Back to Journals » Journal of Inflammation Research » Volume 19
Immunometabolic Reprogramming in Experimental Sepsis: A Driver of Multiple Organ Dysfunction Syndrome
Authors Wu F, Chen Y, Chen L, Wei X, Zhang L, Shen Y
Received 1 November 2025
Accepted for publication 26 February 2026
Published 4 March 2026 Volume 2026:19 577143
DOI https://doi.org/10.2147/JIR.S577143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Anh Ngo
Fan Wu,1 Yantong Chen,1 Lihua Chen,2 Xiaolu Wei,2 Lin Zhang,1 Yi Shen1
1Special Education College, Beijing Union University, Beijing, People’s Republic of China; 2Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China
Correspondence: Yi Shen, Special Education College, Beijing Union University, Beijing, People’s Republic of China, Tel +86 18800127858, Email [email protected]
Abstract: Sepsis is a life-threatening syndrome characterized by infection-induced systemic inflammation and immune dysregulation, commonly resulting in the development of multiple organ dysfunction syndrome (MODS), a leading cause of mortality in clinical practice. In decades, immunometabolic reprogramming has been identified as a critical mechanism that contributes to the progression of sepsis and the associated organ injuries. The review provides a systematic overview of the metabolic alterations in immune cells and organs in experimental models of sepsis. Key features include enhanced glycolysis, impaired mitochondrial function, and disturbed lipid metabolism, all of which are closely associated with organ damage. These metabolic adaptations influence immune responses and cell fate decisions, inter-organ crosstalk, and the development of MODS. A detailed examination is conducted on the temporal progression of pathological changes in established animal models, along with organ-specific metabolic dysfunctions and novel therapeutic targets. It emphasizes the importance of dynamic immunometabolic regulation, tissue-specific responses, and inter-organ interactions in the context of sepsis treatment. The integration of multi-omics technologies, identification of reliable biomarkers, and the development of personalized therapeutic strategies should be used to facilitate clinical translation of mechanistic insights.
Keywords: experimental sepsis, multiple organ dysfunction syndrome, immunometabolic reprogramming, inter-organ crosstalk, glycolysis
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection. Clinically, organ dysfunction is closely linked to increased in-hospital mortality.1 Sepsis is also recognized as one of the leading causes of global mortality and critical illness.2,3 Although these definitions primarily address adult populations, the burden of sepsis in children is comparably severe. It remains a major cause of pediatric mortality worldwide, with significantly higher death rates observed in children who present with dysfunction in at least one non-primary organ system, including the respiratory, cardiovascular, coagulation, or neurological systems.4
Multiple organ dysfunction syndrome (MODS) is a clinical condition defined by progressive and potentially reversible physiological impairment affecting two or more organ systems, typically initiated by acute insults such as sepsis.5 Organ dysfunction resulting from sepsis involves a complex network of inflammatory responses, including endothelial and microvascular impairment, immune and autonomic dysregulation, and alterations in cellular metabolism.6
Immunometabolism, as a central mechanism in sepsis, plays a critical role in promoting immune dysregulation during the septic response.7 A comprehensive understanding of immunometabolic alterations redefines the pathophysiological landscape of sepsis but also reveals how cellular energy dynamics influence immune function and contribute to organ damage, thereby identifying novel therapeutic targets.8 Given the current lack of effective treatments for the complex and life-threatening condition, such mechanistic insights are of urgent importance.
Both clinical and experimental evidence indicate that sepsis is not a single disease entity but a multifaceted syndrome involving interconnected immune dysregulation, microcirculatory impairment, metabolic disturbances, and progressive organ dysfunction. Animal models, by enabling control over experimental variables, serve as essential tools for delineating the causal relationships among these pathological processes. The review therefore focuses on experimental sepsis, with the aim of elucidating how immunometabolic reprogramming contributes to MODS under pathophysiological conditions and identifying potential targets for therapeutic intervention.
Experimental Animal Models of Sepsis
Animal models serve a fundamental function in sepsis research by offering a controlled platform to systematically investigate the metabolic reprogramming of immune cells under stress conditions. These models have significantly advanced the understanding of the complex interactions between immune function and cellular metabolism. In addition, animal models are essential translational tools for assessing the therapeutic potential of immunometabolic modulators in improving clinical outcomes in sepsis.
Given the closer resemblance to clinical presentation, the systemic models of sepsis were compared through the induction method, simulation characteristics, advantages and limitations of various models in Table 1. It is noteworthy that early sepsis models have been predominantly established in rodents, however, alternative animal species such as zebrafish are gaining attention for the utility in studying sepsis pathophysiology and therapeutic responses.9 Large animals, including pigs and dogs, share similar model induction approaches with rodents and offer advantages in evaluating organ function and immune responses with higher physiological fidelity. However, the use is limited by ethical considerations, high operational costs, and demanding maintenance requirements, making them more suitable for hypothesis-driven validation studies. The comparison among species was presented in Table 2. Notably, neonatal sepsis is a leading cause of mortality and disability in newborns, and the use of zebrafish larvae as a preclinical model holds great potential.10
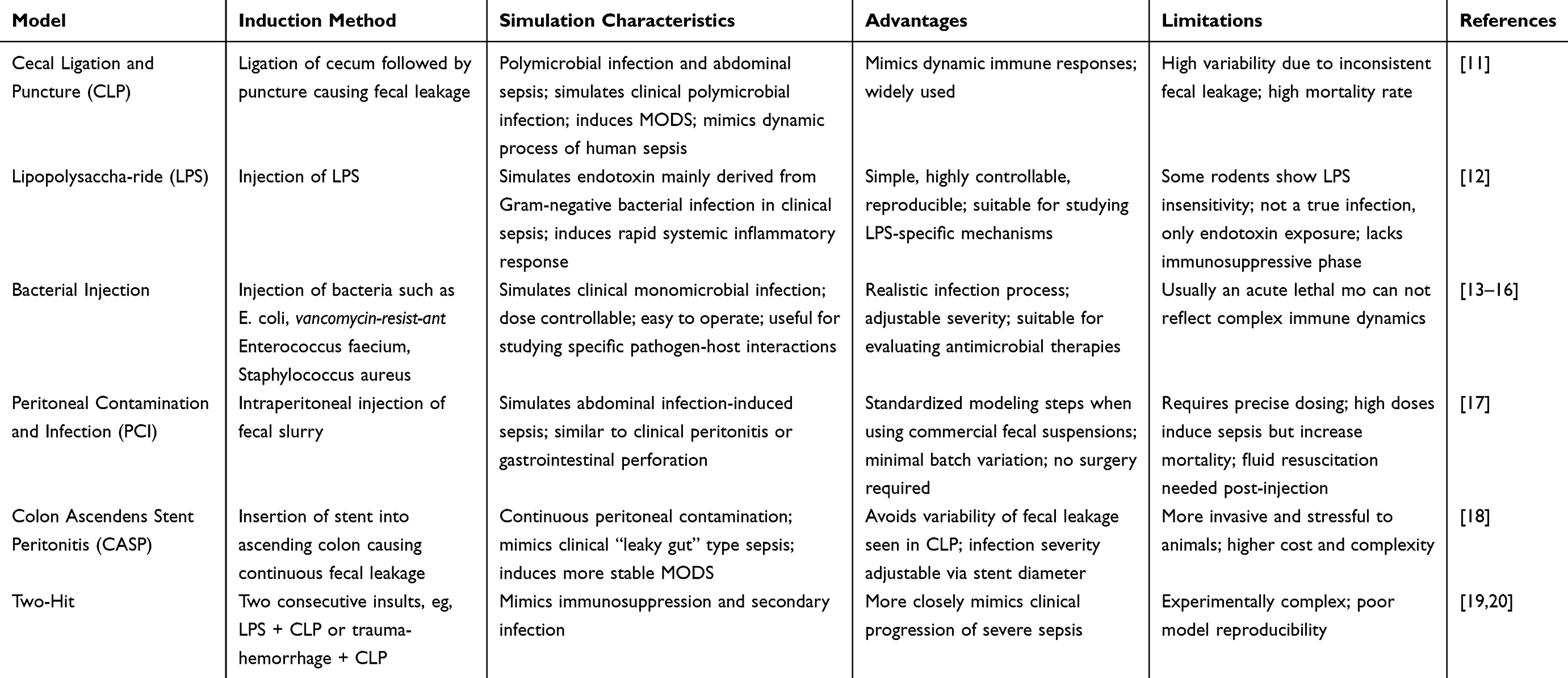 |
Table 1 Experimental Model of Systemic Sepsis |
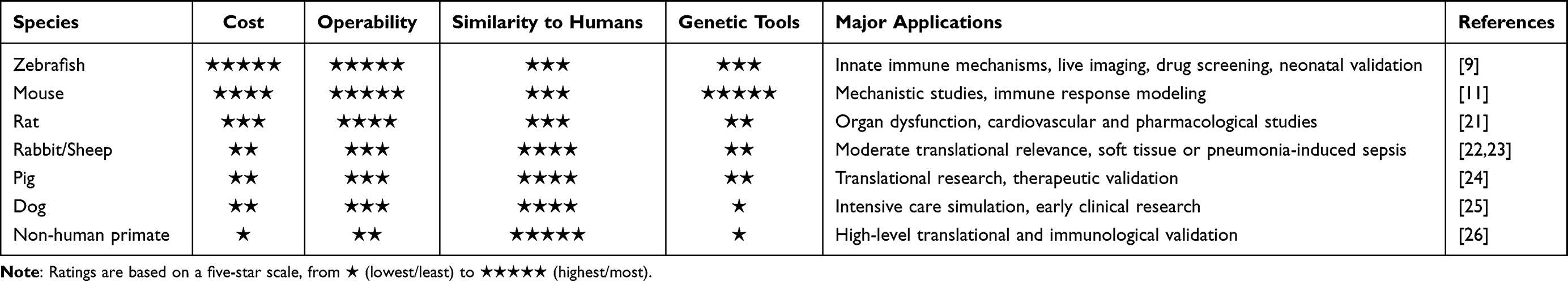 |
Table 2 Comparison of Sepsis Models Across Different Species |
Immunometabolic Reprogramming in Experimental Sepsis
Disease Progression of Experimental Sepsis
Over the past decades, the CLP model has been recognized as the representative experimental model to simulate systemic sepsis,27 for replicating the entire progression from infection-induced systemic inflammatory response syndrome (SIRS) to late-stage immunosuppression known as compensatory anti-inflammatory response syndrome (CARS).28 Its pathology encompasses cytokine storm, organ dysfunction, and metabolic imbalance,29 inducing systemic manifestations similar to human MODS.30–32 The time-dependent characteristics of the model (as illustrated in Figure 1) make it ideal for studying immunometabolic reprogramming, the temporal relationship of organ injuries,32 as well as for evaluating related therapeutic interventions (Table 3).
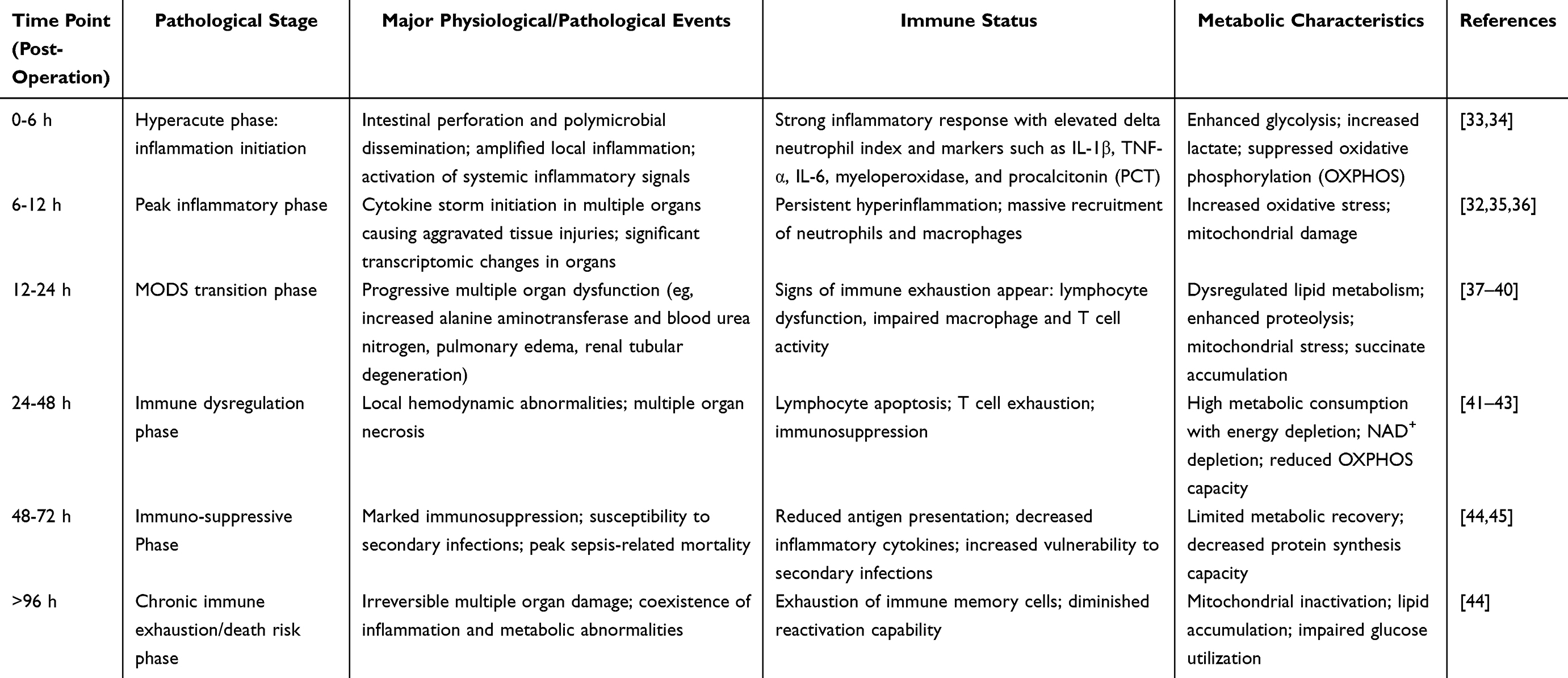 |
Table 3 Time-Dependent Disease Progression Characteristics of CLP Model |
 |
Figure 1 Temporal progression of immunometabolic reprogramming and organ dysfunction in CLP model. The temporal progression of immunometabolic alterations and corresponding pathological changes in CLP model was depicted at defined time points post-insult. The disease course progressed from hyperacute inflammation (0–6 h) and peak inflammation (6–12 h), through MODS transition (12–24 h) and immune dysregulation (24–48 h), to immunosuppression (48–72 h) and chronic immune exhaustion (>96 h). For each stage, key immune cell populations, representative cytokine profiles (eg, TNF-α, IL-6, IL-1β, PCT), and shifts in metabolic pathways (enhancement of glycolysis with lactate accumulation, suppression of oxidative phosphorylation) were illustrated. Chronic immune exhaustion was indicated to be associated with irreversible multi-organ injury, including sepsis-associated encephalopathy, lung injury, respiratory distress syndrome, cardiomyopathy, kidney injury, liver dysfunction, and cholestasis. For the depiction of organs, a darker color indicated a greater degree of necrosis. During the MODS transition, the lung was shown in a lighter color due to pulmonary edema. Finally, in the immunosuppressive stage, mice were depicted in gray to represent a state of marked immune deficiency and a tendency toward increased mortality. |
Immune Cell Metabolism Orchestrates the Functional Transformation
In sepsis, immune cells undergo systemic metabolic reprogramming, known as immunometabolic reprogramming, in response to pathogenic stress, adapting to the highly dynamic inflammatory environment. A hallmark of the reprogramming is a shift from OXPHOS to aerobic glycolysis,46 which is one of the most characteristic features of sepsis-related metabolic dysregulation and strongly associated with hyperinflammation and high mortality.47 To mount an effective defense against pathogens, immune cells require rapid proliferation and biosynthesis of lipids and proteins. The processes are energy-demanding and often activated by pro-inflammatory signaling, resulting in enhanced glycolysis and anabolic pathways, alongside suppression of catabolic processes such as OXPHOS and fatty acid oxidation (FAO).48
Immunometabolic reprogramming shapes immune cell phenotypes and contributes to inter-organ crosstalk disorders through the release of key metabolic intermediates such as lactate, succinate, and reactive oxygen species (ROS).49,50 These mediators further contribute to endothelial injuries,51 coagulation activation,52 and mitochondrial dysfunction,53 all of which are central to the development of MODS.54 Distinct metabolic profiles of immune cell subsets are closely tied to their functional states and may drive the progression of MODS.55,56 The metabolic adaptations support effector functions and determine the direction of the immune response (pro-inflammatory vs immunosuppressive), as a core mechanism in the evolution of MODS.6,57
In established animal models of sepsis, the initial pro-inflammatory phase is characterized by a metabolic shift toward glycolysis, which supports rapid immune cell activation and effector function. Emerging evidence highlights the role of glycolysis-related pathways, such as the lactate-ROS axis and spinster homolog 2 (Spns2)/sphingosine-1-phosphate (S1P) signaling (a regulation of extracellular signaling), in modulating inflammatory responses and maintaining immune homeostasis during early sepsis, thereby limiting both hyperinflammation and subsequent immunosuppression.58 Notably, pharmacological inhibition of glycolysis has been associated with improved survival in LPS models.59 As sepsis progresses, mitochondrial dysfunction,60 collapse of cellular energy metabolism,61 and immune exhaustion62 act as dominant features, collectively contributing to the development of MODS.63–65 While the preceding table summarizes the parallel immune and metabolic states observed at different stages of CLP-induced sepsis, the molecular underpinnings of the immunometabolic transition remain incompletely understood and require further elucidation.
At the early stage of septic stimulation, the host rapidly mounts a robust immune response, characterized by extensive recruitment and activation of immune cells. Classic immune cell types, including T cells, B cells, natural killer (NK) cells, and various myeloid populations, undergo significant phenotypic alterations and display pronounced heterogeneity.66 The dynamic immune landscape is accompanied by a marked elevation of pro-inflammatory cytokines in the peripheral blood, such as TNF-α, IL-1β, IL-6, and IFN-γ.17,67 The early inflammatory events reflect the systemic nature of immune activation and contribute to the progression of immunopathology in sepsis.
Specifically, macrophages exhibit enhanced glycolysis during sepsis, leading to elevated lactate production, which facilitates rapid energy supply necessary to sustain phagocytic activity and inflammatory responses.47,68 The metabolic reprogramming is accompanied by the activation of key signaling pathways across various organs, including HIF-1α,69 mTOR,70 and NF-κB.71 The metabolic reprogramming of macrophages associated with the relevant signals under pro-inflammatory and anti-inflammatory conditions are illustrated in Figure 2. In the hypoxic and inflamed microenvironment induced by sepsis, stabilization of HIF-1α promotes the expression of key glycolytic enzymes and downregulates OXPHOS, thereby accelerating glucose metabolism. The process drives macrophage polarization toward the pro-inflammatory M1 phenotype, characterized by increased secretion of cytokines such as TNF-α and IL-1β.72 Mechanistically, HIF-1α regulates the transcription of multiple glycolytic enzymes such as phosphofructokinase (PFK), increases the expression of glucose transporter 1 (GLUT1) to enhance glucose uptake, inhibits pyruvate entry into mitochondria (which would otherwise fuel OXPHOS), and promotes the conversion of pyruvate to lactate through upregulation of lactate dehydrogenase (LDH). Moreover, the mTORC1-HIF-1α axis is essential for cytokine production and the upregulation of costimulatory molecules CD80 and CD86 in dendritic cells (Jantsch et al, 2008). Thus, modulation of glycolysis and macrophage polarization has emerged as a potential therapeutic avenue in sepsis. For example, the Krüppel-like transcription factor KLF14 has been shown to exert protective effects in septic mice by suppressing the transcription of Hexokinase 2, thereby reducing glycolytic flux and the release of inflammatory cytokines in macrophages.73 Promoting autophagy in alveolar macrophages via the RAGE/PI3K/AKT/mTOR pathway can inhibit NLRP3 inflammasome activation, exerting anti-inflammatory and lung-protective effects.74 Furthermore, the Sphingosine kinase 1 (S1P)/Sphingosine-1-phosphate receptor 3 (S1PR3) signaling axis modulates macrophage polarization and the Warburg effect during sepsis, thereby alleviating excessive inflammation and multi-organ failure.75 Collectively, these findings highlight macrophages as promising targets for therapeutic intervention in sepsis.
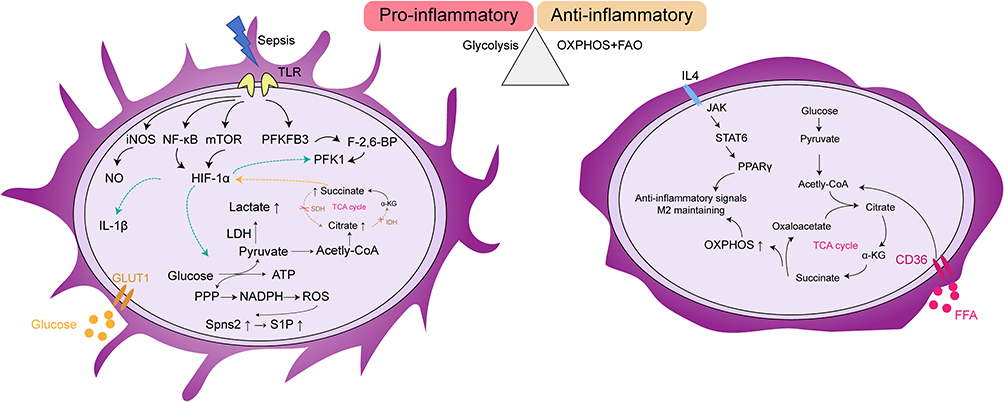 |
Figure 2 Distinct metabolic programs in pro-inflammatory and anti-inflammatory macrophage phenotypes. The metabolic reprogramming of macrophages under pro-inflammatory (left) and anti-inflammatory (right) conditions. In the pro-inflammatory state induced by sepsis, glycolysis is upregulated via toll-like receptor (TLR)-mediated signaling pathways, leading to increased inducible nitric oxide synthase (iNOS) expression, NF-κB activation, mTOR signaling, and hypoxia-inducible factor-1α (HIF-1α). It results in elevated lactate production, accumulation of succinate, and activation of the pentose phosphate pathway (PPP), collectively promoting reactive oxygen species (ROS) generation and pro-inflammatory cytokine release (eg, IL-1β). In the anti-inflammatory state driven by IL-4 stimulation, oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) are enhanced through the JAK-STAT6-PPARγ axis, maintaining M2 macrophage polarization and anti-inflammatory functions. Abbreviations: ATP, Adenosine triphosphate; α-KG, Alpha-ketoglutarate; CD36, Cluster of differentiation 36; FAO, Fatty acid oxidation; F-2,6-BP, Fructose-2,6-bisphosphate; FFA, Free fatty acid; HIF-1α, Hypoxia-inducible factor 1-alpha; IL-1β, Interleukin-1 beta; IL-4, Interleukin-4; iNOS, Inducible nitric oxide synthase; JAK, Janus kinase; LDH, Lactate dehydrogenase; mTOR, Mechanistic target of rapamycin; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; OXPHOS, Oxidative phosphorylation; PFK1, Phosphofructokinase-1; PFKFB3, 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PPARγ, Peroxisome proliferator-activated receptor gamma; PPP, Pentose phosphate pathway; ROS, Reactive oxygen species; S1P, Sphingosine-1-phosphate; Spns2, Spinster homolog 2 (sphingolipid transporter); STAT6, Signal transducer and activator of transcription 6; TCA cycle, Tricarboxylic acid cycle; TLR, Toll-like receptor. |
In both CLP and LPS models, neutrophil metabolic reprogramming is characterized by enhanced glycolysis, increased oxidative stress, and mitochondrial ROS involvement in signaling regulation.76,77 Fatty acids are utilized during developmental stages, while glucose uptake is elevated via upregulation of GLUT1, supporting aerobic glycolysis and redirection toward the pentose phosphate pathway (PPP).78 The processes are essential for chemotaxis, oxidative burst, and neutrophil extracellular traps (NETs) formation and release, which are central to neutrophil-mediated local pathogen control. NETs, an immune defense mechanism, trap and kill pathogens to restrict dissemination, and rely on glycolysis as their main energy source.79 NETs also regulate METTL3-m6A-IGF2BP2-dependent m6A modification of HIF-1α, promoting metabolic reprogramming and ferroptosis in alveolar epithelial cells.76 However, excessive polymorphonuclear neutrophil (PMN) recruitment or NETs formation could drive MODS. Interaction between macrophages and neutrophils further shapes immune signaling in sepsis. For instance, M2-Exos promote LXA4 production, downregulate C-X-C Motif Chemokine Receptor 2(CXCR2) and ROS, thereby inhibiting neutrophil function. Neutrophil-derived exosomal miR-30d-5p induces M1 macrophage polarization and pyroptosis via NF-κB activation, aggravating sepsis-related acute lung injury.80 Beyond macrophages, CD4⁺ T lymphocytes may support early bacterial clearance by modulating neutrophil function, possibly via an IFN-γ-dependent mechanism.81
Deficiency of Adenosine 5’-monophosphate (AMP) -activated protein kinase (AMPK) enhances pyruvate kinase M2 (PKM2)-dependent aerobic glycolysis, leading to the release of high mobility group box 1 (HMGB1), a late mediator of lethal systemic inflammation, in macrophages and monocytes. Activation of AMPK could protect mice from endotoxin shock and polymicrobial sepsis, whereas depletion of AMPKα in myeloid cells promotes disease progression. Moreover, inhibition of PKM2 reduces lactate production, HMGB1 release, and sepsis-related mortality in AMPKα-deficient mice. These findings suggest that disruption of AMPK-dependent immunometabolic pathways may contribute to the development of sepsis and represent a potential target for therapeutic intervention.82
Sepsis, as a systemic inflammatory condition, induces extensive metabolic reprogramming across diverse immune cell populations. Macrophages and neutrophils exhibit well-characterized metabolic changes, while other immune cells such as lymphocytes, dendritic cells, granulocytes, and innate lymphoid cells also show notable functional impairments and metabolic alterations. The immune disturbances contribute to a shift from effective antimicrobial defense toward immune dysfunction, sustained inflammation, and progressive organ damage. Despite growing interest in immunometabolism, the metabolic remodeling of many of these cell types remains poorly understood. Elucidating the relationship between immune cell metabolism and function in sepsis may uncover novel targets for therapeutic intervention.
Around 48 hours after CLP, the host gradually enters an immunosuppressive state, characterized by functional exhaustion, reduced numbers, or depletion of multiple immune cell types, which results in diminished antimicrobial capacity and increased susceptibility to secondary infections and MODS. For instance, CLP mice exhibit a decreased proportion of innate immune cells and macrophages in the bone marrow, along with an increased proportion of T lymphocytes.83
In macrophages, inhibition of the interaction between NF-κB p65 and nuclear factor of activated T-cells 5 could regulate the expression of glycolytic genes and proinflammatory cytokines in immunosuppressive macrophages, thereby promoting a favorable outcome in sepsis.84 Moreover, Mdivi-1, a mitochondrial fission inhibitors alleviates sphingosine-1-phosphate receptor 2 (S1PR2)-induced macrophage immunosuppression and improves outcomes in CLP mice by inhibiting Drp1-dependent mitochondrial fragmentation in macrophages.85
Sepsis-associated sickness behaviors include anorexia, lethargy, altered sleep patterns, and social withdrawal. The systemic mediators involved include adrenal hormones,86 bile acids (BAs),87 adenosine, and microbiota-derived metabolites, especially short-chain fatty acids (SCFAs).88 Other studies have demonstrated that fasting-induced metabolic programs exert beneficial effects during severe infections.89 Fasting regulates leukocyte distribution by promoting monocyte homing to the bone marrow;90 while refeeding leads to a surge of circulating monocytes, which may cause tissue damage in the context of antibacterial host responses.91 It is relevant to sepsis experimental models, since cecal necrosis and leakage, anorexia, and persistent weight loss are characteristic manifestations. Metabolic reprogramming of B cells during sepsis increases ATP production, which is converted to adenosine by CD39 on plasma cells, impairing macrophage bactericidal activity and enhancing IL-10 production, thereby increasing the risk of recurrent infections.92
After LPS stimulation, neutrophils differentiate into multiple subsets that exhibit distinct functional impairments during late sepsis, including inhibition of apoptosis, severe impairment of chemotaxis, and widespread tissue infiltration. LPS mediates programmed death-ligand 1(PD-L1) overexpression, an immune suppression signal, in neutrophils via the p38α-mitogen- and stress-activated protein kinase 1, (MSK1)/-MAPK-activated protein kinase 2 (MK2) pathway. The PD-L1 high subset exerts immunosuppressive effects in a direct contact-dependent manner, including inhibition of T cell activation and induction of T cell apoptosis and transdifferentiation. The process is driven by metabolic reprogramming of neutrophils from mitochondrial oxidative phosphorylation to glycolysis, providing energy and metabolic substrates (eg acetyl-CoA for histone acetylation) necessary for rapid expression of immune regulatory molecules like PD-L1.93
Single-cell resolution sequencing of clinical samples have identified distinct immune cell clusters associated with Persistent Inflammation- Immunosuppression-Catabolism Syndrome (PICS) in sepsis. The clusters show unique phenotypic and functional features of diverse immune cell subsets, including monocytic myeloid-derived suppressor cell-like monocytes, B cells, regulatory T cells, and megakaryocytes.45 Moreover, sepsis-induced immunosuppression is closely linked to the depletion of immature dendritic cells. For example, newly formed immature respiratory dendritic cells are locally programmed by residual inflammatory immune alarms to produce increased IL-10, thereby promoting tolerance.94
However, the distinct metabolic changes and their downstream cascades in specific immune cell populations within experimental sepsis models still require further investigation, as immune cell metabolism reflects their functional state as well as serves as a regulatory causal mechanism. Based on the restoring metabolic homeostasis, such as enhancing mitochondrial function, supplementing NAD⁺, and modulating the balance between glycolysis and lipid metabolism, has long been considered a potential therapeutic strategy to reverse immunosuppression and improve prognosis.
Immunometabolic Alterations Leading to Organ Injuries and Inter-Organ Crosstalk
In the early stage of sepsis, immune cell metabolism shifts toward glycolysis. Although the metabolic reprogramming facilitates rapid pathogen clearance in the short term, its sustained activation and dysregulation leading to abnormal accumulation of metabolic byproducts and a cytokine storm. The process is initiated by inflammasome activation and a robust pro-inflammatory response, characterized by the release of IL-6, TNF-α, and IFN-γ, along with activation of the coagulation system.6 The metabolically inflammatory and toxic substance propagates through the bloodstream to distant organs, inducing mitochondrial dysfunction, oxidative stress, and programmed cell death, ultimately leading to MODS.95
Organ-Specific Metabolic Dysregulation and Injuries
During the progression of sepsis, organs such as heart, lungs, kidneys, brain, and liver become high-risk targets of injuries, exhibiting complex and intertwined patterns of functional impairment. The organ injuries result from cascades of inflammatory cytokines, oxidative stress, and coagulation activation, also closely related to organ-specific changes in the immune microenvironment. Pathological manifestations such as cardiac dysfunction, acute respiratory distress syndrome (ARDS), acute kidney injury, hepatocyte apoptosis, and brain dysfunction collectively form the anatomical basis for the high mortality rate of sepsis (Figure 3). A deeper understanding of these inter-organ injuries and their crosstalk mechanisms should provide theoretical foundations and clinical directions for precision interventions and organ-protective strategies in sepsis.
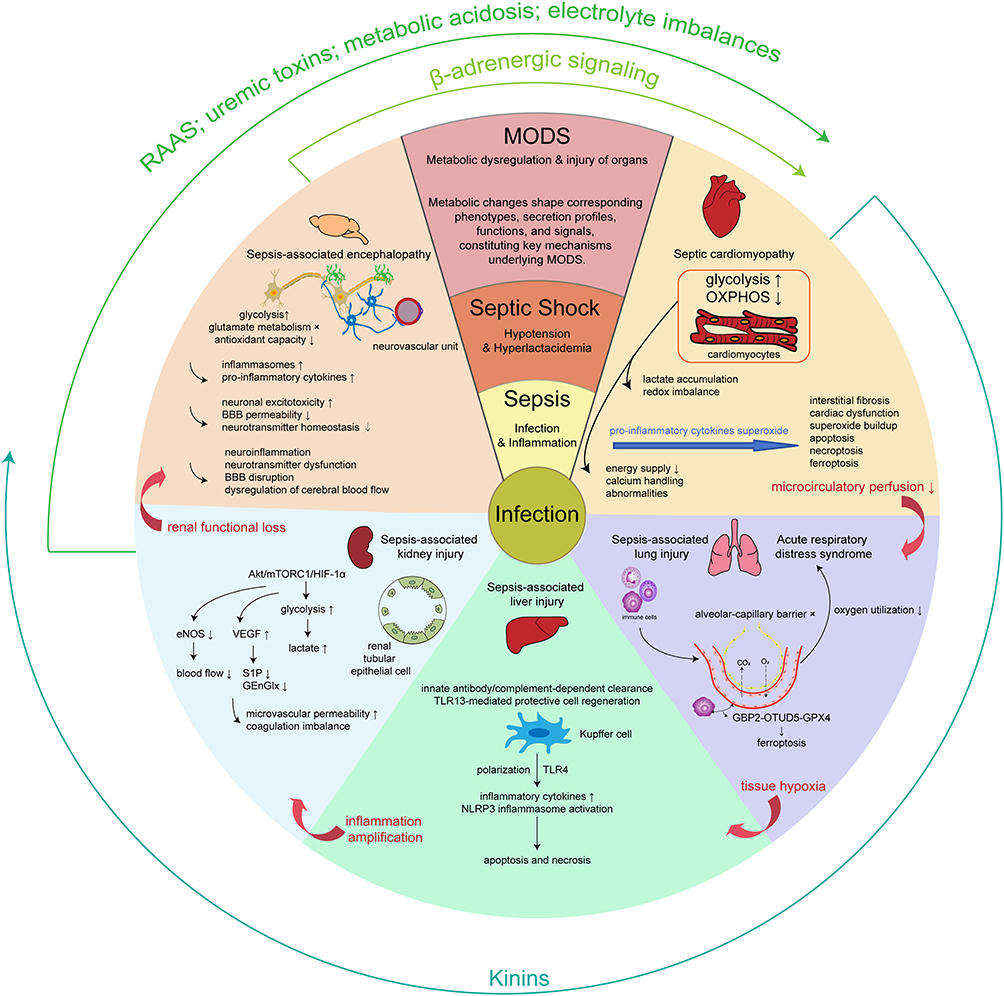 |
Figure 3 Organ-specific injury during sepsis progression. Sepsis-associated organ dysfunction involves disruption of inter-organ crosstalk, where organs form a complex crosstalk network mediated by metabolic products, inflammatory mediators, and immune signals. Metabolic reprogramming is associated with tissue hypoxia, microcirculatory dysfunction, mitochondrial impairment, oxidative stress, and inflammatory amplification, leading to sepsis-associated injuries in the brain, heart, lungs, kidneys, and liver. Key processes include glycolytic shift and accumulation of lactate. Interactions between organs are modulated by systemic factors such as renin-angiotensin-aldosterone system (RAAS) activation, β-adrenergic signaling, kinin release, uremic toxin accumulation, metabolic acidosis, and electrolyte imbalances. It was emphasized that augmented glycolysis, attenuated OXPHOS, and lactate accumulation were universal metabolic features observed across affected organs and immune cell populations. During the progression of sepsis, the detrimental impact of cardiac injury was the reduction of microcirculatory perfusion; pulmonary injury resulted in tissue hypoxia; hepatic injury contributed to inflammation amplification; renal injury led to renal functional loss, including metabolic and homeostatic disturbances caused by reduced glomerular blood flow and coagulation imbalance. Within the central nervous system, SIRS gave rise to diffuse cerebral dysfunction. The above processes were denoted by red arrows. Arrows projecting outward from each organ depicted inter-organ/system crosstalk signals. The regulation of cardiac output was mediated via the β-adrenergic system of nervous system. In the kidney, renin release from the juxtaglomerular apparatus activated the RAAS to elevate blood pressure, although chronic activation predisposed to heart failure. In the setting of impaired renal excretion, accumulation of protein metabolites and gut-derived toxins contributed to cardiomyopathy. Furthermore, metabolic acidosis and electrolyte imbalances caused by renal dysfunction reduced myocardial contractility. Under conditions of myocardial ischemia or stress, kinins exerted cardioprotective effects, improved myocardial perfusion, while its vasodilatory and permeability-enhancing actions, together with its role in modulating inflammation, further affected other organs. Abbreviations: 4E-BP1, Eukaryotic translation initiation factor 4E-binding protein 1; ɑ-KG, Alpha-ketoglutarate; Akt, Protein kinase B; BBB, Blood-brain barrier; eNOS, Endothelial nitric oxide synthase; GBP2, Guanylate-binding protein 2; GEnGlx, Endothelial glycocalyx; GPX4, Glutathione peroxidase 4; HIF-1ɑ, Hypoxia-inducible factor 1ɑ; mTORC1, Mechanistic target of rapamycin complex 1; MTORC1, Mechanistic target of rapamycin complex 1; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; OTUD5, OTU deubiquitinase 5; RAAS, Renin-angiotensin-aldosterone system; S1P, Sphingosine-1-phosphate; TLR13, Toll-like receptor 13; TLR4, Toll-like receptor 4; VEGF, Vascular endothelial growth factor. |
In septic cardiomyopathy, infiltrating macrophages and cardiomyocytes undergo metabolic reprogramming toward glycolysis, accompanied by decreased mitochondrial oxidative phosphorylation capacity, lactate accumulation, and redox imbalance. The metabolic alterations lead to insufficient energy supply and calcium handling abnormalities, thereby triggering septic myocardial dysfunction. Specifically, CLP mice exhibit significant cardiac dysfunction, altered mitochondrial dynamics, reduced cardiac lipid and glucose uptake, impaired fatty acid and glucose oxidation, enhanced myocardial glycolysis, and decreased ATP production.96 The metabolic changes result in CLP-induced interstitial fibrosis, cardiac dysfunction (manifested as reduced ejection fraction, fractional shortening, shortening/relengthening velocity, peak shortening, and electrically stimulated intracellular Ca2⁺ rise, alongside increased left ventricular end systolic diameter and relengthening duration), superoxide (O2−) buildup, apoptosis, necroptosis, and ferroptosis characterized by downregulated glutathione peroxidase 4 (GPX4) and SLC7A11.97
Improving mitochondrial dysfunction could partially alleviate septic cardiomyopathy. In CLP mice, cardiac TLR4-extracellular signal-regulated kinase1/2-dynamin-related protein 1 is activated, leading to mitochondrial fission and dysfunction, leading to cardiac dysfunction, cardiomyocyte apoptosis, recruitment of Mac-2⁺ macrophages, production of superoxide, and pro-inflammatory cytokines.98 Growth arrest–specific 6, an immune homeostasis regulator, alleviates mitochondrial damage, endoplasmic reticulum stress, oxidative stress, and apoptosis through an NLRP3 inflammasome-dependent mechanism, ultimately improving cardiac dysfunction in CLP mice.99
The lung is one of the earliest organs affected in sepsis. In CLP mice, lung injury scores, lung tissue wet-to-dry weight ratios, pulmonary vascular permeability, and levels of inflammatory cytokines including IL-1β, TNF-α, IL-6, IFN-γ, and C-C motif chemokine ligand 3(CCL3) in bronchoalveolar lavage fluid and lung tissue are all elevated, eventually leading to ARDS.100 The process, similar to that occurring in the heart, begins with immune cells releasing large amounts of ROS and pro-inflammatory cytokines, resulting in disruption of the alveolar-capillary barrier. Meanwhile, pulmonary capillary endothelial cells can be divided into distinct subpopulations, such as Plat⁺ capillaries which interact with neutrophils via intercellular adhesion molecule-1 (ICAM-1) adhesion and participate in innate immune responses, and CD74⁺ capillaries that express high levels of major histocompatibility complex (MHC) proteins (an antigen presenting protein) and engage with T cells in adaptive immunity.101 Overall, oxygen consumption in the lung increases, but oxygen utilization efficiency declines, impairing gas exchange function and eventually progressing to acute lung injury or ARDS.100 In the context, inhibition of the macrophage MAPK signaling negative feedback regulator Sprouty4 (Spry4) alleviates inflammation, oxidative stress, and acute lung injury in CLP mice.102 Additionally, lactate accumulated during sepsis promotes acetylation of cold-inducible RNA-binding protein (CIRP) in macrophages, leading to CIRP release, which is internalized by pulmonary vascular endothelial cells (PVECs) via TLR4-mediated endocytosis, exacerbating acute lung injury.103 Moreover, ferroptosis, a programmed cell death process characterized by iron-dependent lipid peroxidation, which is highly dependent on cellular metabolic states, plays a crucial role in lung injuries. Exosome-mediated signaling between macrophages and pulmonary vascular endothelial cells is pivotal in sepsis-induced acute lung injury, with the guanylate-binding protein 2 (GBP2)- OTU deubiquitinase 5 (OTUD5)-GPX4 axis identified as a driver of endothelial ferroptosis and lung damage.104 Activation of the Sirtuin 1 (SIRT1)/NADPH oxidase 4(NOX4) signaling pathway inhibits ferroptosis, thereby mitigating sepsis-induced damage to the lung microvascular endothelial barrier.105
The liver plays a central role in regulating systemic inflammation and metabolism. Kupffer cells, the liver-resident tissue macrophages, act as a crucial component of the innate immune system. Excessive pro-inflammatory activation of Kupffer cells is a key pathogenic mechanism underlying sepsis-induced liver injury. During sepsis, Kupffer cells undergo pronounced phenotypic switching and functional reprogramming, including enhanced innate antibody/complement-dependent clearance, TLR13-mediated protective cell regeneration, and immunometabolic changes regulated by mitochondrial function.106 Treatment with the mitochondrial fission inhibitor Mdivi-1 alleviates CLP-induced liver injury and improves liver pathology and function.43 Extracellular CIRP (eCIRP) promotes M1 polarization of Kupffer cells via the TLR4 pathway, resulting in excessive production of inflammatory cytokines.107 Proteomic and metabolomic analyses of LPS mice liver identified recombinant erythrocyte membrane protein 4.2 (Epb42) and adenosine diphosphate (ADP) as potential key proteins and metabolites driving sepsis-related acute liver injury. Epb42 is a red blood cell membrane skeletal protein involved in maintaining erythrocyte shape and elasticity. Under the hyperinflammatory state of sepsis, impaired erythrocyte deformability and microcirculatory obstruction suggest sinusoidal microcirculatory perfusion dysfunction, exacerbating hepatocyte hypoxia and necrosis. ADP is a critical platelet activation signal molecule that induces platelet aggregation and participates in microthrombosis. Increased ADP release activates sinusoidal endothelial cells and Kupffer cells, worsening hepatic microvascular inflammation and thrombosis, and triggering NLRP3 inflammasome activation to amplify inflammatory responses, leading to hepatocyte apoptosis and necrosis.43 However, it is noteworthy that current studies predominantly focus on hepatic phenotypes and pathological changes, with relatively few investigations addressing the metabolic alterations of Kupffer cells and other immune cells and their relationship to functional outcomes, which highlights an important direction for future research.
The spleen serves as a vital immunological organ during sepsis, contributing to both antigen clearance and the regulation of immune homeostasis. In experimental models of sepsis, the spleen exhibits marked structural damage and metabolic reprogramming. As early as 6 hours after sepsis onset, CD4⁺ T cells in the spleen display distinct transcriptional changes involving T-cell receptor and MAPK signaling pathways, indicating high sensitivity to inflammatory stimuli.108 The splenic interstitial fluid becomes hyperosmotic and its protein composition disrupted, reflecting structural alterations in the local microenvironment in CLP mice.109 As sepsis progresses, immunological dysregulation in the spleen is characterized by increased lymphocyte apoptosis, macrophage dysfunction, and Th1/Th2 imbalance.110 Following acute infection, the accumulation of Siglec-F⁺ neutrophils in the spleen induces the expression of immunosuppressive markers such as PD-1 and lymphocyte-activation gene 3 on T cells, implicating these cells as critical mediators of sepsis-induced immunosuppression. These neutrophils modulate T cell function through cell-to-cell contact and anti-inflammatory cytokines (eg IL-10), serving as key drivers of late-stage sepsis-induced immune paralysis.111 Raman spectroscopy reveals enhanced DNA activity in splenic T lymphocytes within 24 hours of acute inflammation, suggesting a peak in transcriptional activity.112 Moreover, the spleen acts as an extramedullary site of thrombopoiesis under septic conditions, where IL-3 drives the differentiation of myeloid progenitors into megakaryocytes that produce immunomodulatory CD40L⁺ platelets, contributing to host defense and improved survival.113 At the metabolic level, splenic immune cells undergo enhanced glycolysis alongside mitochondrial dysfunction. In LPS-challenged mice, activating transcription factor 4 (ATF4) plays a central role in glycolytic activation, promoting pro-inflammatory responses and alleviating macrophage tolerance during sepsis.61 In the spleens of CLP mice, diminished expression of sterile α and HEAT/armadillo motif-containing protein (SARM), a regulator of immune cell apoptosis, is associated with caspase-3 cleavage and ATP depletion, indicating mitochondrial failure and enhanced apoptosis.103 Furthermore, alpha-ketoglutaric acid (AKG), a key intermediate in the tricarboxylic acid cycle, has been shown to attenuate oxidative stress, restore mitochondrial membrane potential, and balance mitochondrial dynamics (promoting fusion and inhibiting excessive fission) in the spleen of LPS piglets, which enhances autophagic activity in splenic macrophages by modulating mTOR signaling and LC3B expression, thereby alleviating functional impairment in splenic tissues.67
Acute kidney injury (AKI) is a common and severe complication of sepsis, characterized by significant morbidity and mortality. During sepsis, both inflammatory cells and renal parenchymal cells, including macrophages, neutrophils, and renal tubular epithelial cells, undergo metabolic shifts toward aerobic glycolysis to amplify pro-inflammatory responses and enhance cellular tolerance to septic insults. As the disease progresses, these cells revert to OXPHOS, promoting anti-inflammatory responses and facilitating functional recovery. Changes in mitochondrial dynamics and metabolic reprogramming are central to the energy alterations occurring during septic AKI.114 Mitophagy, the selective degradation of damaged mitochondria, has been proved to confer protective effects against AKI via the PTEN-induced kinase 1- parkin RBR E3 ubiquitin protein ligase pathway, where optineurin functions act as an adaptor protein for mitophagy.115 Within the context of septic AKI, immunosuppression manifests as an enhanced anti-inflammatory phenotype with lactate, an end product of glycolysis, playing a key role. Lactate transcends its traditional role as a mere metabolic byproduct to function as an important signaling molecule. Specifically, lactate inhibits TLR4-mediated signaling to mitigate inflammation, thereby promoting M2 macrophage polarization and reducing LPS-induced NF-κB activation. Additionally, lactate affects immune cell migration and cytokine production through export via the monocarboxylate transporter 4. It also triggers “stop migration” signals via lactate transporters SLC5A12 in CD4+ T cells and SLC16A1 in CD8+ T cells.114 Renal tubular epithelial cells respond similarly to immune cells by activating aerobic glycolysis via the Akt/mTORC1/HIF-1α signaling pathway, which promotes the conversion of pyruvate to lactate and, together with pyruvate dehydrogenase kinase (PDHK), inhibits lactate conversion to acetyl-CoA, thereby reducing OXPHOS and related metabolic processes.116,117 Reduced glomerular blood flow attribute to inhibition of endothelial nitric oxide synthase activity in the arteries and glomeruli, while decreased cortical peritubular capillary perfusion is associated with epithelial oxidative stress. Elevated levels of the vascular endothelial growth factor system, decreased circulating sphingosine-1-phosphate, and loss of glomerular endothelial glycocalyx components contribute to increased microvascular permeability. Coagulation imbalances occur across all microvascular segments, representing a significant cause of renal functional loss.118
Sepsis-associated encephalopathy (SAE) is a diffuse brain dysfunction caused by sepsis, presenting with a spectrum of neurological abnormalities ranging from delirium to coma. It is a relatively common complication of sepsis and is associated with poor patient prognosis and high mortality. The pathogenesis involves neuroinflammation, neurotransmitter dysfunction, blood-brain barrier (BBB) disruption, and dysregulation of cerebral blood flow. Notably, astrocytes and microglia undergo metabolic reprogramming characterized by enhanced glycolysis, disrupted glutamate metabolism, and impaired antioxidant capacity. The process activates inflammasomes and leads to the release of pro-inflammatory cytokines such as IL-1β and TNF-α, contributing to increased neuronal excitotoxicity. BBB permeability increases and neurotransmitter homeostasis is disturbed.119 Importantly, inflammation in SAE results from systemic metabolic, inflammatory, and hemodynamic dysregulation driven by SIRS, rather than direct central nervous system infection.120 Although the brain itself is not infected, peripheral inflammatory signals would induce widespread neuroinflammation through neural and humoral pathways, ultimately causing neuronal damage.121 Similar to other immune cells, microglia shift their metabolism from OXPHOS-dominated homeostasis to glycolysis upon septic stimulation. The metabolic switch rapidly meets the energy demands for microglial proliferation, migration, cytokine secretion, and phagocytosis, despite glycolysis being less efficient than OXPHOS. Restoring mitochondrial function and reducing inducible nitric oxide synthase (iNOS) expression could recover impaired brain function.122 The latter is highly expressed in activated glial cells, producing high concentrations of nitric oxide (NO) that damage neuronal mitochondria and membrane structures, inhibit mitochondrial complex IV (cytochrome c oxidase), and perpetuate a vicious cycle, which also compromise the BBB and promote infiltration of inflammatory cells into the brain.123
In summary, immunometabolic reprogramming in experimental sepsis models exhibits overall similarity, with a core feature being a metabolic switch characterized by enhanced glycolysis, impaired mitochondrial activity, and close association with hypoxia-inducible factors and lactate production. It underlies why lactate has served as a clinical biomarker for sepsis over several decades. Changes in immune cell metabolism influence corresponding phenotypes, secretion profiles, clearance functions, and distribution of metabolic products. During the hyperinflammatory phase, these changes induce acute organ damage, while in the immunosuppressive phase, they inhibit tissue repair and exacerbate secondary injuries, representing one of the key mechanisms driving the development and progression of MODS. Therefore, precise intervention targeting immunometabolic processes may represent a promising strategy to improve organ dysfunction in sepsis in the future.
Inter-Organ Crosstalk in Sepsis
Sepsis-associated organ dysfunction involves disruption of inter-organ crosstalk, where organs form a complex crosstalk network mediated by metabolic products, inflammatory mediators, and immune signals. The network collectively drives or mitigates the progression of MODS. For example, injuries in one organ can initiate vicious cycles that exacerbate MODS and cause secondary damage or dysfunction in other organs.124 Although the multi-organ axes influenced by the gut microbiota have been extensively investigated in various disease contexts, the precise mechanisms underlying the inter-organ crosstalk remain incompletely understood.
The cardiovascular, respiratory, and renal systems are closely interconnected and interdependent, with management of cardiovascular dysfunction often focusing on regulating blood pressure and ensuring adequate organ perfusion. For example, refractory hypotension is a hallmark of septic shock. During the process, multiple neuroendocrine mechanisms are activated, such as the renin-angiotensin-aldosterone system (RAAS) and the β-adrenergic nervous system, to maintain cardiac output under decompensated cardiovascular function.125 Additionally, kinins synthesized and secreted by the heart may play a significant role in modulating communication between the cardiovascular system and other organs during sepsis.126,127 Upregulation of TNF-α and IL-6, along with increased levels of uremic toxins such as indoxyl sulfate, are critical mediators of the impact of AKI on distant organs.128 The alterations affect the heart, lungs, central nervous system, hematologic system, liver, gut, and microbiome.129 Furthermore, volume overload, retention of uremic toxins, and excessive activation of the RAAS system due to AKI accelerate heart failure. Accumulation of uremic toxins, metabolic acidosis, and electrolyte imbalances contribute to cardiovascular toxicity and may increase the risk of myocardial ischemia and life-threatening arrhythmias.130 The development of ARDS in sepsis, characterized by diffuse pulmonary inflammation and edema leading to acute respiratory failure, cannot be fully explained by cardiac failure or fluid overload alone. ARDS may result from infection itself or from systemic inflammation.131
Whether in clinical sepsis patients or in the CLP model, differences exist in the microbial composition between the healthy state and sepsis conditions, characterized by the loss of beneficial microbiota and the overgrowth of potential pathogens. Enterococcus, Klebsiella, and Enterobacteriaceae were observed to be enriched in both clinical samples and the CLP model. Enterococcus showed positive correlations with a greater number of genes associated with cell chemotaxis and inflammatory damage, while displaying negative correlations with a larger set of genes involved in lipid metabolism.30 Enterococcus is also known to reshape the gut’s metabolic environment via the arginine deiminase pathway, thereby promoting the growth of various pathogens, such as Escherichia coli and Clostridioides difficile, and potentially contributing to secondary infections.132,133 In addition, Bacteroides has been identified as a key bacterium associated with adverse outcomes in sepsis, affecting gut barrier permeability or H2S production during the course of the disease. The decrease in gut bacterial diversity is reflected by a marked depletion and alteration of intestinal metabolites, where metabolites involved in aromatic amino acid metabolism are the most severely affected.134 Tryptophan can also be metabolized by various gut microbes into indole derivatives, which are capable of activating the aryl hydrocarbon receptor (AhR) or pregnane X receptor (PXR) in intestinal epithelial cells or lymphocytes, thereby promoting anti-inflammatory responses, antimicrobial peptide secretion, and xenobiotic metabolism.135 Following gut barrier disruption, do the composition and metabolism of intestinal microbiota change, but the microbial barrier also becomes disorganized, losing its protective function. When gut microbes and their products, such as LPS and peptidoglycan, enter the portal circulation, they further exacerbate hepatic burden and inflammatory responses. Impaired circulatory perfusion, subsequent ischemia, inflammatory injury, and neuroendocrine dysregulation act in concert, ultimately leading to gut barrier failure.136 The process subsequently induces secondary alterations in the gut microbiota production of bioactive mediators, including short-chain fatty acids (acetate, propionate, and butyrate), succinate, and serotonin, which exert systemic effects on distant organs and modulate inflammatory signaling pathways.137
Given the characteristic gut barrier disruption in the CLP model and the pronounced gastrointestinal alterations observed in clinical sepsis patients, the “gut-X” axes have become a central focus and hotspot in sepsis research, including the Gut-Liver Axis, Gut-Brain Axis, Gut-Lung Axis, Gut-Kidney Axis, Gut-Heart Axis, Gut-Immune Axis, and Gut-Liver-Brain Axis, emphasizing the bidirectional regulatory interactions between the gut and distal organs or systems. Among these, the Gut-Liver-Brain Axis, as a composite axis, highlights the intricate interplay between the gut microbiota and barrier, hepatic immune and metabolic functions, inflammatory mediators and metabolites, and the nervous system, and is increasingly being investigated. The axis is particularly relevant for patients with cognitive dysfunction and poor long-term outcomes. Under physiological conditions, the gut microbiota-liver-brain axis maintains homeostasis through bidirectional communication; however, in sepsis, The network is disrupted. Such disruption exacerbates gastrointestinal and hepatic dysfunction but also contributes to cognitive impairment and severe disease progression.49
The liver is a key regulator of systemic homeostasis, responsible for the clearance of metabolic byproducts, modulation of immune responses, and secretion of hepatokines. During sepsis, disruption of the gut barrier and dysbiosis facilitate the translocation of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to the liver and systemic circulation. The liver plays a critical role in modulating immune defense during systemic infection through mechanisms including bacterial clearance, lipopolysaccharide detoxification, release of cytokines and acute-phase proteins, and regulation of inflammatory metabolism.138 Under physiological conditions, small amounts of intestinal LPS passing through the portal vein are processed and detoxified by the liver. When sepsis-induced inflammation and circulatory disturbances exceed the liver capacity, the defensive intraluminal mechanisms that prevent massive LPS translocation into the systemic circulation gradually fail.139 Lipopolysaccharide-binding protein (LBP) binds to the lipid A moiety of LPS, forming an LPS-LBP complex, which is recognized by TLR4 and myeloid differentiation factor 2 (MD-2), thereby triggering a cascade of inflammatory responses. In addition, gut-derived PAMPs and DAMPs may serve as key triggers, leading to dysregulated immune responses or excessive inflammation, impaired hepatic clearance of pathogenic bacteria, and metabolic disturbances.138 For example, the accumulation of metabolites such as lactate, ammonia, and bilirubin in the circulation can lead to hepatic encephalopathy and hepatorenal syndrome. Cytokine storms and immune dysregulation may precipitate acute respiratory distress syndrome. Abnormalities in coagulation factors and the fibrinolytic system can result in disseminated intravascular coagulation, impaired microcirculatory perfusion in the kidney, lung, and brain, and ischemic tissue injury. In summary, these inter-organ crosstalk mechanisms contribute to cardiovascular dysfunction, systemic inflammation, and insulin resistance, thereby accelerating the progression of MODS.140
Within the Gut-Liver-Brain Axis, neural, endocrine, and immune pathways constitute a network that mediates bidirectional communication and regulation with the central nervous system. Hepatic vagal sensory afferent nerves indirectly sense the gut microenvironment and transmit these sensory signals to the nucleus tractus solitarius in the brainstem, ultimately influencing vagal parasympathetic nerves and enteric neurons. Surgical or chemical disruption of these hepatic afferent nerves has been shown to reduce the abundance of colonic peripheral regulatory T (pTreg) cells, which is attributed to decreased expression of aldehyde dehydrogenase (ALDH) and impaired retinoic acid synthesis by intestinal antigen-presenting cells.141 In addition, the Gut-Liver-Brain coordinated regulation involves the formation of the cholinergic anti-inflammatory pathway, sensing of gut- and liver-derived molecules by afferent neuron terminals, modulation via the hypothalamic-pituitary-adrenal (HPA) axis through glucocorticoids, engagement of serotonin receptors, and regulation through immune pathways.142 In summary, all these organ responses feed back to the neuro-humoral-immune system, while additional signals such as HIF-1, cytokines, lactate, and neurotransmitters serve as biomarkers of sepsis severity and modulate organ function.
On this basis, beyond immune mediators and endocrine signals, neurotransmitters play a critical role in mediating inter-organ communication and regulation during sepsis, modulating immune responses, vascular endothelial function, and the progression of multi-organ injury, thereby offering novel insights into sepsis-associated MODS. Dopamine, a classical central neurotransmitter, has been shown to significantly inhibit the expression of aconitate decarboxylase 1 (ACOD1) induced by bacterial LPS, which is a key immunometabolic regulator, and its overactivation exacerbates inflammatory responses in sepsis.143 By suppressing ACOD1, dopamine may mitigate sepsis-associated immune dysregulation and organ injury.144 Endothelial dysfunction, a central feature of MODS, is also modulated by neurotransmitters such as dopamine, which can regulate vascular tone, microcirculation, and inflammatory mediator release, thereby affecting endothelial permeability and organ perfusion.145 In addition, the central nervous system communicates with peripheral organs via neuro-immune axes, such as the vagus nerve reflex; imbalances in neurotransmitters like glutamate may induce prolonged neuroinflammation and cognitive dysfunction through NMDA receptor-mediated pathways, worsening MODS.146 Programmed cell death pathways, including PANoptosis, represent key mechanisms in organ injury,147 and neurotransmitters may influence these processes via modulation of mitochondrial function (eg, HIF-1α) or immunometabolic signaling.49 Collectively, neurotransmitters contribute to sepsis-induced MODS through immune modulation, endothelial protection, central-peripheral organ communication, and regulation of cell death, providing critical insights into inter-organ crosstalk and potential therapeutic strategies.
From Mechanistic Insights to Translational Challenges in Therapeutic Targeting
MODS induced by sepsis remains a leading cause of clinical mortality, with effective interventions still extremely limited. Various experimental models have revealed the central role of immunometabolic reprogramming in the development of MODS, while metabolic crosstalk networks between organs serve as key channels amplifying tissue injuries. Although mechanistic studies have advanced considerably, numerous obstacles persist in translating these experimental findings into clinical applications.
Intervention Strategies Remain at the Target Island Stage
Current interventions largely focus on inhibiting single molecules or modulating isolated metabolic pathways, eg suppressing lactate production, restoring mitochondrial function, or regulating macrophage metabolic polarization. However, such strategies often fail to address the highly dynamic and spatiotemporally heterogeneous pathological environment of sepsis. In fact, immunometabolic pathways may exhibit biphasic effects depending on the timing and organ context (eg, early pro-inflammatory responses help pathogen clearance, whereas late-phase activation may induce immunoparalysis). Premature or delayed intervention could therefore worsen the disease.
In the early stage of sepsis, the immune response is highly activated, and cellular metabolism rapidly shifts towards glycolysis to support the rapid proliferation and activation of immune cells. The therapeutic strategy during this phase should focus on controlling excessive inflammation and preventing immune-mediated tissue damage. One such approach is the use of multifunctional nanoparticles that scavenge multiple inflammatory mediators can alleviate cytokine storms, exhibiting both antibacterial effects and the ability to clear various inflammatory mediators, thereby effectively mitigating acute lung injury observed in the CLP model.148 Similarly, as the M1 polarization of macrophages intensified, it led to ferroptosis and exacerbated the acute lung injury symptoms induced by CLP.149 When glycolysis in macrophages was inhibited, the secretion of inflammatory cytokines was consequently reduced, demonstrating a protective effect in the LPS model.73 However, premature immunosuppression may suppress effective pathogen clearance, delay optimal treatment, and exacerbate the spread of infection. Diethylhexyl phthalate (DEHP), an environmental immunotoxicant that disrupts the immune system, has been shown to aggravate immunosuppression and organ injury in the CLP model.150
As sepsis progresses, the immune system enters a state of immune suppression, characterized by a decline in immune cell function and an increased susceptibility to secondary infections. During this phase, metabolic dysregulation often leads to diminished tissue repair and further organ dysfunction. At this point, therapeutic strategies should focus on restoring immune function and promoting metabolic recovery. Research has shown that immune checkpoint inhibitors (such as anti-PD-1 antibodies) can help restore T cell function during the immunosuppressive phase, improving immune responses and enhancing pathogen clearance.151
Moreover, restoring metabolic balance is crucial in the treatment of immune suppression. Some studies suggest that interventions aimed at supplementing NAD+, improving mitochondrial function, and activating fatty acid oxidation pathways can reverse immune suppression (Delano and Ward, 2016). For instance, supplementation with NMN (nicotinamide mononucleotide) has been shown in mouse models to restore mitochondrial function, enhance T cell activity, and improve immune responses.143,152 Similarly, activation of fatty acid oxidation has been proven to promote immune cell function and reduce organ damage.153
Clinically, the effectiveness of sepsis treatment depends not only on the therapeutic interventions themselves but, more critically, on the precise timing of their implementation, as both premature and delayed interventions may exacerbate immune dysregulation and worsen patient outcomes. Substantial evidence indicates that early and appropriate administration of antibiotics remains the cornerstone for improving survival; however, current guidelines are limited in patient stratification and time-sensitive decision-making, highlighting the urgent need to integrate dynamic risk assessment with real-time physiological monitoring to optimize the therapeutic window for antibiotic initiation.154 With the growing understanding of immune-metabolic reprogramming in sepsis, therapeutic strategies are shifting from single-target interventions toward stage-specific, dynamically adjusted multimodal approaches. During the initial phase of the disease, interventions primarily target the limitation of excessive inflammation to prevent immune dysregulation, while in the later phase, they aim to reestablish immune competence and metabolic homeostasis, facilitating tissue repair and the restoration of organ function.155 In this context, although corticosteroids are widely used, effectiveness is not uniform across patients. Treatment outcomes can be affected by individual factors, such as relative adrenal insufficiency (RAI), and prolonged administration may result in immunosuppression and metabolic disturbances.156,157 Antibiotics play a pivotal role in sepsis management by controlling infection and reducing mortality, yet antibiotic administration is associated with challenges such as overuse, antimicrobial resistance, and adverse effects.158,159 Consequently, growing interest has emerged in exploring additional therapeutic approaches. Selenium and its organic form, selenomethionine, demonstrate potential immunometabolic protective effects through modulation of oxidative stress, mitochondrial function, and inflammatory responses.160,161 Mesenchymal stem cells and derived extracellular vesicles have been shown to alleviate immune dysregulation and improve outcomes in animal models; however, clinical translation remains limited by challenges related to standardization and safety.162 Notably, nanotechnology provides novel platforms for the targeted delivery of trace elements, bioactive molecules, and cell-derived therapeutics, with the potential to achieve more efficient and low-toxicity precision interventions.163 Current research trends emphasize personalized treatment guided by clinical phenotyping and real-time immunometabolic monitoring, with a focus on the systemic regulatory roles of key immunometabolic nodal proteins, including mTOR, HIF-1α, and the SIRT family, in inter-organ crosstalk.164 Overall, future sepsis management should build upon early anti-infective therapy by integrating time-resolved metabolic monitoring, targeted immunometabolic modulation, and advanced delivery technologies, thereby facilitating the clinical translation of multimodal, stage-specific intervention strategies.
The Significant Gap Between Animal Models and Clinical Practice
Although existing experimental models simulate human infectious sepsis with considerable physiological similarity, they cannot fully replicate the complexity of human sepsis. The stability and individual variability of CLP model, including the number of perforations or degree of fecal leakage, significantly influence inflammatory severity. Although the current experimental protocols are highly detailed, variations in body weight and species of experimental animals result in inevitable discrepancies in the exact location of cecal ligation and puncture. Moreover, in the experimental process, excessive force applied during extrusion of intestinal contents, as well as anatomical variations in the vasculature on the cecal surface, may lead to stage-specific differences. The LPS model allows precise control over the magnitude of systemic inflammation, yet it fails to capture the dynamic complexity of host-pathogen interactions, microbial translocation, and progressive infection. Pathogen-specific bacteremia model can be employed to investigate the effects of defined microorganisms; however, it may overlook the interplay among multiple microbial species. Immune metabolic features differ markedly between rodents and humans, including T cell subset proportions and macrophage polarization lineages.164 Therefore, for studies on immune responses and inflammatory diseases, at least those related to sepsis, both human and experimental animal data should be considered. Most studies overlook common comorbidities (eg, diabetes, obesity, aging), which are prevalent in clinical settings, male young adult rats are generally selected, which reduces variability but overlooks the confounding effects of these clinically prevalent conditions. Differences in endpoints further complicate translation. Animal experiments usually focus on survival or tissue inflammation, while clinical practice emphasizes organ support duration, biomarker dynamics, and functional recovery. Hence, it is urgent to develop more translationally relevant humanized models, organoid systems, and large animal models that mimic clinical scenarios, as well as to encourage incorporation of individual heterogeneity and temporal analyses in animal experiments. However, each prospective direction faces its own challenges. Humanized mice are xenotransplantation models in which a functional human immune system is established through the expansion of engrafted human cells.165 It has been demonstrated that humanized mice experience more severe weight loss within 5 days after CLP induction, thereby validating the translational relevance, given that cachexia is a recognized clinical manifestation of sepsis. In contrast to wild-type counterparts, the emergence of T-cell anergy and Fas expression constitutes a human-specific response to septic challenge.110 The key applications of humanized mouse models have focused on elucidating the mechanisms of apoptosis, the interactions of staphylococcal superantigens with the immune system, the development of vasculitis in hemorrhagic fever and meningitis, and the short- and long-term epigenetic reprogramming of leukocytes.166 However, further limitations to the use of humanized mice in sepsis research are their substantial cost and restricted availability. These models are costly to acquire and maintain, and the reconstitution of a functional immune system after stem cell engraftment typically requires several weeks.
Integrative Multi-Omics Analysis of Key Biomarkers and Systemic Mechanisms in Sepsis
Beyond lactate, a key biomarker in sepsis, preclinical studies have identified multiple additional signals and factors worthy of investigation. Lactate-associated proteins, including monocarboxylate transporters (MCT1, MCT4) and the receptor GPR81, play critical roles in regulating immune cell function and may serve as complementary biomarkers.167 Lactylation, a post-translational modification, has been implicated in sepsis-induced myocardial depression, suggesting that enzymes and metabolites within the lactate metabolic pathway may hold biomarker potential.168 Epigenetic regulators, such as lactate-mediated m6A RNA modifications, contribute to sepsis-associated acute lung injury and may represent novel research targets.169 Immunometabolic molecules, including the lactate-ROS axis and the sphingosine-1-phosphate (S1P) signaling pathway, reflect early hyperinflammatory responses and impaired macrophage antibacterial function, respectively, and could serve as early warning signals.170 Vascular endothelial injury markers, such as glycocalyx components, may additionally indicate microvascular dysfunction in sepsis.58,171
Advances in rapid detection technologies have further enhanced the utility of biomarkers. Near-infrared fluorescence (NIRF) lactate assays and electrochemical sensing platforms enable faster bedside monitoring compared with conventional methods.172,173 Integrative multi-omics analyses, encompassing transcriptomics, metabolomics, proteomics, single-cell omics, and extracellular vesicle profiling, improve diagnostic specificity while providing a comprehensive view of immunometabolic regulation, organ injury, pharmacological mechanisms, and potential therapeutic targets in sepsis.174,175 Systemic network mapping across multiple organs and cell types facilitates understanding of inter-organ crosstalk mechanisms, such as the liver–heart and lung–gut axes, and enables identification of key regulatory nodes and metabolic switches, including NAD⁺ metabolism, ubiquitination, and the tryptophan-kynurenine pathway. Time-resolved dynamic atlases, combined with pseudotime analysis and dynamic metabolic flux simulations, reveal the sequential progression of pathological events.176 Furthermore, artificial intelligence and machine learning tools can integrate these complex omics datasets to construct biomarker panels predictive of disease progression and therapeutic response, laying a foundation for precision medicine.177
Overall, integrating multi-mechanism biomarkers with high-throughput multi-omics analyses, rapid detection technologies, and AI-driven analytical tools holds promise for enabling early warning, dynamic monitoring, and precision intervention in sepsis, thereby facilitating the translation of preclinical findings into clinical applications.
In vivo Biosensors Enable Real-Time Dynamic Monitoring of Sepsis Metabolic States
Traditional sepsis monitoring relies on static measurements, such as intermittent blood gas analysis, lactate, PCT, and C-reactive protein (CRP), which inadequately capture the rapidly fluctuating metabolic and immune response dynamics. Recently, implantable or wearable biosensors have emerged that enable continuous real-time monitoring of parameters including lactate, glucose, pH, and inflammatory mediators, which has been an innovative direction. Coupled with artificial intelligence, these technologies hold promise for novel approaches to assess disease evolution and guide personalized interventions.178,179 Moreover, non-invasive biosensors for small animals remain in early development stages and urgently require further advancement. The sensors could capture key signals, such as IL-3,180 IL-6, IL-8, IL-10, IP-10, TNF-related apoptosis-inducing ligand, d-dimer, CRP, and granulocyte colony-stimulating factor,181 exhibiting excellent specificity and sensitivity. In experimental animal models, a paper-based biosensor for the detection of sepsis in the FIP mouse model has been shown to detect an immediate increase in matrix metalloproteinase-9 levels within 1 hour, although blood samples still need to be extracted.182 However, currently available biosensors for rodents remains mostly invasive, such as implantable sensors monitoring temperature or electrocardiography.177,178 The devices require surgical implantation and a relatively long recovery period, and expensive. Non-invasive sensors for small animals, similar to wearable human devices that monitor heart rate or blood pressure as auxiliary medical devices, are still in the preliminary stage and remain to be developed.
Summary and Outlook
Sepsis-induced MODS is a complex and challenging pathological process in clinical practice. In recent years, immune metabolic reprogramming has been increasingly recognized as a central mechanism in sepsis and associated organ injuries. Inter-organ crosstalk through metabolic products, inflammatory mediators, and immune signals to form a complex crosstalk network, jointly participating in the onset and progression of MODS. Although animal models and multi-omics technologies have greatly enriched our understanding of the immune metabolic mechanisms in sepsis, significant challenges remain in translating these basic research findings into clinical applications, including pathological heterogeneity, model limitations, and the lack of precise temporal intervention. Researchers can try to overcome the limitations by integrating systems biology and dynamic metabolic monitoring to reveal immune metabolic networks across spatial and temporal dimensions and to identify key regulatory nodes across organs in the future. The application of innovative in vivo biosensors and artificial intelligence technologies will open new avenues for real-time monitoring of metabolic states in sepsis, precise disease assessment, and personalized treatment. Moreover, the development of more clinically relevant humanized models and diverse animal models is also crucial.
Overall, precise intervention in immune metabolism and inter-organ crosstalk, combined with multidisciplinary technologies, holds promise as an effective strategy to improve sepsis outcomes and reduce mortality. With the continuous deepening of our understanding of immune metabolic networks in sepsis, the prospects for translational medicine are encouraging.
Generative AI Statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
FW: Writing-original draft, Conceptualization. YTC: Writing-original draft, Formal analysis, Methodology, Software. LHC: Writing-original draft, Data curation, Investigation, Funding acquisition. XLW: Writing-review and editing, Resources, Validation. LZ: Writing-review and editing, Project administration, Supervision. YS: Writing-review and editing, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Scientific and technological innovation project of China Academy of Chinese Medical Sciences (CI2023C020YL, CI2023E001TS09).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–25. doi:10.1001/jama.2016.0287
2. Vincent JL, Marshall JC, Namendys-Silva SA, et al; ICON investigators. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med. 2014;2(5):380–386. doi:10.1016/S2213-2600(14)70061-X
3. Fleischmann C, Scherag A, Adhikari NK, et al; International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis. Curr Estimates Limitations Am J Respiratory Crit Care Med. 2016;193(3):259–272. doi:10.1164/rccm.201504-0781OC
4. Schlapbach LJ, Watson RS, Sorce LR, et al; Society of Critical Care Medicine Pediatric Sepsis Definition Task Force (2024). International consensus criteria for pediatric sepsis and septic shock. JAMA. 2024;331;8:665–674. doi:10.1001/jama.2024.0179
5. Srdić T, Đurašević S, Lakić I, et al. From molecular mechanisms to clinical therapy: understanding sepsis-induced multiple organ dysfunction. Int J Mol Sci. 2024;25(14):7770. doi:10.3390/ijms25147770
6. Pool R, Gomez H, Kellum JA. Mechanisms of organ dysfunction in sepsis. Critic Care Clin. 2018;34(1):63–80. doi:10.1016/j.ccc.2017.08.003
7. Kumar V. Immunometabolism: another road to sepsis and its therapeutic targeting. Inflammation. 2019;42(3):765–788. doi:10.1007/s10753-018-0939-8
8. Koutroulis I, Batabyal R, McNamara B, Ledda M, Hoptay C, Freishtat RJ. Sepsis immunometabolism: from defining sepsis to understanding how energy production affects immune response. Critic Care Explorat. 2019;1(11):e0061. doi:10.1097/CCE.0000000000000061
9. He J, Xu P, Chen R, et al. Exploiting the Zebrafish model for sepsis research: insights into pathophysiology and therapeutic potentials. Drug Des Devel Ther. 2024;18:5333–5349. doi:10.2147/DDDT.S500276
10. Keij FM, Koch BEV, Lozano Vigario F, et al. Zebrafish larvae as experimental model to expedite the search for new biomarkers and treatments for neonatal sepsis. J Clin Transl Sci. 2021;5(1):e140. doi:10.1017/cts.2021.803
11. Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nature Protocols. 2009;4(1):31–36. doi:10.1038/nprot.2008.214
12. Pérez-Hernández EG, Delgado-Coello B, Luna-Reyes I, Mas-Oliva J. New insights into lipopolysaccharide inactivation mechanisms in sepsis. Biomed Pharmacothe. 2021;141:111890. doi:10.1016/j.biopha.2021.111890
13. He XH, Ouyang DY, Xu LH. Injection of escherichia coli to induce sepsis. Methods Mol Biol. 2021;2321:43–51. doi:10.1007/978-1-0716-1488-4_5
14. Ru X, Chen S, Chen D, Shao Q, Shao W, Ye Q. Simulating the clinical manifestations and disease progression of human sepsis: a monobacterial injection approach for animal modeling. Virulence. 2024;15(1):2395835. doi:10.1080/21505594.2024.2395835
15. Yuan X, Song X, Zhang X, et al. Unraveling host-pathogen dynamics in a murine Model of septic peritonitis induced by vancomycin-resistant Enterococcus faecium. Virulence. 2024;15(1):2367659. doi:10.1080/21505594.2024.2367659
16. Jin SH, Sun JJ, Liu G, et al. Nrf2/PHB2 alleviates mitochondrial damage and protects against Staphylococcus aureus-induced acute lung injury. MedComm. 2023;4(6):e448. doi:10.1002/mco2.448
17. Seemann S, Zohles F, Lupp A. Comprehensive comparison of three different animal models for systemic inflammation. J Biomed Sci. 2017;24(1):60. doi:10.1186/s12929-017-0370-8
18. Ai H, Li B, Meng F, Ai Y. CASP-model sepsis triggers systemic innate immune responses revealed by the systems-level signaling pathways. Front Immunol. 2022;13:907646. doi:10.3389/fimmu.2022.907646
19. Tran DT, Jeong YY, Kim JM, Bae HB, Son SK, Kwak SH. The anti-inflammatory role of bilirubin on “Two-Hit. Sepsis Animal Model Int J Mol Sci. 2020;21(22):8650. doi:10.3390/ijms21228650
20. Xu D, Horst K, Wang W, et al. The influence of macrophage-activating lipopeptide 2 (MALP-2) on local and systemic inflammatory response in a murine two-hit model of hemorrhagic shock and subsequent sepsis. Inflammation. 2021;44(2):481–492. doi:10.1007/s10753-020-01329-3
21. Cai L, Rodgers E, Schoenmann N, Raju RP. Advances in rodent experimental models of sepsis. Int J Mol Sci. 2023;24(11):9578. doi:10.3390/ijms24119578
22. Zamani K, Irajian G, Zahedi Bialvaei A, et al. Passive immunization with anti- chimeric protein PilQ/PilA -DSL region IgY does not protect against mortality associated with pseudomonas aeruginosa sepsis in a rabbit model. Mol Immunol. 2022;141:258–264. doi:10.1016/j.molimm.2021.11.021
23. Murakami K, Bjertnaes LJ, Schmalstieg FC, et al. A novel animal model of sepsis after acute lung injury in sheep. Crit Care Med. 2002;30(9):2083–2090. doi:10.1097/00003246-200209000-00022
24. Lyons NB, Proctor KG. Are the outcomes of a pig endotoxemia model applicable to human sepsis? Crit Care Med. 2023;51(8):1102–1104. doi:10.1097/CCM.0000000000005904
25. Shaw JH, Wolfe RR. A conscious septic dog model with hemodynamic and metabolic responses similar to responses of humans. Surgery. 1984;95(5):553–561.
26. Langley RJ, Tipper JL, Bruse S, et al. Integrative “omic” analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am J Respir Crit Care Med. 2014;190(4):445–455. doi:10.1164/rccm.201404-0624OC
27. Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19(4):198–208. doi:10.1016/j.tim.2011.01.001
28. Singh A, Nakade UP, Sharma A, et al. Caecal ligation and puncture develops time dependent progression of sepsis with multiple organs damage and vascular dysfunctions in mice. Toxicol Int. 2021;371–384. doi:10.18311/ti/2021/v28i4/28025
29. Fang H, Gong C, Fu J, et al. Evaluation of 2 rat models for sepsis developed by improved cecal ligation/puncture or feces intraperitoneal-injection. Med Sci Monit. 2020;26. doi:10.12659/MSM.919054
30. Sun S, Wang D, Dong D, et al. Altered intestinal microbiome and metabolome correspond to the clinical outcome of sepsis. Crit Care. 2023;27(1):127. doi:10.1186/s13054-023-04412-x
31. Capcha JMC, Moreira RS, Rodrigues CE, Silveira MAD, Andrade L, Gomes SA. Using the cecal ligation and puncture model of sepsis to induce rats to multiple organ dysfunction. Bio-Protocol. 2021;11(7):e3979. doi:10.21769/BioProtoc.3979
32. Rumienczyk I, Kulecka M, Ostrowski J, et al. Multi-organ transcriptome dynamics in a mouse model of cecal ligation and puncture-induced polymicrobial sepsis. J Inflamm Res. 2021;14:2377–2388. doi:10.2147/JIR.S307305
33. Lee H, Lim JM, Lee J, Kim SK, Lee T. Positive role of delta neutrophil index (DNI) as a prodiagnostic marker in cecal ligation and puncture (CLP)-induced sepsis murine model. Medicina. 2022;58(3):369. doi:10.3390/medicina58030369
34. Qin F, Tan H, Yang Y, Xu L, Yang X. Upregulation of Cullin1 neddylation promotes glycolysis and M1 polarization of macrophage via NF-κB p65 pathway in sepsis. Funct Integrat Genomics. 2024;24(6):204. doi:10.1007/s10142-024-01483-z
35. Yang K, Fan M, Wang X, et al. Lactate induces vascular permeability via disruption of VE-cadherin in endothelial cells during sepsis. Sci Adv. 2022;8(17):eabm8965. doi:10.1126/sciadv.abm8965
36. Zhang Y, Xing J. Effect of transforming growth factor-β1 on monocyte Toll-like receptor 4 expression in septic rats. World J Emerg Med. 2011;2(3):228–231. doi:10.5847/wjem.j.1920-8642.2011.03.013
37. Song L, Zou Y, Cao Z. Comparison of two different models of sepsis induced by cecal ligation and puncture in rats. J Surg Res. 2018;229:277–282. doi:10.1016/j.jss.2018.03.058
38. Ham SD, Abraham MN, Deutschman CS, Taylor MD. Single-cell RNA sequencing reveals immune education promotes T cell survival in mice subjected to the cecal ligation and puncture sepsis model. Front Immunol. 2024;15:1366955. doi:10.3389/fimmu.2024.1366955
39. Fu XZ, Wang Y. Interferon-γ regulates immunosuppression in septic mice by promoting the Warburg effect through the PI3K/AKT/mTOR pathway. Mol Med. 2023;29(1):95. doi:10.1186/s10020-023-00690-x
40. Deng D, Li X, Liu C, et al. Systematic investigation on the turning point of over-inflammation to immunosuppression in CLP mice model and their characteristics. Int Immunopharmacol. 2017;42:49–58. doi:10.1016/j.intimp.2016.11.011
41. Wang Z, Holthoff JH, Seely KA, et al. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol. 2012;180(2):505–516. doi:10.1016/j.ajpath.2011.10.011
42. Ahmad A, Druzhyna N, Szabo C. Delayed treatment with sodium hydrosulfide improves regional blood flow and alleviates cecal ligation and puncture (CLP)-induced septic shock. Shock Augusta Ga. 2016;46(2):183–193. doi:10.1097/SHK.0000000000000589
43. Zhang J, Wang X, Peng Y, et al. Combined metabolomic and proteomic analysis of sepsis related acute liver injury and its pathogenesis research. Int Immunopharmacol. 2024;130:111666. doi:10.1016/j.intimp.2024.111666
44. Islam MM, Watanabe E, Salma U, et al. Immunoadjuvant therapy in the regulation of cell death in sepsis: recent advances and future directions. Front Immunol. 2024;15. doi:10.3389/fimmu.2024.1493214
45. Sun X, Luo W-C, Huang S, et al. Immune-cell signatures of persistent inflammation, immunosuppression, and catabolism syndrome after sepsis. Med. 2025. doi:10.1016/j.medj.2024.12.003
46. Nolt B, Tu F, Wang X, et al. Lactate and immunosuppression in sepsis. Shock Augusta Ga. 2018;49(2):120–125. doi:10.1097/SHK.0000000000000958
47. Xiao M, Liu D, Xu Y, Mao W, Li W. Role of PFKFB3-driven glycolysis in sepsis. Ann Med. 2023;55(1):1278–1289. doi:10.1080/07853890.2023.2191217
48. Pan T, Sun S, Chen Y, et al. Immune effects of PI3K/Akt/HIF-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care. 2022;26(1):29. doi:10.1186/s13054-022-03893-6
49. Borges A, Bento L. Organ crosstalk and dysfunction in sepsis. Ann Intens Care. 2024;14(1):147. doi:10.1186/s13613-024-01377-0
50. Feng K, Dai W, Liu L, et al. Identification of biomarkers and the mechanisms of multiple trauma complicated with sepsis using metabolomics. Front Public Health. 2022;10:923170. doi:10.3389/fpubh.2022.923170
51. Ince C, Mayeux PR, Nguyen T, et al; ADQI XIV Workgroup. The endothelium in sepsis. Shock Augusta Ga. 2016;45(3):259–270. doi:10.1097/SHK.0000000000000473
52. Hua T, Yao F, Wang H, Liu W, Zhu X, Yao Y. Megakaryocyte in sepsis: the trinity of coagulation, inflammation and immunity. Crit Care. 2024;28(1):442. doi:10.1186/s13054-024-05221-6
53. Peters VBM, Arulkumaran N, Melis MJ, et al. Butyrate supplementation exacerbates myocardial and immune cell mitochondrial dysfunction in a rat model of faecal peritonitis. Life. 2022;12(12):2034. doi:10.3390/life12122034
54. Arts RJ, Gresnigt MS, Joosten LA, Netea MG. Cellular metabolism of myeloid cells in sepsis. J Leukocyte Biol. 2017;101(1):151–164. doi:10.1189/jlb.4MR0216-066R
55. Song L, Jiang W, Lin H, Yu J, Liu K, Zheng R. Post-translational modifications in sepsis-induced organ dysfunction: mechanisms and implications. Front Immunol. 2024;15:1461051. doi:10.3389/fimmu.2024.1461051
56. Kaneki M. Metabolic inflammatory complex in sepsis: septic cachexia as a novel potential therapeutic target. Shock Augusta Ga. 2017;48(6):600–609. doi:10.1097/SHK.0000000000000906
57. Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi:10.1038/nature01326
58. Fang C, Ren P, Bian G, et al. Enhancing Spns2/S1P in macrophages alleviates hyperinflammation and prevents immunosuppression in sepsis. EMBO Rep. 2023;24(8):e56635. doi:10.15252/embr.202256635
59. Wu Y, He Y, Liu C, et al. Histone deacetylase inhibitor (SAHA) reduces mortality in an endotoxemia mouse model by suppressing glycolysis. Int J Mol Sci. 2023;24(15):12448. doi:10.3390/ijms241512448
60. Hu D, Sheeja Prabhakaran H, Zhang YY, Luo G, He W, Liou YC. Mitochondrial dysfunction in sepsis: mechanisms and therapeutic perspectives. Critical Care. 2024;28(1):292. doi:10.1186/s13054-024-05069-w
61. Liu Y, Yang H, Luo N, et al. An Fgr kinase inhibitor attenuates sepsis-associated encephalopathy by ameliorating mitochondrial dysfunction, oxidative stress, and neuroinflammation via the SIRT1/PGC-1α signaling pathway. J Transl Med. 2023;21(1):486. doi:10.1186/s12967-023-04345-7
62. Görg B, Wettstein M, Metzger S, Schliess F, Häussinger D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology. 2005;41(5):1065–1073. doi:10.1002/hep.20662
63. Kumar S, Srivastava VK, Kaushik S, Saxena J, Jyoti A. Free radicals, mitochondrial dysfunction and sepsis-induced organ dysfunction: a mechanistic insight. Curr Pharm Des. 2024;30(3):161–168. doi:10.2174/0113816128279655231228055842
64. Lira Chavez FM, Gartzke LP, van Beuningen FE, et al. Restoring the infected powerhouse: mitochondrial quality control in sepsis. Redox Biol. 2023;68:102968. doi:10.1016/j.redox.2023.102968
65. Zou R, Tao J, Qiu J, et al. DNA-PKcs promotes sepsis-induced multiple organ failure by triggering mitochondrial dysfunction. J Adv Res. 2022;41:39–48. doi:10.1016/j.jare.2022.01.014
66. Yao RQ, Li ZX, Wang LX, et al. Single-cell transcriptome profiling of the immune space-time landscape reveals dendritic cell regulatory program in polymicrobial sepsis. Theranostics. 2022;12(10):4606–4628. doi:10.7150/thno.72760
67. Wang Z, Dayang EZ, Zwiers PJ, et al. Recruitment of neutrophils in glomeruli in early mouse sepsis is associated with E-selectin expression and activation of endothelial nuclear factor kappa-light-chain-enhancer of activated B cells and mitogen-activated protein kinase pathways. J Leukocyte Biol. 2024;116(6):1479–1497. doi:10.1093/jleuko/qiae146
68. Lu S, Li R, Deng Y, et al. GDF15 ameliorates sepsis-induced lung injury via AMPK-mediated inhibition of glycolysis in alveolar macrophage. Respir Res. 2024;25(1):201. doi:10.1186/s12931-024-02824-z
69. Meng Y, Kong KW, Chang YQ, Deng XM, Yang T. Histone methyltransferase SETD2 inhibits M1 macrophage polarization and glycolysis by suppressing HIF-1α in sepsis-induced acute lung injury. Med Microbiol Immunol. 2023;212(5):369–379. doi:10.1007/s00430-023-00778-5
70. Zhou C, Liang C, Zhang R, Wang Y, Luo S, Pan J. TREM2 improves coagulopathy and lung inflammation in sepsis through the AKT-mTOR pathway. Int Immunopharmacol. 2025;150:114330. doi:10.1016/j.intimp.2025.114330
71. Luo J, Wang J, Zhang J, et al. Nrf2 deficiency exacerbated CLP-induced pulmonary injury and inflammation through autophagy- and NF-κB/PPARγ-mediated macrophage polarization. Cells. 2022;11(23):3927. doi:10.3390/cells11233927
72. Li N, Gong Y, Zhu Y, et al. Exogenous acetate attenuates inflammatory responses through HIF-1α-dependent glycolysis regulation in macrophage. Cell Mol Life Sci. 2024;82(1):21. doi:10.1007/s00018-024-05521-8
73. Yuan Y, Fan G, Liu Y, et al. The transcription factor KLF14 regulates macrophage glycolysis and immune function by inhibiting HK2 in sepsis. Cell Mol Immunol. 2022;19(4):504–515. doi:10.1038/s41423-021-00806-5
74. Qin Y, Li W, Liu J, et al. Andrographolide ameliorates sepsis-induced acute lung injury by promoting autophagy in alveolar macrophages via the RAGE/PI3K/AKT/mTOR pathway. Int Immunopharmacol. 2024;139:112719. doi:10.1016/j.intimp.2024.112719)
75. Wang D, Bi X, Zhao L, et al. Targeting SphK1/S1PR3 axis ameliorates sepsis-induced multiple organ injury via orchestration of macrophage polarization and glycolysis. Biochimica et biophysica acta. Mol Cell Res. 2025;1872(1):119877. doi:10.1016/j.bbamcr.2024.119877
76. Zhang H, Wu D, Wang Y, et al. METTL3-mediated N6-methyladenosine exacerbates ferroptosis via m6A-IGF2BP2-dependent mitochondrial metabolic reprogramming in sepsis-induced acute lung injury. Clin Transl Med. 2023;13(9):e1389. doi:10.1002/ctm2.1389
77. Du F, Ding Z, Rönnow CF, Rahman M, Schiopu A, Thorlacius H. S100A9 induces reactive oxygen species-dependent formation of neutrophil extracellular traps in abdominal sepsis. Exp Cell Res. 2022;421(2):113405. doi:10.1016/j.yexcr.2022.113405
78. Gleeson LE, Sheedy FJ. Metabolic reprogramming and inflammation: fuelling the host response to pathogens. Semin Immunopathol. 2016;28(5):450–468. doi:10.1016/j.smim.2016.10.007
79. Tan C, Gu J, Chen H, et al. Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site via CXCR2 in sepsis. Shock Augusta Ga. 2020;53(1):114–123. doi:10.1097/SHK.0000000000001334
80. Jiao Y, Zhang T, Zhang C, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care. 2021;25(1):356. doi:10.1186/s13054-021-03775-3
81. Martignoni A, Tschöp J, Goetzman HS, et al. CD4-expressing cells are early mediators of the innate immune system during sepsis. Shock Augusta Ga. 2008;29(5):591–597. doi:10.1097/shk.0b013e318157f427
82. Huang J, Liu K, Zhu S, et al. AMPK regulates immunometabolism in sepsis. Brain Behav Immun. 2018;72:89–100. doi:10.1016/j.bbi.2017.11.003
83. Zhao T, Li Y, Liu B, et al. Inhibition of histone deacetylase 6 restores innate immune cells in the bone marrow in a lethal septic model. J Trauma Acute Care Surg. 2016;80(1):34–41. doi:10.1097/TA.0000000000000897
84. Xu J, Gao C, He Y, et al. NLRC3 expression in macrophage impairs glycolysis and host immune defense by modulating the NF-κB-NFAT5 complex during septic immunosuppression. Mol Ther. 2023;31(1):154–173. doi:10.1016/j.ymthe.2022.08.023
85. Yu X, Hu X, Wang D, et al. Macrophage S1PR2 drives sepsis-induced immunosuppression by exacerbating mitochondrial fragmentation. Am J Respir Cell Mol Biol. 2025;72(6):615–626. doi:10.1165/rcmb.2024-0161OC
86. Dos Santos-Junior NN, da Costa LHA, Catalão CHR, Alves Rocha MJ. Corticosterone and adrenocorticotrophic hormone secretion is recovered after immune challenge or acute restraint stress in sepsis survivor animals. Neuroimmunomodulation. 2022;29(4):306–316. doi:10.1159/000520746
87. Chang S, Kim YH, Kim YJ, et al. Taurodeoxycholate increases the number of myeloid-derived suppressor cells that ameliorate sepsis in mice. Front Immunol. 2018;9:1984. doi:10.3389/fimmu.2018.01984
88. Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi:10.1038/nrd2638
89. Wing EJ, Young JB. Acute starvation protects mice against Listeria monocytogenes. Infect Immun. 1980;28(3):771–776. doi:10.1128/iai.28.3.771-776.1980
90. Goldberg EL, Dixit VD. Bone marrow: an immunometabolic refuge during energy depletion. Cell Metab. 2019;30(4):621–623. doi:10.1016/j.cmet.2019.08.022
91. Janssen H, Kahles F, Liu D, et al. Monocytes re-enter the bone marrow during fasting and alter the host response to infection. Immunity. 2023;56(4):783–796.e7. doi:10.1016/j.immuni.2023.01.024
92. Nascimento DC, Viacava PR, Ferreira RG, et al. Sepsis expands a CD39+ plasmablast population that promotes immunosuppression via adenosine-mediated inhibition of macrophage antimicrobial activity. Immunity. 2021;54(9):2024–2041.e8. doi:10.1016/j.immuni.2021.08.005
93. Qi X, Yu Y, Sun R, et al. Identification and characterization of neutrophil heterogeneity in sepsis. Crit Care. 2021;25(1):50. doi:10.1186/s13054-021-03481-0
94. Bouras M, Asehnoune K, Roquilly A. Contribution of dendritic cell responses to sepsis-induced immunosuppression and to susceptibility to secondary pneumonia. Front Immunol. 2018;9:2590. doi:10.3389/fimmu.2018.02590
95. Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14(7):417–427. doi:10.1038/s41581-018-0005-7
96. Xu Y, Zhang X, Tang X, et al. Dexmedetomidine post-treatment exacerbates metabolic disturbances in septic cardiomyopathy via α2A-adrenoceptor. Biomed Pharmacothe. 2024;170:115993. doi:10.1016/j.biopha.2023.115993
97. Li FJ, Hu H, Wu L, et al. Ablation of mitophagy receptor FUNDC1 accentuates septic cardiomyopathy through ACSL4-dependent regulation of ferroptosis and mitochondrial integrity. Free Radic Biol Med. 2024;225:75–86. doi:10.1016/j.freeradbiomed.2024.09.039
98. Wu F, Zhang YT, Teng F, Li HH, Guo SB. S100a8/a9 contributes to sepsis-induced cardiomyopathy by activating ERK1/2-Drp1-mediated mitochondrial fission and respiratory dysfunction. Int Immunopharmacol. 2023;115:109716. doi:10.1016/j.intimp.2023.109716
99. Ji T, Liu Q, Yu L, et al. GAS6 attenuates sepsis-induced cardiac dysfunction through NLRP3 inflammasome-dependent mechanism. Free Radic Biol Med. 2024;210:195–211. doi:10.1016/j.freeradbiomed.2023.11.007
100. Xuan W, Wu X, Zheng L, et al. Gut microbiota-derived acetic acids promoted sepsis-induced acute respiratory distress syndrome by delaying neutrophil apoptosis through FABP4. Cell Mol Life Sci. 2024;81(1):438. doi:10.1007/s00018-024-05474-y
101. Yang R, Zheng T, Xiang H, Liu M, Hu K. Lung single-cell RNA profiling reveals response of pulmonary capillary to sepsis-induced acute lung injury. Front Immunol. 2024;15:1308915. doi:10.3389/fimmu.2024.1308915
102. Chen R, Cao C, Liu H, et al. Macrophage Sprouty4 deficiency diminishes sepsis-induced acute lung injury in mice. Redox Biol. 2022;58:102513. doi:10.1016/j.redox.2022.102513
103. Gong T, Wang Q-D, Loughran PA, et al. Mechanism of lactic acidemia-promoted pulmonary endothelial cells death in sepsis: role for CIRP-ZBP1-PANoptosis pathway. Military Med Res. 2024;11(1):71. doi:10.1186/s40779-024-00574-z
104. Li Z, Bu Y, Wang C, et al. Extracellular vesicle-packaged GBP2 from macrophages aggravates sepsis-induced acute lung injury by promoting ferroptosis in pulmonary vascular endothelial cells. Redox Biol. 2025;82:103614. doi:10.1016/j.redox.2025.103614
105. Wu Z, Xi Q, Zhao Q, Zhu S. GDF11 overexpression alleviates sepsis-induced lung microvascular endothelial barrier damage by activating SIRT1/NOX4 signaling to inhibit ferroptosis. Shock Augusta Ga. 2024;62(2):245–254. doi:10.1097/SHK.0000000000002391
106. Fantini F, Da Dalt L, Moregola A, et al. Modulation of mitochondrial dynamism in Kupffer cells impacts systemic metabolism. European Atherosclerosis J. 2024;3(3):91. doi:10.56095/eaj.v3i3.81
107. Shimizu J, Murao A, Lee Y, Aziz M, Wang P. Extracellular CIRP promotes Kupffer cell inflammatory polarization in sepsis. Front Immunol. 2024;15:1411930. doi:10.3389/fimmu.2024.1411930
108. McDunn JE, Turnbull IR, Polpitiya AD, et al. Splenic CD4+ T cells have a distinct transcriptional response six hours after the onset of sepsis. J Am College Surg. 2006;203(3):365–375. doi:10.1016/j.jamcollsurg.2006.05.304
109. Svendsen Ø, Stangeland L, Elvevoll B, et al. 0729. Interstitial changes in spleen during sepsis. Intensive Care Med Experim. 2014;2(Suppl S1):P51. doi:10.1186/2197-425X-2-S1-P51
110. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. doi:10.1038/nrdp.2016.45
111. Chen J, Zhang J, Chen S, et al. CD121b-positive neutrophils predict immunosuppression in septic shock. Front Immunol. 2025;16:1565797. doi:10.3389/fimmu.2025.1565797
112. Osadare IE, Xiong L, Rubio I, et al. Raman spectroscopy profiling of splenic T-cells in sepsis and endotoxemia in mice. Int J Mol Sci. 2023;24(15):12027. doi:10.3390/ijms241512027
113. Valet C, Magnen M, Qiu L, et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J Clin Invest. 2022;132(7):e153920. doi:10.1172/JCI153920
114. Liu C, Wei W, Huang Y, Fu P, Zhang L, Zhao Y. Metabolic reprogramming in septic acute kidney injury: pathogenesis and therapeutic implications. Metabolism. 2024;158:155974. doi:10.1016/j.metabol.2024.155974
115. Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. doi:10.1016/j.redox.2020.101767
116. Kalakeche R, Hato T, Rhodes G, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22(8):1505–1516. doi:10.1681/ASN.2011020203
117. Dellepiane S, Marengo M, Cantaluppi V. Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit Care. 2016;20:61. doi:10.1186/s13054-016-1219-3
118. Molema G, Zijlstra JG, van Meurs M, Kamps JAAM. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat Rev Nephrol. 2022;18(2):95–112. doi:10.1038/s41581-021-00489-1
119. Gao S, Jiang Y, Chen Z, et al. Metabolic reprogramming of microglia in sepsis-associated encephalopathy: insights from neuroinflammation. Curr Neuropharmacol. 2023;21(9):1992–2005. doi:10.2174/1570159X21666221216162606
120. Manabe T, Heneka MT. Cerebral dysfunctions caused by sepsis during ageing. Nat Rev Immunol. 2022;22(7):444–458. doi:10.1038/s41577-021-00643-7
121. Ren C, Yao RQ, Zhang H, Feng YW, Yao YM. Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflammation. 2020;17(1):14. doi:10.1186/s12974-020-1701-3
122. Haileselassie B, Joshi AU, Minhas PS, Mukherjee R, Andreasson KI, Mochly-Rosen D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J Neuroinflammation. 2020;17(1):36. doi:10.1186/s12974-019-1689-8
123. Pytel P, Alexander JJ. Pathogenesis of septic encephalopathy. Curr Opin Neurol. 2009;22(3):283–287. doi:10.1097/WCO.0b013e32832b3101
124. Arteel GE. Liver-lung axes in alcohol-related liver disease. Clin Mol Hepatol. 2020;26(4):670–676. doi:10.3350/cmh.2020.0174
125. Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98(3):1627–1738. doi:10.1152/physrev.00038.2017
126. Ferrero KM, Koch WJ. Metabolic crosstalk between the heart and fat. Korean Circulation J. 2020;50(5):379–394. doi:10.4070/kcj.2019.0400
127. Jahng JW, Song E, Sweeney G. Crosstalk between the heart and peripheral organs in heart failure. Exp Mol Med. 2016;48(3):e217. doi:10.1038/emm.2016.20
128. Li X, Yuan F, Zhou L. Organ crosstalk in acute kidney injury: evidence and mechanisms. J Clin Med. 2022;11(22):6637. doi:10.3390/jcm11226637
129. White LE, Chaudhary R, Moore LJ, Moore FA, Hassoun HT. Surgical sepsis and organ crosstalk: the role of the kidney. J Surg Res. 2011;167(2):306–315. doi:10.1016/j.jss.2010.11.923
130. Lee SA, Cozzi M, Bush EL, Rabb H. Distant organ dysfunction in acute kidney injury: a review. Am J Kidney Dis. 2018;72(6):846–856. doi:10.1053/j.ajkd.2018.03.028
131. Quílez ME, López-Aguilar J, Blanch L. Organ crosstalk during acute lung injury, acute respiratory distress syndrome, and mechanical ventilation. Curr Opin Crit Care. 2012;18(1):23–28. doi:10.1097/MCC.0b013e32834ef3ea
132. Keogh D, Tay WH, Ho YY, et al. Enterococcal metabolite cues facilitate interspecies niche modulation and polymicrobial infection. Cell Host Microbe. 2016;20(4):493–503. doi:10.1016/j.chom.2016.09.004
133. Smith AB, Jenior ML, Keenan O, et al. Enterococci enhance clostridioides difficile pathogenesis. Nature. 2022;611(7937):780–786. doi:10.1038/s41586-022-05438-x)
134. Chen Q, Liang X, Wu T, et al. Integrative analysis of metabolomics and proteomics reveals amino acid metabolism disorder in sepsis. J Transl Med. 2022;20(1):123. doi:10.1186/s12967-022-03320-y
135. Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. 2018;23(6):716–724. doi:10.1016/j.chom.2018.05.003
136. Chawla LS, Fink M, Goldstein SL, et al; ADQI XIV Workgroup. The epithelium as a target in sepsis. Shock Augusta Ga. 2016;45(3):249–258. doi:10.1097/SHK.0000000000000518
137. Miller WD, Keskey R, Alverdy JC. Sepsis and the microbiome: a vicious cycle. J Infect Dis. 2021;223(12 Suppl 2):S264–S269. doi:10.1093/infdis/jiaa682
138. Zhang X, Liu H, Hashimoto K, Yuan S, Zhang J. The gut-liver axis in sepsis: interaction mechanisms and therapeutic potential. Crit Care. 2022;26(1):213. doi:10.1186/s13054-022-04090-1
139. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14(3):141–153. doi:10.1038/nri3608
140. Kim TH, Hong DG, Yang YM. Hepatokines and Non-Alcoholic Fatty Liver Disease: Linking Liver Pathophysiology to Metabolism.Biomedicines. 2021;9(12):1903. doi:10.3390/biomedicines9121903
141. Teratani T, Mikami Y, Nakamoto N, et al. The liver-brain-gut neural arc maintains the Treg cell niche in the gut. Nature. 2020;585(7826):591–596. doi:10.1038/s41586-020-2425-3
142. Wang X, Wen X, Yuan S, Zhang J. Gut-brain axis in the pathogenesis of sepsis-associated encephalopathy. Neurobiol Dis. 2024;195:106499. doi:10.1016/j.nbd.2024.106499
143. Wang N, Liu J, Wu R, et al. A neuroimmune pathway drives bacterial infection. Sci Adv. 2025;11(18):eadr2226. doi:10.1126/sciadv.adr2226
144. Wang H, Kang R, Tang D. Dopamine as an endogenous regulator of innate immunity in sepsis. Front Immunol. 2025;16:1625368. doi:10.3389/fimmu.2025.1625368
145. Tang F, Zhao XL, Xu LY, Zhang JN, Ao H, Peng C. Endothelial dysfunction: pathophysiology and therapeutic targets for sepsis-induced multiple organ dysfunction syndrome. Biomed Pharmacothe. 2024;178:117180. doi:10.1016/j.biopha.2024.117180
146. Bardaghi Z, Rajabian A, Beheshti F, Arabi MH, Hosseini M, Salmani H. Memantine, an NMDA receptor antagonist, protected the brain against the long-term consequences of sepsis in mice. Life Sci. 2023;323:121695. doi:10.1016/j.lfs.2023.121695
147. Tang F, Wu S, Zou Z, Li X, Qiao L. The core role of central nervous system in sepsis-related organ damage. Front Immunol. 2025;16:1694003. doi:10.3389/fimmu.2025.1694003
148. Li Z, Feng Y, Zhang S, et al. A multifunctional nanoparticle mitigating cytokine storm by scavenging multiple inflammatory mediators of sepsis. ACS Nano. 2023;17(9):8551–8563. doi:10.1021/acsnano.3c00906
149. Ling X, Wei S, Ling D, et al. Irf7 regulates the expression of Srg3 and ferroptosis axis aggravated sepsis-induced acute lung injury. Cell Mol Biol Lett. 2023;28(1):91. doi:10.1186/s11658-023-00495-0
150. Linghu D, Zhu Z, Zhang D, et al. Diethylhexyl phthalate induces immune dysregulation and is an environmental immune disruptor. J Hazard Mater. 2024;480:136244. doi:10.1016/j.jhazmat.2024.136244
151. Triantafyllou E, Gudd CL, Mawhin MA, et al. PD-1 blockade improves Kupffer cell bacterial clearance in acute liver injury. J Clin Invest. 2021;131(4):e140196. doi:10.1172/JCI140196
152. Cao T, Ni R, Ding W, et al. Nicotinamide mononucleotide as a therapeutic agent to alleviate multi-organ failure in sepsis. J Transl Med. 2023;21(1):883. doi:10.1186/s12967-023-04767-3
153. Gómez H. Reprogramming metabolism to enhance kidney tolerance during sepsis: the role of fatty acid oxidation, aerobic glycolysis, and epithelial de-differentiation. Nephron. 2023;147(1):31–34. doi:10.1159/000527392
154. Gross BJ, Donahue A, Ford JS, et al. Mortality and antibiotic timing in deep learning-derived surviving sepsis campaign risk groups: a multicenter study. Crit Care. 2025;29(1):302. doi:10.1186/s13054-025-05493-6
155. Pei F, Yao RQ, Ren C, et al; International Federation of the Shock Societies (IFSS). Expert consensus on the monitoring and treatment of sepsis-induced immunosuppression. Military Med Res. 2022;9(1):74. doi:10.1186/s40779-022-00430-y
156. Louaguenouni Y, Mansart A, Annane D, Fattal E, Fay F. Nanoparticle-based approaches for sepsis treatment: current trends and perspectives. J Control Rel. 2025;386:114139. doi:10.1016/j.jconrel.2025.114139
157. Wu CH, Guo L, Hao D, et al. Relative adrenal insufficiency is a risk factor and endotype of sepsis - A proof-of-concept study to support a precision medicine approach to guide glucocorticoid therapy for sepsis. Front Immunol. 2023;13:1110516. doi:10.3389/fimmu.2022.1110516
158. Kullberg RFJ, Haak BW, Chanderraj R, Prescott HC, Dickson RP, Wiersinga WJ. Empirical antibiotic therapy for sepsis: save the anaerobic microbiota. Lancet Respir Med. 2025;13(1):92–100. doi:10.1016/S2213-2600(24)00257-1
159. Hechtman RK, Kipnis P, Cano J, Seelye S, Liu VX, Prescott HC. Heterogeneity of benefit from earlier time-to-antibiotics for sepsis. Am J Respir Crit Care Med. 2024;209(7):852–860. doi:10.1164/rccm.202310-1800OC
160. Liu Y, Zhao X, Xu Z, Li Z, Wang Y. Protective role of selenium in sepsis: mechanisms and potential therapeutic strategies. Open Med. 2025;20(1):20251296. PMID: 41216203; PMCID: PMC12596874. doi:10.1515/med-2025-1296
161. Li T, Zhao X, Xu Z, et al. Selenomethionine attenuates sepsis-induced skeletal muscle atrophy by inhibiting ROS/NLRP3 signaling. Shock. 2025. PMID: 40961381. doi:10.1097/SHK.0000000000002690
162. Wang D, Xu L, Liu Y, et al. Role of mesenchymal stem cells in sepsis and their therapeutic potential in sepsis‑associated myopathy (Review). Int J Mol Med. 2024;54(5):92. PMID: 39219272; PMCID: PMC11374154. doi:10.3892/ijmm.2024.5416
163. Liu Y, Liu J, Wang D, et al. Advancements, challenges, and future prospects of nanotechnology in sepsis therapy. Int J Nanomed. 2025;20:7685–7714. PMID: 40546802; PMCID: PMC12180596. doi:10.2147/IJN.S488026
164. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. doi:10.1126/science.1250684
165. Seok J, Warren HS, Cuenca AG, et al; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110(9):3507–3512. doi:10.1073/pnas.1222878110
166. Stripecke R, Münz C, Schuringa JJ, et al. Innovations, challenges, and minimal information for standardization of humanized mice. EMBO Mol Med. 2020;12:e8662. doi:10.15252/emmm.201708662
167. Laudanski K. Humanized mice as a tool to study sepsis-more than meets the eye. Int J Mol Sci. 2021;22(5):2403. doi:10.3390/ijms22052403
168. Zhang T, Chen L, Kueth G, et al. Lactate’s impact on immune cells in sepsis: unraveling the complex interplay. Front Immunol. 2024;15:1483400. doi:10.3389/fimmu.2024.1483400
169. Zhang TN, Huang XM, Li L, et al. Lactylation of HADHA promotes sepsis-induced myocardial depression. Circulation Res. 2025;137(4):e65–e87. doi:10.1161/CIRCRESAHA.124.325708
170. Wu D, Spencer CB, Ortoga L, Zhang H, Miao C. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. 2024;74:103194. doi:10.1016/j.redox.2024.103194
171. Lu Z, Fang P, Li S, et al. Lactylation of histone H3k18 and Egr1 promotes endothelial glycocalyx degradation in sepsis-induced acute lung injury. Adv Sci. 2025;12(7):e2407064. doi:10.1002/advs.202407064
172. Fang P, Li S, Lu ZQ, et al. Histone lactylation exacerbates acute lung injury in septic mice by promoting ferroptosis in pulmonary microvascular endothelial cells. Burns Trauma. 2025;13:tkaf056. doi:10.1093/burnst/tkaf056
173. Guirguis N, Machuca-Parra AI, Matoori S. Portable near-infrared fluorometer for a liposomal blood lactate assay. ACS Pharmacol Transl Sci. 2023;6(6):907–912. doi:10.1021/acsptsci.3c00055
174. Kaur M, Bhat SH, Tiwari R, et al. Rapid electrochemical detection of bacterial sepsis in cirrhotic patients: a microscaffold-based approach for early intervention. Anal Chem. 2024;96(12):4925–4932. doi:10.1021/acs.analchem.3c05754
175. Póvoa P, Coelho L, Dal-Pizzol F, et al. How to use biomarkers of infection or sepsis at the bedside: guide to clinicians. Intensive Care Med. 2023;49(2):142–153. doi:10.1007/s00134-022-06956-y
176. Mi Y, Burnham KL, Charles PD, et al. High-throughput mass spectrometry maps the sepsis plasma proteome and differences in patient response. Sci Trans Med. 2024;16(750):eadh0185. doi:10.1126/scitranslmed.adh0185
177. Na AY, Lee H, Min EK, et al. Novel time-dependent multi-omics integration in sepsis-associated liver dysfunction. Genomics Proteomics Bioinform. 2023;21(6):1101–1116. doi:10.1016/j.gpb.2023.04.002
178. Gao Y, Chen H, Wu R, Zhou Z. AI-driven multi-omics profiling of sepsis immunity in the digestive system. Front Immunol. 2025;16:1590526. doi:10.3389/fimmu.2025.1590526
179. Huang P, Liu Y, Li Y, et al. Metabolomics- and proteomics-based multi-omics integration reveals early metabolite alterations in sepsis-associated acute kidney injury. BMC Med. 2025;23(1):79. doi:10.1186/s12916-025-03920-7
180. Gu Y, Li Z, Li H, et al. Exploring the efficacious constituents and underlying mechanisms of sini decoction for sepsis treatment through network pharmacology and multi-omics. Phytomedicine. 2024;123:155212. doi:10.1016/j.phymed.2023.155212
181. Xu LL, Zhou Z, Schäuble S, et al. Multi-omics and -organ insights into energy metabolic adaptations in early sepsis onset. Adv Sci. 2025;e04418. doi:10.1002/advs.202504418
182. Xu P, Tao Z, Zhang C. Integrated multi-omics and artificial intelligence to explore new neutrophils clusters and potential biomarkers in sepsis with experimental validation. Front Immunol. 2024;15:1377817. doi:10.3389/fimmu.2024.1377817
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.