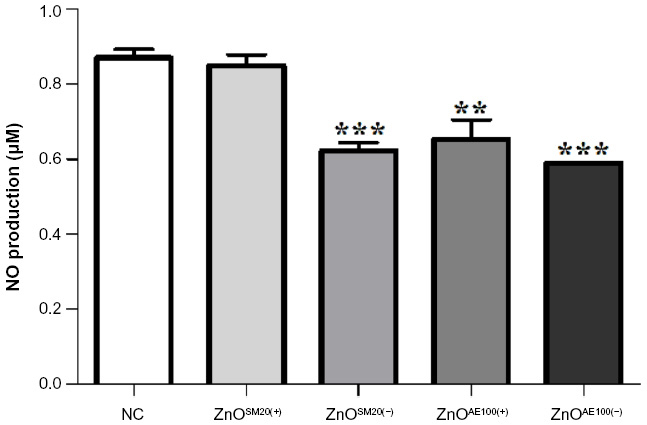 | Figure 4 Detection of NO induced by ZnO NPs on mouse splenocytes. Splenocytes were isolated and cultured. NO production in the supernatant was measured by the Griess reaction. |
Serum cytokine level
To explore immune status change induced by ZnO NPs we examined serum pro- and anti-inflammatory cytokine (IL-1β, IL-6, TNF-α, IL-12p70, IFN-γ, and IL-10) levels at the endpoint treatment. We found that overall, the level of serum cytokines in ZnO-fed mice was lower than saline-fed mice (Figure 5). In particular, serum levels of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-12p70) in ZnO NP-fed mice were ZnOSM20(+) > ZnOSM20(−) > ZnOAE100(+) > ZnOAE100(−) in descending order. Accordingly, the level of IL-10, an anti-inflammatory cytokine in ZnO NP-fed mice tended to be significantly lower than PBS-fed mice. By contrast, there was no significant change of cytokine level in ZnOSM20(+)-fed mice. In terms of size and charge effect on serum cytokine level, the size and charge were correlated to serum cytokine level. Further, the size appears more correlated to the serum cytokine than the charge. Taken together, our data show that oral intake of ZnO NPs in mice could drop the level of serum cytokines, importantly implying the possible suppression of immune status of C57BL/6 mice fed with ZnO NPs.
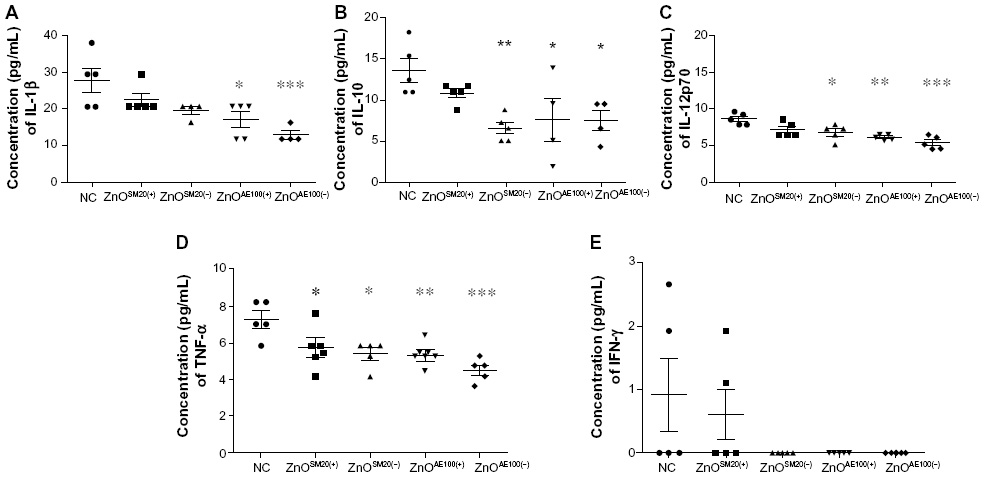 | Figure 5 ZnO NPs suppress cytokine release. After 14 days exposure, mice were retro-orbital bled, and serum was isolated by centrifugation and examined for levels of IL-1β (A), IL-10 (B), IL-12p70 (C), TNF-α (D), and IFN-γ (E) using Multiplex Bead Array System (Bio-Rad Laboratories, Hercules, CA, USA). |
Discussion
Convincing evidence has shown that ZnO NPs would have a potential to be accumulated to several organs such as liver, spleen, kidneys, brain or heart after deliberately placed in the body.32 In this regard, ZnO NPs would encounter diverse immune cells in the human body, thus damaging immune tissues and organs. However, the immunotoxicity of ZnO NPs in relation to the size and charge of ZnO NPs is unclear. To address this issue, we assessed the immunotoxicity of ZnO NPs in vitro and in vivo and explored their underlying mechanism. First, the in vitro study indicates that ZnO NPs induce cytotoxicity to immunocytes via cell viability assay, which is influenced by the size and charge of ZnO NPs. To validate this, we examined cell viability using colorimetric (CCK-8 assay) in Raw 264.7, macrophage cell line.
Cell viability assay showed that ZnO NPs with different size and electrostatic charge have differential cytotoxicity to Raw 264.7 cells (Figure 1). Interestingly, the positively charged NPs exerted higher cytotoxicity against Raw 264.7 cells than the negatively charged ones. ZnO NP-induced cytotoxicity could be affected by the release of Zn2+ ions through the dissolution of ZnO NPs within aqueous culture media.33–35 In terms of EC50, we found that ZnO NPs would cause cytotoxicity at lower doses (Table 1). This difference might be due to inherent features of metal oxides: free Zn2+ ions and oxidative radical-generated oxide, and ZnO NP surface charge, would synergistically exert cytotoxicity to eukaryotic cells. In this experiment, another merit is the validity of in vitro toxicity test using the Raw 264.7 cell line. While, this cell line is a well-known standard cell line for in vitro toxicity testing, in vitro toxicity testing for nanoparticles has not been established. Collectively, we have demonstrated that macrophage Raw 264.7 cells exposed to ZnO NPs show different sized- and charged-dependent cytotoxicity, as was observed by others.19,36,37 Further, the dose-dependent decrease in cell viability by exposure with ZnO NPs might be related to the dissolution and release of Zn2+ ions, as previously observed in in-vitro studies.33–35,38–40 This Zn2+ ion would affect cellular differentiation and cellular metabolism solely or via the interaction with proteins in vitro and in vivo.
Since, in vitro culture cannot reflect the complexity of an in vivo system, which is preferred for the toxicological evaluation, we next examined the immunotoxicological parameters induced by oral intake of ZnO NPs in mice. Our in vivo study indicates that the different sized- and charged-ZnO NPs could cause immunotoxicity in ZnO NP-fed mice, of which nature is an immunosuppression. This stems from our immunotoxicological data. Prior to immunotoxicological evaluation, we checked the weight-related parameters (bodyweight and coefficient of spleen to bodyweight). Of these, only the reduced bodyweight gain was noted (data not shown), suggesting the potential immunotoxicity of ZnO NPs in vivo. Since in vitro data showed ZnO-induced toxicity in immunocytes, bodyweight change coupled with in vitro data might be considered for predicting potential immunotoxicity of nanoparticles.
To explore the effect of ZnO NPs on the CMI, we measured the DTH reaction to ZnO NPs, splenocyte proliferation in response to Con A and LPS, NK-cell activity, and splenocyte phenotyping. Overall, no significant change was observed in DTH responses (Table 3) and T- and B-cell proliferation (Figure 3). By contrast, we found a slight change in splenocyte phenotypes (Table 2). Since CD4+ cells can help the proliferation and differentiation of Th cells, and CD8+ cells can exert cytotoxicity and regulate CD4+ cells, the ratio of CD4+/CD8+ can reflect the overall immune status in the host. Of note, there was the decreased percentage of CD4+ Th cell subpopulation in ZnOAE100(+)-fed mice as compared with control mice (Table 2). Consistently, the ratio of CD4+/CD8+ was reduced, which might imply systemic immune suppression. These CMI variables do not reveal specific function of the innate arm in CMI. Moreover, CD16 (FcγRIII) alone can be a marker for NK, as this surface marker is shed from the surface after activation of NK-cells.41,42 Only ZnOAE100(+)-fed mice showed a decreased level of CD16 among other groups fed with ZnO. Collective evidence showed that loss of CD16 on NK-cells is associated with reduced antibody-mediated cellular cytotoxicity and weaker antitumor response.43 Therefore, levels of CD16 could influence NK-cell function. To address this, we checked NK-cell activity. Astonishingly, NK-cell activity at the ratio of 100:1 was significantly decreased in all ZnO NP-fed mice compared with control (Figure 2) and most prominent on the ZnOAE100(+)-fed group. However, further research is needed to prove this inference, such as development of tumors. NK-cells were defined as lymphocytes mediating cytotoxicity against certain tumors and virus-infected target cells. In this point, the suppression of NK-cell activity may reveal the weakened innate defense in CMI, consequently getting vulnerable to opportunistic dangers such as infection, cancer, and stress. Given this, we hypothesized that if these data would be valid, humoral immune mediators should be co-regulated toward the immune suppression. To verify this, we selected two humoral immune parameters (NO and cytokines).
First, we checked NO production from splenocytes of ZnO-fed mice. NO is a reactive nitrogen species which plays an important role in the destruction and suppression of many intracellular pathogenic organisms.44,45 NO acts as an effector molecule in macrophage-mediated cytotoxicity.46 However, the NO production of macrophages was regulated and required to be optimized by CD4+ T-cells. We found that three oral intakes of ZnO NPs, except ZnOSM20(+), significantly decreased NO production of splenocytes (Figure 4). Viewed together, this is in line with the suppression of NK-cell activity and the ratio of CD4+/CD8+ observed.
Since NO could contribute to the regulation of immune reaction by modifying the release of cytokines,47–49 finally we quantified the serum cytokine release in ZnO-fed mice. To further analyze the several facets of in vivo immunotoxicity in ZnO NP-fed mice, we selected five kinds of pro- and anti-inflammatory cytokines (IL-1β, TNF-α, IL-12p70, IFN-γ, and IL-10). Cytokine assay showed that the overall level of serum cytokines in ZnO-fed mice decreased as compared with control (Figure 5). Of these cytokines, in line with the concentration in serum, IL-1β and IL-10 were prominent in detecting the cytokine change in ZnO NP-fed mice. These cytokines were the representative pro- and anti-inflammatory cytokines, respectively. The decreased level of these opposite cytokines in ZnO NP-fed mice might be ascribed to nonspecific immune suppression or even an imbalance between pro- and anti-inflammatory cytokine networks. The limitation of this study is the lack of different dose usage in vivo. To gauge this phenomena in detail, further work is underway.
Conclusion
Synthesizing these immunotoxicological data in vivo and in vitro, our results indicate that different sized and charged ZnO NPs would cause in vitro and in vivo immunotoxicity, of which nature is a minor immunosuppression. This has important implications for individuals who may be chronically exposed to ZnO NPs. Further, this study might offer the possibility of the new immune parameters such as cytokine and NO for gauging immunotoxicity for nanoparticles.
Acknowledgment
This research was supported by a Grant (10182MFDS991) from the Ministry of Food and Drug Safety 2011. This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2013R1A1A2012665).
Disclosure
The authors report no conflicts of interest in this work.
References
Duguet E, Vasseur S, Mornet S, Devoisselle JM. Magnetic nanoparticles and their applications in medicine. Nanomedicine (Lond). 2006;1(2): 157–168. | |
Salata O. Applications of nanoparticles in biology and medicine. J Nanobiotechnology. 2004;2(1):3. | |
Sanvicens N, Marco MP. Multifunctional nanoparticles – properties and prospects for their use in human medicine. Trends Biotechnol. 2008;26(8):425–433. | |
Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761–769. | |
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | |
Hanley C, Layne J, Punnoose A, et al. Preferential killing of cancer cells and activated human T cells using ZnO nanoparticles. Nanotechnology. 2008;19(29):295103. | |
Zhang H, Chen B, Jiang H, Wang C, Wang H, Wang X. A strategy for ZnO nanorod mediated multi-mode cancer treatment. Biomaterials. 2011;32(7):1906–1914. | |
Hooper HL, Jurkschat K, Morgan AJ, et al. Comparative chronic toxicity of nanoparticulate and ionic zinc to the earthworm Eisenia veneta in a soil matrix. Environ Int. 2011;37(6):1111–1117. | |
Jang J, Lim DH, Choi IH. The impact of nanomaterials in immune system. Immune Netw. 2010;10(3):85–91. | |
Matsumura M, Takasu N, Nagata M, Nakamura K, Kawai M, Yoshino S. Effect of ultrafine zinc oxide (ZnO) nanoparticles on induction of oral tolerance in mice. J Immunotoxicol. 2010;7(3):232–237. | |
Pasupuleti S, Alapati S, Ganapathy S, Anumolu G, Pully NR, Prakhya BM. Toxicity of zinc oxide nanoparticles through oral route. Toxicol Ind Health. 2012;28(8):675–686. | |
Zheng YF, Li Z, Wang YD. In vitro and in vivo biocompatibility studies of Zno nanoparticles. Int J Mod Phys B. 2009;23(06–07):1566–1571. | |
Bae HC, Ryu HJ, Jeong SH, et al. Oxidative stress and apoptosis induced by ZnO nanoparticles in HaCaT cells. Mol Cell Toxicol. 2012;7(4):333–337. | |
Lee SH, Pie J-E, Kim Y-R, Lee HR, Son SW, Kim M-K. Effects of zinc oxide nanoparticles on gene expression profile in human keratinocytes. Mol Cellular Toxicol. 2012;8(2):113–118. | |
Heng BC, Zhao X, Tan EC, et al. Evaluation of the cytotoxic and inflammatory potential of differentially shaped zinc oxide nanoparticles. Arch Toxicol. 2011;85(12):1517–1528. | |
Roy R, Tripathi A, Das M, Dwivedi PD. Cytotoxicity and uptake of zinc oxide nanoparticles leading to enhanced inflammatory cytokines levels in murine macrophages: comparison with bulk zinc oxide. J Biomed Nanotechnol. 2011;7(1):110–111. | |
Yanagisawa R, Takano H, Inoue K, et al. Titanium dioxide nanoparticles aggravate atopic dermatitis-like skin lesions in NC/Nga mice. Exp Biol Med (Maywood). 2009;234(3):314–322. | |
Liu Y, Jiao F, Qiu Y, et al. The effect of Gd@C82(OH)22 nanoparticles on the release of Th1/Th2 cytokines and induction of TNF-alpha mediated cellular immunity. Biomaterials. 2009;30(23–24):3934–3945. | |
Hanley C, Thurber A, Hanna C, Punnoose A, Zhang J, Wingett DG. The influences of cell type and ZnO nanoparticle size on immune cell cytotoxicity and cytokine induction. Nanoscale Res Lett. 2009;4(12): 1409–1420. | |
Heng BC, Zhao X, Xiong S, Ng KW, Boey FY, Loo JS. Toxicity of zinc oxide (ZnO) nanoparticles on human bronchial epithelial cells (BEAS-2B) is accentuated by oxidative stress. Food Chem Toxicol. 2010;48(6):1762–1766. | |
Lipovsky A, Nitzan Y, Gedanken A, Lubart R. Antifungal activity of ZnO nanoparticles – the role of ROS mediated cell injury. Nanotechnology. 2011;22(10):105101. | |
Song W, Zhang J, Guo J, et al. Role of the dissolved zinc ion and reactive oxygen species in cytotoxicity of ZnO nanoparticles. Toxicol Lett. 2010;199(3):389–397. | |
Moos PJ, Chung K, Woessner D, Honeggar M, Cutler NS, Veranth JM. ZnO particulate matter requires cell contact for toxicity in human colon cancer cells. Chem Res Toxicol. 2010;23(4):733–739. | |
Sasidharan A, Chandran P, Menon D, Raman S, Nair S, Koyakutty M. Rapid dissolution of ZnO nanocrystals in acidic cancer microenvironment leading to preferential apoptosis. Nanoscale. 2011;3(9):3657–3669. | |
Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis. 2012;17(8): 852–870. | |
Di Gioacchino M, Petrarca C, Lazzarin F, et al. Immunotoxicity of nanoparticles. Int J Immunopathol Pharmacol. 2011;24 Suppl 1: S65–S71. | |
Sohaebuddin SK, Thevenot PT, Baker D, Eaton JW, Tang L. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part Fibre Toxicol. 2010;7:22. | |
Yang H, Liu C, Yang D, Zhang H, Xi Z. Comparative study of cytotoxicity, oxidative stress and genotoxicity induced by four typical nanomaterials: the role of particle size, shape and composition. J Appl Toxicol. 2009;29(1):69–78. | |
Padmavathy N, Vijayaraghavan R. Enhanced bioactivity of ZnO nanoparticles – an antimicrobial study. Sci Tech Adv Mater. 2008;9(3): 035004. | |
Dwivedi PD, Misra A, Shanker R, Das M. Are nanomaterials a threat to the immune system? Nanotoxicology. 2009;3(1):19–26. | |
Kim K-M, Kim T-H, Kim H-M, et al. Colloidal behaviors of ZnO nanoparticles in various aqueous media. Toxicol Environ Health Sci. 2012;4(2):121–131. | |
Yeh TK, Chen JK, Lin CH, et al. Kinetics and tissue distribution of neutron-activated zinc oxide nanoparticles and zinc nitrate in mice: effects of size and particulate nature. Nanotechnology. 2012;23(8):085102. | |
Cho WS, Duffin R, Howie SE, et al. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn2+ dissolution inside lysosomes. Part Fibre Toxicol. 2011;8:27. | |
George S, Pokhrel S, Xia T, et al. Use of a rapid cytotoxicity screening approach to engineer a safer zinc oxide nanoparticle through iron doping. ACS Nano. 2010;4(1):15–29. | |
Xia T, Kovochich M, Liong M, et al. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano. 2008;2(10): 2121–2134. | |
Feltis BN, O’Keefe SJ, Harford AJ, et al. Independent cytotoxic and inflammatory responses to zinc oxide nanoparticles in human monocytes and macrophages. Nanotoxicology. 2012;6(7):757–765. | |
Baek M, Kim MK, Cho HJ, et al. Factors influencing the cytotoxicity of zinc oxide nanoparticles: particle size and surface charge. J Phys Conf Ser. 2011;304(1):012044. | |
Horie M, Fujita K, Kato H, et al. Association of the physical and chemical properties and the cytotoxicity of metal oxide nanoparticles: metal ion release, adsorption ability and specific surface area. Metallomics. 2012;4:350–360. | |
Kunzmann A, Andersson B, Thurnherr T, Krug H, Scheynius A, Fadeel B. Toxicology of engineered nanomaterials: focus on biocompatibility, biodistribution and biodegradtion. Biochim Biophys Acta. 2011;1810(3):361–373. | |
Plascencia-Villa G, Starr CR, Armstrong LS, Ponce A, Yacaman MJ. Imaging interactions of metal oxide nanoparticles with macrophage cells by ultra-high resolution scanning electron microscopy techniques. Integr Biol. 2012;4:1358–1366. | |
Hanna J, Mandelboim O. When killers become helpers. Trends Immunol. 2007;28:201–206. | |
Anderson-Wilman B, Gehrmann U, Cansu Z, et al. Effects of subtoxic concentrations of TiO2 and ZnO nanoparticles on human lymphocytes, dendritic cells and exosome production. Toxicol Appl Pharmacol. 2012;264(1):94–103. | |
Lowe DB, Shearer MH, Jumper CA, Bright RK, Kennedy RC. Fc gamma receptors play a dominant role in protective tumor immunity against a virus encoded tumor-specific antigen in a murine model of experimental metastases. J Virol. 2007;81(3):1313–1318. | |
Green SJ, Scheller LF, Marletta MA, et al. Nitric oxide: cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol Lett. 1994;43(1–2):87–94. | |
Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54(4):469–487. | |
Holan V, Krulova M, Zajicova A, Pindjakova J. Nitric oxide as a regulatory and effector molecule in the immune system. Mol Immunol. 2002;38(12–13):989–995. | |
Marcinkiewicz J, Chain BM. Differential regulation of cytokine production by nitric oxide. Immunology. 1993;80(1):146–150. | |
Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric oxide and wound repair: role of cytokines? Nitric Oxide. 2002;7(1):1–10. | |
Tavares Murta BM, Machado JS, Zaparoli M, Lara VC, Murta EF. The relationship of host immune cells, cytokine and nitric oxide production to tumor cells in ovarian carcinoma. Sao Paulo Med J. 1999;117(2):87–92. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.