Back to Journals » ImmunoTargets and Therapy » Volume 3
Immunologic special forces: anti-pathogen cytotoxic T-lymphocyte immunotherapy following hematopoietic stem cell transplantation
Received 21 February 2014
Accepted for publication 5 April 2014
Published 18 June 2014 Volume 2014:3 Pages 97—106
DOI https://doi.org/10.2147/ITT.S40082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Michael D Keller, Catherine M Bollard
Program for Cell Enhancement and Technologies for Immunotherapy, Sheikh Zayed Institute for Pediatric Surgical Innovation, and Center for Cancer and Immunology Research, Children’s National Health System, Washington, DC, USA
Abstract: Anti-pathogen adoptive T-cell immunotherapy has been proven to be highly effective in preventing or controlling viral infections following hematopoietic stem cell transplantation. Recent advances in manufacturing protocols allow an increased number of targeted pathogens, eliminate the need for viral transduction, broaden the potential donor pool to include pathogen-naïve sources, and reduce the time requirement for production. Early studies suggest that anti-fungal immunotherapy may also have clinical benefit. Future advances include further broadening of the pathogens that can be targeted and development of T-cells with resistance to pharmacologic immunosuppression.
Keywords: immunotherapy, stem cell transplantation, T-cell, virus, fungus
Introduction
Since the advent of hematopoietic stem cell transplantation (HSCT), infections have remained a leading cause of morbidity and mortality in patients.1–4 Although advances in prophylactic therapy have reduced the early burden of viral and fungal infections, therapeutic options for breakthrough infections are complicated by toxicities, and for many viral infections there are no effective treatments.5–9 It has been well established that T-cell reconstitution is the most important factor in preventing viral infection following HSCT, and factors that influence the speed of T-cell recovery also impact the risk of viral infection in this period.2,3 As transplantation protocols have progressed to allow an increasing number of donor sources for transplantation, clinicians have had to balance the risks of graft versus host disease (GVHD) when using a T-cell replete graft versus delayed T-cell engraftment when using T-cell depletion or a naïve donor source such as cord blood.10,11
Given the importance of T-cells to antiviral immunity, use of donor lymphocyte infusions from the stem cell donor was discovered to be an effective salvage therapy for viral infections in HSCT recipients prior to T-cell recovery.12 However, the high rate of potentially fatal GVHD has relegated this treatment to a course of last resort. However, subsequent advances in immunobiology and culturing techniques have permitted great progress in improving the safety and efficacy of cytotoxic T-lymphocyte (CTL) immunotherapy following HSCT. These include: an improved knowledge of conserved T-cell epitopes for various pathogens,13–15 improvements in ex vivo culture of T-cells and antigen-presenting cells,16–18 and rapid tests to evaluate the effector function and major histocompatibility complex (MHC) restriction of T-cells.19,20 In essence, CTL therapy allows clinicians to bypass the months required for T-cell engraftment and a subsequent primary immune response to a pathogen.
Although trials utilizing antiviral CTLs represent the bulk of the studies to date, preclinical studies and early clinical trials of antifungal CTLs have also shown promise. Adoptive immunotherapy targeting tumor targets is also a burgeoning field, and has recently been reviewed.21 In this review, we summarize the methodologies and results of recent and current trials of anti-pathogen CTL therapy, and recap recent preclinical advances that provide the framework for future CTL clinical studies.
Methodologies of CTL production
In CTL production protocols to date, two concepts are essential, ie, harnessing pathogen-specific T-cells, and the exclusion of alloreactive T-cells. This has been accomplished previously by either direct selection of donor cells, or stimulation and ex vivo culture of donor T-cells from peripheral blood mononuclear cells (PBMCs).
Direct selection relies on cell sorting of donor PBMCs, usually after a short stimulation with the antigen of interest.22 Selection can be achieved via multimer selection (selecting for T-cells with a T-cell receptor of known antigen specificity), or by column selection of interferon-gamma-producing T-cells following a brief stimulation with an antigen of interest (Figure 1). It has the advantage of a minimal time requirement for product manufacturing, and uses existing Good Manufacturing Practice-compliant sorting technologies. However, this technique requires leukapheresis of donors in order to collect sufficient cells for clinical use. Additionally, it requires that there be detectable pathogen-specific T-cells in the periphery, and thus it would not be a viable option for manufacturing of CTLs from pathogen-naïve donors nor for pathogens that induce a poor memory response. Multimer selection has the disadvantage of selecting only CD8+ T-cells of limited specificity and MHC restriction, which could allow pathogen evasion and possibly impair CTL persistence.23 Additionally, previous studies have suggested that residual binding of multimers may impact T-cell function in vitro,24 although the clinical impact of this effect is unclear. The recent development of reversible streptamer technology for selection bypasses this potential risk.25 Interferon-gamma selection allows inclusion of polyclonal antigen-specific CD4+ and CD8+ T-cells, and allows selection of a wider range of antigen-specific cells in the final product.
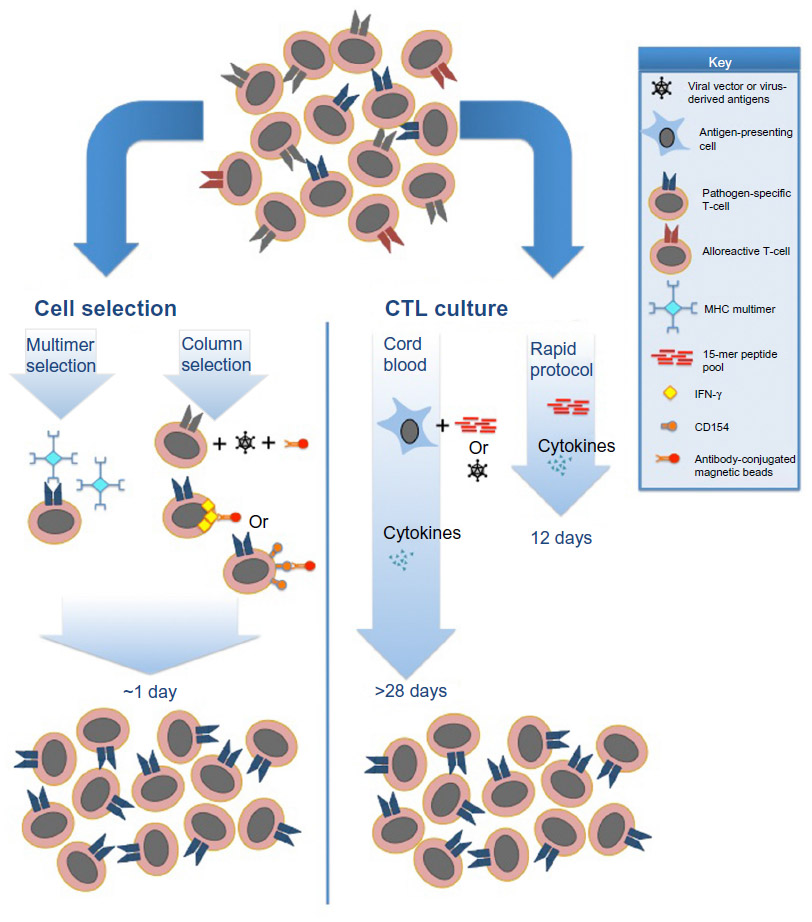 | Figure 1 Current Good Manufacturing Practice-compliant approaches for generation of antipathogen CTL products. |
Alternatively, stimulation and ex vivo culture permits expansion of single or multiple pathogen-specific CTLs. Culture has several advantages over cell selection, including generation of polyclonal CTLs, and expansion of cells to clinically useful volumes from a small volume of blood.26 These advantages come at the expense of the culture and processing time required for CTL stimulation and expansion, which can vary from 10 days to more than 3–4 weeks, depending on the donor source. Loss of the ability of cells to self-renew and impaired persistence in vivo has been a longstanding concern with the use of prolonged ex vivo culture and expansion.27 However, clinical trials to date have demonstrated prolonged persistence in spite of ex vivo culture.28 Additionally, studies have demonstrated that ex vivo culturing with pathogen-specific stimuli eliminates alloreactivity,15 likely due to cell death or inability to compete with pathogen-specific T-cells, and residual alloreactivity in manufactured CTLs has been shown to be clinically insignificant.29 Early trials of CTL therapy depended on the use of virus-infected antigen-presenting cells, such as cytomegalovirus (CMV) lysates, CMV-infected fibroblasts, or Epstein Barr virus (EBV)-lymphoblastoid cell lines as a stimulant for expansion of donor-derived memory T-cells.30–32 Subsequent knowledge of dominant and highly conserved antigens such as CMV-pp65 and Adenovirus (Adv) hexon and penton have permitted the replacement of live virus with antigen stimulation using either 15-mer peptide pools spanning viral proteins, or with transduction of DNA plasmids encoding viral antigens into antigen-presenting cells.33,34 New methods to rapidly grow and manipulate antigen-presenting cells have also enabled the use of a wider population of donors and targeting of a greater number of pathogens in a single CTL culture.16,35 Optimization of cytokine cocktails for CTL culture has also allowed improved yields and targeted cellular phenotypes. In the recent rapid CTL protocol, interleukin (IL)-4 and IL-7 were shown to produce CD4+ T-cells with a predominantly Th1 phenotype, whereas IL-2 and IL-15 seem to favor proliferation of natural killer cells at the expense of T-cells.34 Finally, studies have shown that central memory T-cells (characterized by expression of chemokine receptors CCR7, CD62L, and CD45RA) have superior persistence in vivo following adoptive transfer, and may be the ideal cell population for adoptive immunotherapy.36,37 Consequently, studies using both selection and culture methods have demonstrated the development of central memory T-cells in the resulting CTL products.25,34
Clinical studies of anti-viral CTLs
Clinical studies utilizing cell selection
Cell selection has been used in several prior studies to treat patients following HSCT (Table 1). Cobbold et al published the first clinical report in which CD8+ CMV-specific CTLs were isolated via tetramer selection.38 Complete or partial clinical responses were achieved in nine patients who received infusions, although there were limited data on long-term persistence of infused CTLs. Feuchtinger et al utilized interferon-gamma column selection (Gamma capture assay; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) to produce CMV-CTL, resulting in partial to complete responses in 15 of 18 patients who were given a single dose.39 Peggs et al also used interferon-gamma selection to produce CMV-CTL, using either recombinant pp65 or an overlapping peptide pool of 15-mers covering the pp65 protein as stimulants.40 They were successful in protecting seven patients who were prophylactically treated, while in vivo expansion of CMV-CTLs was detected in 11 patients infused who had detectable CMV.40 Schmitt et al produced CMV-CTL from HSCT donors utilizing reversible streptamers with MHC-restricted pp65 peptides.25 These products were used to successfully treat two patients who developed CMV reactivation during treatment of GVHD after HSCT.
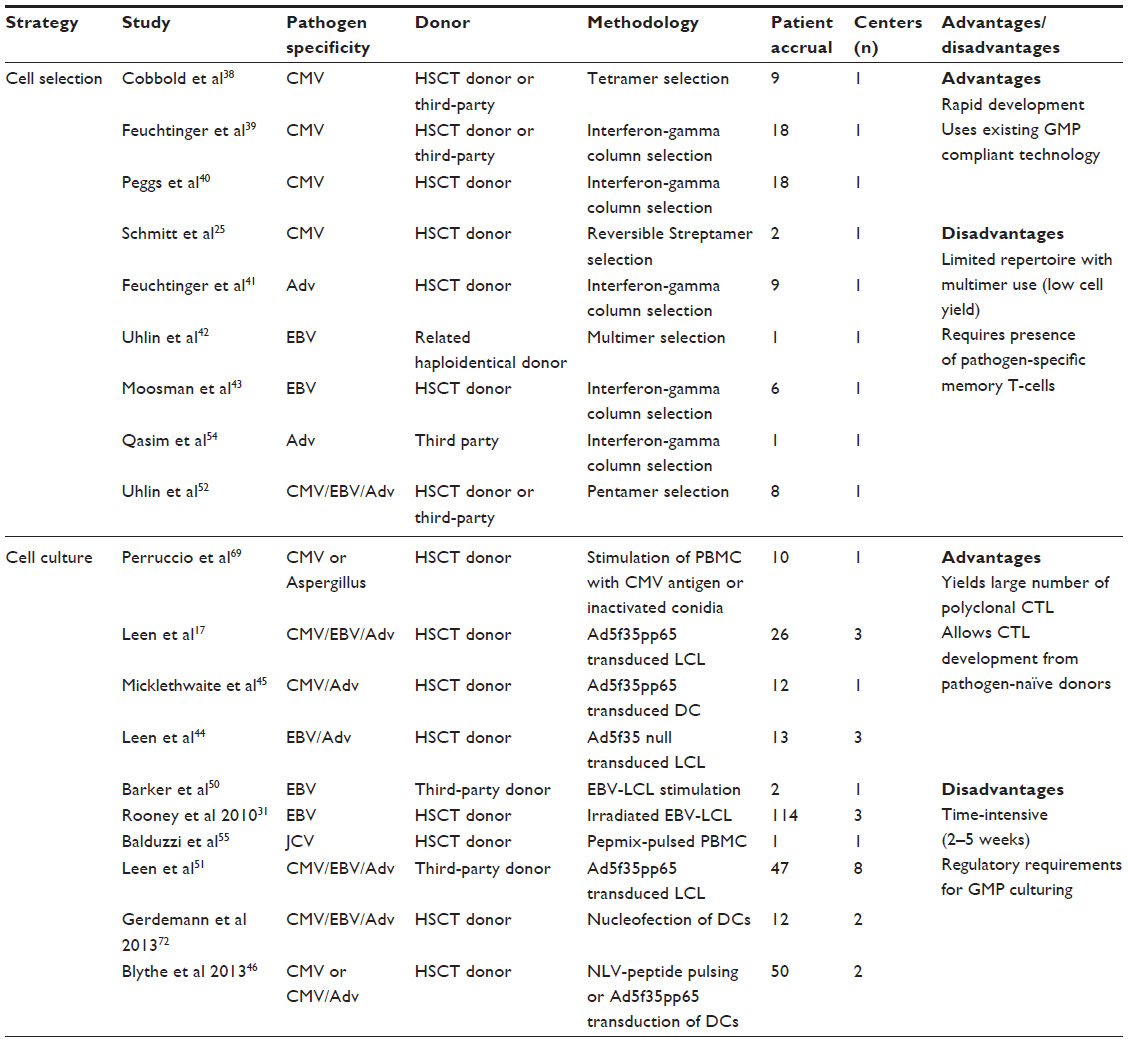 | Table 1 Previous clinical trials of pathogen-specific T-cell therapy |
Fewer clinical studies have been performed using these techniques to produce Adv-specific or EBV-specific CTL. Feuchtinger et al successfully produced Adv CTL by interferon-gamma selection for treatment of nine patients with treatment-refractory Adv infections.41 In vivo CTL expansion was demonstrated in five of six patients tested, and four patients had clearance of disease. Uhlin et al used human leukocyte antigen (HLA)-A2-specific pentamers to produce EBV-CTL from the haploidentical mother of a patient who underwent cord blood transplantation and subsequently developed EBV-induced post-transplant lymphoproliferative disease.42 A complete clinical response was obtained following two doses of CTLs. Moosmann et al treated six patients with EBV-induced post-transplant lymphoproliferative disease with EBV-CTL developed by interferon-gamma selection, and achieved complete responses in three patients with early disease, but no response in three patients with advanced, multiorgan disease.43 Of note, in all studies utilizing cell selection, no significant GVHD occurred, and clinical impacts were seen in spite of very low cell doses (<5×104 cells/kg in most studies).
Clinical studies utilizing cell culture
CTL production utilizing ex vivo cell culture has been the most common methodology to date, and accounts for the majority of patients treated in clinical trials of antipathogen adoptive immunotherapy (Table 1) over the past decade. Walter et al were among the first to show that stimulation of donor PBMC by CMV extracts resulted in expansion of CMV-specific CTLs, which lost alloreactivity after several weeks of ex vivo culture but retained antiviral activity.32 There have been many advancements in ex vivo CTL culture since then, which have decreased both the manufacturing time and cost. Early notable strides involved the culture and manipulation of antigen-presenting cells for CTL culture. Rooney et al successfully used irradiated EBV-lymphoblastoid cell lines (EBV-LCL) to generate EBV-specific CTL, which were effective as prophylaxis or treatment for EBV-induced post-transplant lymphoproliferative disease in 114 patients.28,31 Of note, the first 26 patients received gene-marked CTLs, and follow-up studies showed persistence of the gene-marked cells as long as 105 months following infusion.
The development of clinical grade Adv vector Ad5f35pp65, which contains immunodominant CMV antigen pp65, permitted transduction of either donor-derived dendritic cells or EBV-LCL for use as antigen-presenting cells for CTL culture. Leen et al used this strategy to produce triviral (CMV, EBV, Adv-specific) CTLs, which were utilized in a dose-escalation trial to treat 26 patients.17 No adverse effects were seen at doses ranging from 5×106 to 1×108 cells/m2, and all patients were effectively protected against CMV, EBV, and Adv disease. However, although EBV-specific and CMV-specific CTLs showed persistence by interferon-gamma ELISPOT, Adv-specific CTLs were not detectable except in the setting of infection. A follow-up trial utilized Ad5f35-transduced EBV-LCL to produce EBV-specific and Adv-specific CTL, which were infused into 13 patients as prophylaxis or treatment of EBV and Adv following HSCT.44 Although the products provided effective protection against EBV and Adv in vivo, Adv-specific CTLs were again not detectable except in the setting of Adv infection, suggesting that even at levels below the limits of detection by interferon-gamma ELISPOT, the Adv-specific CTL provided protection and was able to undergo expansion in the setting of viral infection. Ad5f35pp65-transduced dendritic cells were similarly used by Micklethwaite et al to produce CMV-specific and Adv-specific CTLs, which were clinically effective in 12 patients who received infusions following HSCT.45 Only two subsequent episodes of CMV reactivation occurred in the setting of administration of prednisone at levels as low as 0.5 mg/kg/day. Blyth et al similarly treated 50 patients following HSCT with triviral (CMV, EBV, Adv-specific) CTLs which were derived by a mix of methods: ten were produced by pulsing donor dendritic cells with the HLA-A2-restricted CMV peptide NLVPMVATV and 40 were produced using Ad5f35pp65-transduced donor dendritic cells.46 Only five of the 50 patients developed CMV reactivations following CTL infusions, and one of these five required antiviral pharmacotherapy after being treated with steroids for acute GVHD.
Further protocol advances have validated the use of 15-mer peptide pools encompassing immunodominant viral antigens in place of viral transduction of antigen-presenting cells, thus removing the potential safety and regulatory barriers associated with use of viral vectors.33 The use of gas-permeable rapid-expansion (G-Rex) bioreactors has further simplified CTL culture.47 Gerdemann et al combined these two advances to develop a rapid protocol that yields CTL at clinical volumes in 10–12 days, and provided effective antiviral protection in ten patients who were infused following HSCT.34 The ongoing ARMS (Administration of Rapidly Generated Multivirus-Specific Cytotoxic T-Lymphocytes for the Prophylaxis and Treatment of EBV, CMV, Adv, human herpesvirus 6 [HHV6], and BK virus infections post Allogeneic Stem Cell Transplant; NCT01570283) study has further modified this rapid protocol to produce five virus-specific CTL from a monoculture.
Gerdemann et al have further modified the rapid CTL protocol by utilizing nucleofection of DNA plasmids containing viral epitopes into donor-derived dendritic cells.48 The resulting CTL cultures showed antiviral activity in vitro by interferon-gamma ELISPOT and Cr51 cytotoxicity assays comparable with that of similar products derived via stimulation with 15-mer peptide pools for the same viral epitopes.
Adverse events following administration of ex vivo cultured CTL products in 381 infusions for 180 patients on 18 protocols were recently reviewed by the groups at Baylor College of Medicine.49 Twenty-four mild adverse events were reported within 6 hours of infusion, with nausea and vomiting being most common, and 22 nonserious adverse events (fever, chills, nausea) occurring within 24 hours. No significant GVHD was attributable to CTL infusion. The only significant complications of CTL therapy have been rare reports of systemic inflammatory responses following EBV-CTL therapy in patients with bulky EBV+ lymphoma. Blyth et al reported that seven cases of acute GVHD occurred following CTL infusion, although some were attributable to corticosteroid weaning prior to CTL infusion, and additionally the authors noted that the degree of HLA mismatch was greater in patients who received CTL therapy versus controls.46
Recent developments
Third-party CTL use, expanded viral targets, T-cell receptor gene transfer, and CTL manufacture from pathogen-naïve donors
Until recently, the selection or culture of antipathogen CTLs was dependent on the presence of pathogen-specific memory T-cells in donor blood. These protocols failed to help recipients of pathogen-naïve stem cell products, a population that has been well described to be at increased risk of viral infection following HSCT.
One answer to this problem is the use of “off-the-shelf” CTLs derived from third-party donors. This approach has been successfully used in several prior studies.50,51 Barker et al successfully treated two patients with refractory EBV-induced post-transplant lymphoproliferative disease following cord blood transplantation with third-party EBV-specific CTLs.50 Leen et al utilized a bank of 32 CTL lines with characterized activity against EBV, CMV, and Adv to identify matched lines for 50 patients with refractory viral infections.51 These infusions resulted in antiviral responses in 74%, 78%, and 67% of those with CMV, Adv, and EBV, respectively. This represents a dramatic improvement from the standard therapy response rate in eight patients for whom a matched line could not be found, who had a response rate of 13% and a mortality rate of 75%. In spite of only partial HLA matching (1–4 loci), only two patients developed grade I GVHD. The lower rate of response against EBV relative to CMV and Adv may be reflective of a greater breadth of immunodominant epitopes that differ by MHC types, which complicates the task of selecting the ideal third-party line with both antiviral activity and proper MHC restriction.
Third-party CTL treatment has also been successful using selection methodology. Uhlin et al used pentamer selection to produce anti-viral CTL specific for CMV, EBV, or Adv from related third-party donors for six patients with refractory viral infections (four with CMV, and one each with EBV and Adv).52 Five of six patients had partial or complete responses. Notably, an infant with severe combined immunodeficiency was treated prior to cord blood transplantation with CMV-CTL derived from her mother, with a ten-fold reduction in her CMV DNA level. Wy and Qasim used interferon-gamma selection to manufacture Adv-CTL from related third-party donors to treat two patients who underwent HSCT and subsequently developed Adv viremia.53 Although treatment successfully cleared the Adv infection in one patient, she developed grade III skin and liver GVHD.54 Curiously, cytogenetic studies of liver tissue showed infiltration with T-cells from the original HSCT donor but not the CTL donor. The authors postulated that this was due to a “bystander” effect of CTLs on the HSCT donor cells; however, such an effect has not been seen in larger trials utilizing third-party CTL therapy.
A small number of other viruses have been targeted via adoptive immunotherapy. John Cunningham virus (JCV) is an ubiquitous polyoma virus which can cause progressive multifocal leukoencephalopathy, a devastating neurologic disease, in patients who are profoundly immunocompromised, including recipients of HSCT or solid organ transplants and patients with advanced human immunodeficiency virus or primary immunodeficiency disorders. Balduzzi et al described the use of donor-derived JCV-specific CTL in a 14-year-old patient who developed progressive multifocal leukoencephalopathy in the setting of prolonged steroid treatment for GVHD following HSCT.55 These CTL were manufactured using 15-mer peptide pools encompassing the JCV antigens VP1 and LT, and were cultured for 26 days. The patient received two doses of JCV-specific CTLs, and had a remarkable and sustained improvement, including clearance of JCV-DNA from the cerebrospinal fluid and substantial improvements in his neurologic status.
Although not a frequent problem following HSCT, human papillomavirus (HPV) is not an uncommon late complication of HSCT, particularly in patients treated for primary immunodeficiency disorders. HPV has also been evaluated in preclinical studies as a potential target for CTL therapy. Ramos et al have described the use of peptide pools spanning the HPV E6 and E7 proteins to generate HPV-specific CTLs from patients with oropharyngeal or cervical cancer, many of which arise due to HPV16 infection.56 The resulting CTLs showed specific activity against HPV E6 and E7, and also showed antitumor activity against CaSki, an HPV16 cervical cancer cell line.
Several studies have explored the possibility of transducing CTL with a T-cell antigen receptor of known viral specificity.57–59 This offers a novel strategy to develop CTL from pathogen-naïve donors, but imposes the additional regulatory requirements of transgenic technology. Additionally, the use of a single antiviral T-cell antigen receptor may risk antigenic escape by the pathogen. Nonetheless, a current trial of transgenic CTL utilizing a retroviral vector with a CMV-specific T-cell antigen receptor is being conducted in the UK by Emma Morris (principal investigator).60
An important landmark in the field of adoptive immunotherapy has been the successful development of virus-specific CTLs from virus-naïve donors. Hanley et al first demonstrated that CTL could be produced in a 20% fraction from cord blood using donor-derived dendritic cells and an EBV-lymphoblastoid cell line as antigen-presenting cells, and Ad5f35pp65 transduction as a source of CMV and Adv antigens.16 The resulting cell lines had specific antiviral activity against CMV, EBV, and Adv in interferon-gamma ELISPOT analysis as well as Cr51 cytotoxicity assays, with no evidence of alloreactivity. Curiously, epitope mapping showed that the immunodominant epitopes recognized by cord blood-derived CTLs differed from CTLs manufactured from CMV-seropositive and EBV-seropositive adult donors, with the HLA-A2 restricted epitope NLVPMVATV notably absent in the cord blood-derived lines. Despite this finding, CTLs manufactured from cord blood have been used successfully in 12 cord blood transplant recipients to date in the ongoing ACTCAT (Safety, Toxicity and MTD of One Intravenous IV Injection of Donor CTLs Specific for CMV and Adenovirus; NCT00880789) trial.
Most recently, Hanley et al have successfully manufactured multiviral CTLs from CMV-naïve adult donors.35 To do so, CMV-CTLs were produced from CD45RA+ naïve T-cells isolated via column selection, and stimulated by donor dendritic cells pulsed with CMV 15-mer peptide pools. Preclinical data suggest that they have similar antiviral activity, and the current MUSTAT (Multivirus-Specific Cytotoxic T-Lymphocytes for the Prophylaxis and Treatment of EBV, CMV, and Adenovirus Infections post Allogeneic Stem Cell Transplant; NCT01945814) trial will seek to compare the clinical efficacy of CTLs derived from CMV-seropositive versus CMV-naïve donors.
Anti-fungal CTLs
Fungal infections are a well described risk after HSCT. The importance of Th17 immunity in controlling Candida infections has been well demonstrated by forms of primary immunodeficiency such as Hyperimmunoglobulin E syndrome and chronic mucocutaneous candidiasis, as well as human immunodeficiency virus infection.61,62 The importance of T-cell immunity in defense against invasive aspergillosis and mucormycosis is less clear, while their ties to innate defense (most notably neutrophil function) are well established. Interestingly, a recent study of patients with chronic granulomatous disease showed that they have abundant Aspergillus-specific T-cells with increased interferon-gamma production compared with healthy controls.63 Despite these uncertainties, fungal infections may be a valid target for treatment via adoptive immunotherapy after HSCT.
Several preclinical studies have been successful in developing CTLs with activity against Candida, Aspergillus, and Rhizopus species (Table 2). Beck et al successfully produced Aspergillus-specific CTLs by stimulation of PBMCs with antigens from Aspergillus extracts, followed by interferon-gamma selection and culture.64 The resulting population was predominantly CD4+ memory (CD45RO+) cells, but demonstrated interferon-gamma production in response to several species of Aspergillus as well as Penicillium. The authors also showed that these T-cells enhanced hyphal damage by neutrophils and antigen-presenting cells in vitro. Tramsen et al similarly used interferon-gamma selection following stimulation with cellular extracts from Candida albicans, Aspergillus fumigatus, and Rhizopus oryzae to produce multifungal-specific CTL lines, which were also almost exclusively CD4+ CD45RO+ HLA-DR+.65 These lines displayed pathogen-specific activation markers (interferon-gamma CD154, tumor necrosis factor-alpha) and also enhanced oxidative activity of neutrophils when coincubated with antigen and antigen-presenting cells and tested via the 123-dihydrorhodamine assay. Khanna et al described a novel selection method based on upregulation of CD154 to produce multipathogen-specific T-cells against CMV, EBV, Adv, Candida, and Aspergillus.66 Donor PBMCs were incubated with peptide libraries from CMV-pp65, EBV-LMP2, Adv-Hexon, Candida MP65, and a 15-mer peptide from Aspergillus CRF1. Following 14 days of culture, the authors showed pathogen-specific interferon-gamma production, proliferation, and cytotoxicity in vitro. Although these results are intriguing, there are very limited data regarding the relative importance of MP65 and CRF1 in antifungal immunity.67,68
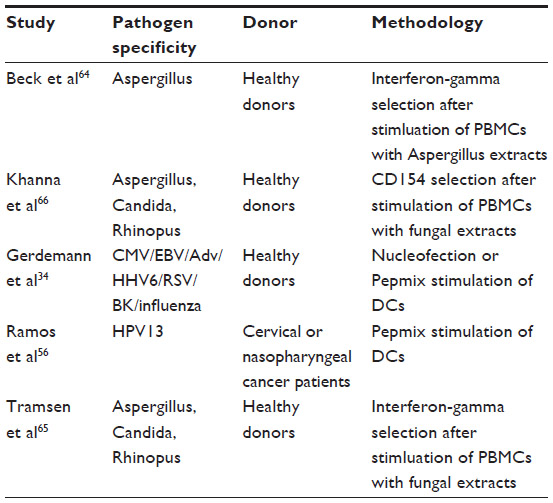 | Table 2 Preclinical studies of novel antipathogen T-cell therapies |
As of the time of this review, only one clinical trial of anti-fungal CTLs has been published. Perruccio et al developed CTLs via stimulation of donor PBMCs with inactivated conidia from A. fumigatus, followed by several weeks of culture, resulting in clonal CD4+ CTLs with anti-Aspergillus activity by interferon-gamma ELISPOT.69 Clinical use of these lines in patients with pulmonary aspergillosis resulted in survival of eight of nine patients treated, compared with a survival rate of 7/13 in patients with aspergillosis who did not receive infusions. There was no difference in the length of antifungal therapy required for survivors in the two groups.
Although these studies are intriguing, several key issues require attention before anti-fungal CTL trials begin to catch up with their antiviral brethren. First, a better understanding of the immunodominant T-cell targets for various fungal species is needed. Second, standardized Good Manufacturing Practice-compliant fungal antigen sources are necessary to allow consistency and valid comparisons between future clinical trials.
Future of CTL therapy
Expanding the breadth of monoculture CTL lines: is there an antigen limit?
As manufacture of CTLs expands to include more pathogens in a single culture, the possibility of antigenic competition between the different pathogen-specific T-cells has caused many to question the limits of CTL monocultures. This concern has certainly been validated in attempts to produce multivirus-specific CTLs from donors who are CMV-naïve, in which the resulting culture is dominated by memory-derived EBV-specific and Adv-specific T-cells. Although the relative proportions of individual virus-specific CTLs decrease as the number of antigens increases, this has not seemed to impact the efficacy of these products in clinical trials. Recent studies have challenged the upper antigen limit of CTL monoculture, as Gerdemann et al successfully produced CTLs specific for seven viruses (CMV, EBV, Adv, BK, HHV6, respiratory syncytial virus (RSV), and influenza) utilizing peptide pools for 15 antigens, and demonstrated specific activity against all targeted viruses via interferon-gamma ELISPOT.34 As additional preclinical studies attempt to add further pathogens to monoculture, it remains to be seen whether an increased number of targets will compromise specific CTL function or persistence in vivo.
Engineering resistance to immunosuppression
The need for immunosuppressive medications is common in recipients of HSCT, and unfortunately the use of these drugs also suppresses CTL products. Most existing protocols require recipients to be receiving less than 0.5 mg/kg/day prednisone and at least 30 days out from any anti-T-cell serotherapy in order to receive a CTL infusion. Calcineurin inhibitors such as cyclosporin A, tacrolimus, or sirolimus would similarly impact the clinical benefits of CTL at therapeutic doses.
One answer to this problem is to produce genetically modified CTLs that have resistance to immunosuppressive medications. Several recent studies have successfully demonstrated the viability of this concept. De Angelis et al produced EBV-specific CTLs with resistance to tacrolimus by knockdown of FKBP12 via a retrovirally-transduced specific siRNA.70 Transduction of CTLs did not impact antiviral activity, and the cells showed activity in a mouse EBV-lymphoma model in the presence of tacrolimus. Brewin et al similarly produced EBV-specific CTLs with resistance to both cyclosporin A and tacrolimus by direct mutation of calcineurin.71 The mutation had no impact on the phenotype or antiviral activity of the CTL in vitro, and mutated cells showed a growth advantage in the presence of calcineurin inhibitors.
Although similarly modified cells have not been used clinically to date, they have great potential in treating both HSCT and solid organ transplant recipients. Future extension of these studies could potentially allow production of CTLs with resistance to monoclonal biologic agents such as alemtuzumab.
Conclusion
With several hundred patients having been treated successfully, antipathogen CTLs have been established as a safe and highly effective therapy following HSCT. Further studies to identify preserved viral T-cell epitopes, probe the antigen limits in CTL monoculture, and test the clinical efficacy of immunosuppressive-resistant CTLs will further broaden the usefulness of this therapy. As rapid advances in protocols and multiple available methods of manufacture broaden the availability of this therapy, in time CTL therapy may become the standard of care following HSCT.
Acknowledgment
This work was supported by a National Institute of Child Health and Human Development award to MDK (K12-HD-001399) and Cancer Prevention and Research Institute of Texas (R01 RP100469) and National Cancer Institute (P01 CA148600-02) awards to CMB.
Disclosure
The authors report no conflicts of interest.
References
Boeckh M, Leisenring W, Riddell SR, et al. Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood. 2003;101(2):407–414. | |
Brunstein CG, Weisdorf DJ, DeFor T, et al. Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood. 2006;108(8):2874–2880. | |
Myers GD, Krance RA, Weiss H, et al. Adenovirus infection rates in pediatric recipients of alternate donor allogeneic bone marrow transplants receiving either antithymocyte globulin (ATG) or alemtuzumab (Campath). Bone Marrow Transplant. 2005;36(11):1001–1008. | |
Neofytos D, Horn D, Anaissie E, et al. Epidemiology and outcome of invasive fungal infection in adult hematopoietic stem cell transplant recipients: analysis of Multicenter Prospective Antifungal Therapy (PATH) Alliance registry. Clin Infect Dis. 2009;48(3):265–273. | |
Avery R. Update in management of ganciclovir-resistant cytomegalovirus infection. Curr Opin Infect Dis. 2008;21(4):433–437. | |
Biron KK. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006;71(2–3):154–163. | |
Nichols WG, Corey L, Gooley T, et al. Rising pp65 antigenemia during preemptive anticytomegalovirus therapy after allogeneic hematopoietic stem cell transplantation: risk factors, correlation with DNA load, and outcomes. Blood. 2001;97(4):867–874. | |
Ljungman P, Deliliers GL, Platzbecker U, et al. Cidofovir for cytomegalovirus infection and disease in allogeneic stem cell transplant recipients. The Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2001;97(2):388–392. | |
Kuehnle I, Huls MH, Liu Z, et al. CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood. 2000;95(4):1502–1505. | |
Komanduri KV, St John LS, de Lima M, et al. Delayed immune reconstitution after cord blood transplantation is characterized by impaired thymopoiesis and late memory T-cell skewing. Blood. 2007;110(13):4543–4551. | |
Rubinstein P, Carrier C, Scaradavou A, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med. 1998;339(22):1565–1577. | |
Papadopoulos EB, Ladanyi M, Emanuel D, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330(17):1185–1191. | |
Slezak SL, Bettinotti M, Selleri S, Adams S, Marincola FM, Stroncek DF. CMV pp65 and IE-1 T cell epitopes recognized by healthy subjects. J Transl Med. 2007;5:17. | |
Leen AM, Christin A, Khalil M, et al. Identification of hexon-specific CD4 and CD8 T-cell epitopes for vaccine and immunotherapy. J Virol. 2008;82(1):546–554. | |
Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol. 2012;9(9):510–519. | |
Hanley PJ, Cruz CR, Savoldo B, et al. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114(9):1958–1967. | |
Leen AM, Myers GD, Sili U, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12(10):1160–1166. | |
Sili U, Huls MH, Davis AR, et al. Large-scale expansion of dendritic cell-primed polyclonal human cytotoxic T-lymphocyte lines using lymphoblastoid cell lines for adoptive immunotherapy. J Immunother. 2003;26(3):241–256. | |
Kern F, Faulhaber N, Frommel C, et al. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur J Immunol. 2000;30(6):1676–1682. | |
Hanley PJ, Shaffer DR, Cruz CR, et al. Expansion of T cells targeting multiple antigens of cytomegalovirus, Epstein-Barr virus and adenovirus to provide broad antiviral specificity after stem cell transplantation. Cytotherapy. 2011;13(8):976–986. | |
Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39(1):49–60. | |
Sellar RS, Peggs KS. The role of virus-specific adoptive T-cell therapy in hematopoietic transplantation. Cytotherapy. 2012;14(4):391–400. | |
Hansen SG, Powers CJ, Richards R, et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science. 2010;328(5974):102–106. | |
Neudorfer J, Schmidt B, Huster KM, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods. 2007;320(1–2):119–131. | |
Schmitt A, Tonn T, Busch DH, et al. Adoptive transfer and selective reconstitution of streptamer-selected cytomegalovirus-specific CD8+ T cells leads to virus clearance in patients after allogeneic peripheral blood stem cell transplantation. Transfusion. 2011;51(3):591–599. | |
Bollard CM, Kuehnle I, Leen A, Rooney CM, Heslop HE. Adoptive immunotherapy for posttransplantation viral infections. Biol Blood Marrow Transplant. 2004;10(3):143–155. | |
Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115(6):1616–1626. | |
Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–935. | |
Melenhorst JJ, Leen AM, Bollard CM, et al. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood. 2010;116(22):4700–4702. | |
Peggs KS, Verfuerth S, Pizzey A, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362(9393):1375–1377. | |
Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–935. | |
Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333(16):1038–1044. | |
Trivedi D, Williams RY, O’Reilly RJ, Koehne G. Generation of CMV-specific T lymphocytes using protein-spanning pools of pp65-derived overlapping pentadecapeptides for adoptive immunotherapy. Blood. 2005;105(7):2793–2801. | |
Gerdemann U, Keirnan JM, Katari UL, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther. 2012;20(8):1622–1632. | |
Hanley PJ, Cruiz RY, Melenhorst J, et al. Naïve T-cell-derived CTL recognize atypical epitopes of CMVpp65 with higher avidity than CMV-seropositive donor-derived CTL – a basis for treatment of post-transplant viral infection by adoptive transfer of T-cells from virus-naïve donors. Cytotherapy. 2013;15(4):S9. | |
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. | |
Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175(9):5895–5903. | |
Cobbold M, Khan N, Pourgheysari B, et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med. 2005;202(3):379–386. | |
Feuchtinger T, Opherk K, Bethge WA, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. 2010;116(20):4360–4367. | |
Peggs KS, Thomson K, Samuel E, et al. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis. 2011;52(1):49–57. | |
Feuchtinger T, Matthes-Martin S, Richard C, et al. Safe adoptive transfer of virus-specific T-cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation. Br J Haematol. 2006;134(1):64–76. | |
Uhlin M, Okas M, Gertow J, Uzunel M, Brismar TB, Mattsson J. A novel haplo-identical adoptive CTL therapy as a treatment for EBV-associated lymphoma after stem cell transplantation. Cancer Immunol Immunother. 2010;59(3):473–477. | |
Moosmann A, Bigalke I, Tischer J, et al. Effective and long-term control of EBV PTLD after transfer of peptide-selected T cells. Blood. 2010;115(14):2960–2970. | |
Leen AM, Christin A, Myers GD, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. 2009;114(19):4283–4292. | |
Micklethwaite KP, Clancy L, Sandher U, et al. Prophylactic infusion of cytomegalovirus-specific cytotoxic T lymphocytes stimulated with Ad5f35pp65 gene-modified dendritic cells after allogeneic hemopoietic stem cell transplantation. Blood. 2008;112(10):3974–3981. | |
Blyth E, Clancy L, Simms R, et al. Donor-derived CMV-specific T cells reduce the requirement for CMV-directed pharmacotherapy after allogeneic stem cell transplantation. Blood. 2013;121(18):3745–3758. | |
Vera JF, Brenner LJ, Gerdemann U, et al. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex). J Immunother. 2010;33(3):305–315. | |
Gerdemann U, Christin AS, Vera JF, et al. Nucleofection of DCs to generate multivirus-specific T cells for prevention or treatment of viral infections in the immunocompromised host. Mol Ther. 2009;17(9):1616–1625. | |
Cruz CR, Hanley PJ, Liu H, et al. Adverse events following infusion of T cells for adoptive immunotherapy: a 10-year experience. Cytotherapy. 2010;12(6):743–749. | |
Barker JN, Doubrovina E, Sauter C, et al. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116(23):5045–5049. | |
Leen AM, Bollard CM, Mendizabal AM, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121(26):5113–5123. | |
Uhlin M, Gertow J, Uzunel M, et al. Rapid salvage treatment with virus-specific T cells for therapy-resistant disease. Clin Infect Dis. 2012;55(8):1064–1073. | |
Wy Ip W, Qasim W. Management of adenovirus in children after allogeneic hematopoietic stem cell transplantation. Adv Hematol. 2013;2013: 176418. | |
Qasim W, Derniame S, Gilmour K, et al. Third-party virus-specific T cells eradicate adenoviraemia but trigger bystander graft-versus-host disease. Br J Haematol. 2011;154(1):150–153. | |
Balduzzi A, Lucchini G, Hirsch HH, et al. Polyomavirus JC-targeted T-cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient. Bone Marrow Transplant. 2011;46(7):987–992. | |
Ramos CA, Narala N, Vyas GM, et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies. J Immunother. 2013;36(1):66–76. | |
Schub A, Schuster IG, Hammerschmidt W, Moosmann A. CMV-specific TCR-transgenic T cells for immunotherapy. J Immunol. 2009;183(10):6819–6830. | |
Xue SA, Gao L, Ahmadi M, et al. Human MHC Class I-restricted high avidity CD4 T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology. 2013;2(1):e22590. | |
Frumento G, Zheng Y, Aubert G, et al. Cord blood T cells retain early differentiation phenotype suitable for immunotherapy after TCR gene transfer to confer EBV specificity. Am J Transplant. 2013;13(1):45–55. | |
MRC. CMV TCR Gene Therapy: A Phase I/II Safety, Toxicity and Feasibility Study of Adoptive Immunotherapy in Allo-HSCT. Available from: http://gtr.rcuk.ac.uk/project/644662DD-8EE2-4A77-82F2-24DBD79B1B87. Project reference: G0701703. Accessed June 11, 2014. | |
Milner JD, Holland SM. The cup runneth over: lessons from the ever-expanding pool of primary immunodeficiency diseases. Nat Rev Immunol. 2013;13(9):635–648. | |
Kim CJ, McKinnon LR, Kovacs C, et al. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J Immunol. 2013;191(5):2164–2173. | |
Cruz CR, Lam S, Hanley PJ, et al. Robust T cell responses to aspergillosis in chronic granulomatous disease: implications for immunotherapy. Clin Exp Immunol. 2013;174(1):89–96. | |
Beck O, Topp MS, Koehl U, et al. Generation of highly purified and functionally active human TH1 cells against Aspergillus fumigatus. Blood. 2006;107(6):2562–2569. | |
Tramsen L, Schmidt S, Boenig H, et al. Clinical-scale generation of multi-specific anti-fungal T cells targeting Candida, Aspergillus and mucormycetes. Cytotherapy. 2013;15(3):344–351. | |
Khanna N, Stuehler C, Conrad B, et al. Generation of a multipathogen-specific T-cell product for adoptive immunotherapy based on activation-dependent expression of CD154. Blood. 2011;118(4):1121–1131. | |
Gomez MJ, Maras B, Barca A, La Valle R, Barra D, Cassone A. Biochemical and immunological characterization of MP65, a major mannoprotein antigen of the opportunistic human pathogen Candida albicans. Infect Immun. 2000;68(2):694–701. | |
Jolink H, Meijssen IC, Hagedoorn RS, et al. Characterization of the T-cell-mediated immune response against the Aspergillus fumigatus proteins Crf1 and catalase 1 in healthy individuals. J Infect Dis. 2013;208(5):847–856. | |
Perruccio K, Tosti A, Burchielli E, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood. 2005;106(13):4397–4406. | |
De Angelis B, Dotti G, Quintarelli C, et al. Generation of Epstein-Barr virus-specific cytotoxic T lymphocytes resistant to the immunosuppressive drug tacrolimus (FK506). Blood. 2009;114(23):4784–4791. | |
Brewin J, Mancao C, Straathof K, et al. Generation of EBV-specific cytotoxic T cells that are resistant to calcineurin inhibitors for the treatment of posttransplantation lymphoproliferative disease. Blood. 2009;114(23):4792–4803. | |
Gerdemann U, Katari UL, Papadopoulou A, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther. 2013;21(11):2113–2121. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.