Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Immunization of AGE-modified albumin inhibits diabetic nephropathy progression in diabetic mice
Authors Mashitah MW, Azizah N, Samsu N ![]() , Indra MR, Bilal M, Yunisa MV, Arisanti AD
, Indra MR, Bilal M, Yunisa MV, Arisanti AD
Received 9 April 2015
Accepted for publication 15 May 2015
Published 4 August 2015 Volume 2015:8 Pages 347—355
DOI https://doi.org/10.2147/DMSO.S86332
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ming-Hui Zou
Musthika Wida Mashitah,1 Nurona Azizah,1 Nur Samsu,2 Muhammad Rasjad Indra,1 Muhammad Bilal,3 Meti Verdian Yunisa,4 Amildya Dwi Arisanti4
1Department of Biomedicine, Faculty of Medicine, Brawijaya University, Malang, 2Department of Internal Medicine, Division of Nephrology and Hypertension, Saiful Anwar General Hospital, Malang, 3Department of Medicine, Faculty of Medicine, Brawijaya University, Malang, 4Department of Nursing, Faculty of Medicine, Brawijaya University, Malang, Indonesia
Background: Diabetic nephropathy (DN) is a serious vascular complication of diabetes and an important cause of end-stage renal disease. One mechanism by which hyperglycemia causes nephropathy is through the formation of advanced glycation end products (AGE). Development of vaccination would be a promising therapy for the future, while to date, anti-AGE therapy is based on medicines that are needed to be consumed lifelong. This study aimed to find out the effect of immunization of AGE-modified albumin against DN pathogenesis in streptozotocin-induced diabetic in mice.
Methods: We used 24 BALB/c male mice as experimental animals, which were divided into six groups, two nondiabetic groups (negative control and AGE-modified bovine serum albumin [BSA] preimmunized groups) and four streptozotocin-induced diabetic groups (diabetic control group and diabetic preimmunized groups for AGE-BSA, Keyhole limpet hemocyanin (KLH), and AGE-BSA-KLH, respectively).
Results: Diabetic preimmunized groups for AGE-BSA, KLH, and AGE-BSA-KLH showed amelioration in renal function and histopathology compared with the diabetic control group. Preimmunization also maintained nephrin intensity and decreased serum AGE level, kidney AGE deposition, and kidney cells apoptosis.
Conclusion: AGE-BSA and AGE-BSA-KLH immunizations inhibit the progression of DN. Our results strengthen the evidence that the anti-AGE antibodies have a protective role against diabetic vascular complication, especially DN. This study provides a basis for the development of DN-based immunotherapy with AGE immunization as a potential candidate.
Keywords: immunization, advanced glycation end products (AGE), AGE-modified albumin, anti-AGE antibody, diabetic nephropathy
Introduction
Diabetic nephropathy (DN) is a serious vascular complication of diabetes and an important cause of end-stage renal disease. The increase in the prevalence of diseases that cause chronic kidney disease, including diabetes, is predicted to increase the prevalence of chronic kidney disease worldwide,1 whereas, the global diabetes prevalence is predicted to rise from 171 million in 2000 to 366 in 2030.2 In Indonesia, DN is the second common cause of hemodialysis patients in 2000.3 Indonesia is the country with the highest frequency of asymptomatic diabetic kidney disease among the countries that were followed in the Developing Education on Microalbuminuria for Awareness of Renal and Cardiovascular Risk in Diabetes (DEMAND) study in 2009.4
One mechanism on how hyperglycemia causes nephropathy is through the formation of advanced glycation end products (AGE).5,6 Long-term hyperglycemia induces protein glycation, named Maillard reaction, and generates AGE.7 Both AGE in tissue and circulation contribute to DN progression. The ability AGE cross-link with extracellular matrix (ECM) exacerbates glomerulosclerosis. Interaction of circulating AGE with receptor for AGE (RAGE) on mesangial cells, tubular, and podocytes will increase the production of intracellular reactive oxygen species and upregulation of transcription factor of nuclear factor kappa B, which in turn increase some growth factors and inflammatory cytokines. Therefore, nephropathy inductions are characterized by glomerular hyperfiltration, hyperpermeability, proteinuria, inflammation, ECM accumulation, thickening of glomerular basement membrane (GBM), mesangial expansion, glomerular hypertrophy, decreasing nephrin expression, increasing renal apoptotic cell death, glomerulosclerosis, and tubulointerstitial fibrosis.8–10
Development of vaccination would be a promising therapy for future treatment, while to date, anti-AGE therapy is based on medicines that are needed to be consumed lifelong.7,11 Basically, AGE consists of a wide variety of glycated proteins and has antigenic properties that stimulate the formation of autoantibodies in diabetic condition in vivo. However, its role remains unclear, whether it is protective or it exacerbates vascular complication.12 The previous study demonstrated that diabetic preimmunized mice with AGE-bovine serum albumin (BSA) increased anti-AGE antibody and decreased AGE level in serum.13 Moreover, for better antigenic response induction, immunized protein often coupled with Keyhole limpet hemocyanin (KLH) for its protein carrier. Furthermore, it has been proven previously that reactive immunization of KLH itself suppressed and mitigated DN in diabetic rat.14 Here, this study further examines the role of AGE-BSA and AGE-BSA-KLH immunization against DN pathogenesis and then provide a basic immunotherapy development for DN prevention.
Materials and methods
Reagents and animals
AGE-BSA was obtained from BioVision (Milpitas Boulevard, Milpitas, CA, USA); KLH, Complete Freund’s Adjuvant (CFA), Incomplete Freund’s Adjuvant (IFA), and streptozotocin (STZ) were obtained from Sigma-Aldrich Co (St Louis, MO, USA). Rabbit polyclonal anti-AGE antibody was procured from Abcam (Cambridge, MA, USA); rabbit anti-nephrin polyclonal antibody and rabbit anti-caspase-3 polyclonal antibody were procured from Bioss Inc. (Woburn, MA, USA). AGE-BSA and KLH were coupled using glutaraldehyde conjugation method. Healthy male BALB/c mice, 6–8 weeks of age, weighing 20–25 g, were purchased from Center of Laboratory Animal Breeding in Brawijaya University. Mice were housed individually with alternating 12-hour cycles of light and dark, fed a standard chow diet, and had free access of water. Mice were euthanized using ether. All work with mice was approved by the Ethics Committee of Medical Faculty, Brawijaya University, Malang, Indonesia.
Experimental design
The mice were divided into six groups of four animals each. Group I consisted of normal control mice (negative control). Group II received AGE-BSA preimmunization without diabetes induction. Group III, as diabetic control mice (positive control), received diabetes induction using STZ without preimmunization previously. Group IV, V, and VI received AGE-BSA, KLH, and AGE-BSA-KLH preimmunizations, respectively, followed by diabetes induction using STZ. Mice were adapted in the Pharmacology Laboratory for 7 days before experiment. Mice were immunized by intraperitoneal (ip) injection of 50 μg AGE-BSA, KLH, or AGE-BSA-KLH in phosphate-buffered saline (PBS) emulsified in CFA (1:1). All mice received boosters twice every 14 days later with the same dosage in IFA. Diabetes was induced by a single dose of 100 mg/kg STZ ip injection in 50 mM citrate buffer (pH 4.5) after 2 weeks from the last booster. To induce nephropathy, on the 7th week after the first STZ injection, mice received one dose of 40 mg/kg STZ ip injection per day for 5 consecutive days.15 Body weight was determined using Ohaus digital scale to get accurate STZ dose. Nonfasting blood glucose was measured from the tail vein by glucose meter (Nesco) every 7 days after the last STZ injection for assessing diabetes state. On the 5th week after last STZ injection, mice were euthanized with ether. Urine samples were taken from the bladder for measuring proteinuria by spectrophotometer with 660 nm absorbance according to the Lowry method. Blood samples were taken from the cardiac aorta to measure AGE and anti-AGE level in serum. Kidneys were harvested immediately for histological and immunohistochemistry evaluation.
Renal histological analysis
The kidneys were fixed in 10% buffered formalin, embedded in paraffin, cut into 3 μm sections, and stained with hematoxylin and eosin. The sections were viewed with a light microscope (Olympus) under 400× magnification. Forty glomeruli per sample sections were photographed randomly. Glomerular area (GA) and mesangial area (MA) were assessed with ImageJ 1.48 software. Glomerular volume (GV) was calculated using formula GV =1.2545 (GA)1.5. Mesangial expansion was calculated in percent by measuring MA proportion per GA or (MA/GA) × 100%.16
Measurement of serum AGE and anti-AGE level
Blood samples were incubated for 2–3 hours at room temperature (RT) and then centrifuged at 3,000 rpm for 15 minutes. Supernatant was collected to obtain serum, and the serum was stored in aliquot at −40°C to prevent repeated thawing. AGE and anti-AGE antibody levels were measured by indirect and direct ELISA, respectively, as previously described.17
Immunohistochemical assay
Paraffin embedded 3 μm tissue sections were deparaffinized with xylene and gradually dehydrated in ethanol. The sections were incubated with 4% hydrogen peroxidase (15 minutes at RT) to block endogenous peroxidase activity. Nonspecific binding of antibodies was blocked by incubating slides with 3% fetal bovine serum (FBS) + 0.25% Triton X-100 (1 hour at RT). The sections were stained using rabbit anti-AGE polyclonal antibody (dilution 1:1,000), rabbit anti-nephrin polyclonal antibody (dilution 1:250), and rabbit anti-caspase-3 polyclonal antibody (dilution 1:250) (90 minutes at RT), which were dissolved in PBS + 3% FBS + 0.25% Triton X-100 for expression of AGE, nephrin, and caspase-3, respectively. The slides then were incubated with biotinylated secondary antibody (1 hour at RT), followed by streptavidin horseradish peroxidase (40 minutes at RT). Slides were visualized using diaminobenzidine (DAB) and counterstained with hematoxylin. Slides were mounted and observed under light microscope (Olympus) with 400× magnification. Twenty glomeruli per sample section were photographed randomly. The AGE deposition, nephrin, and caspase-3 expressions were assessed by calculating the DAB-stained area proportion per GA using IHC Toolbox application of ImageJ 1.48 software. The DAB-stained area was calculated in percent by the formula = (DAB-stained area/GA) × 100%.
Statistical analysis
Statistics were conducted by SPSS 17.0 software. All data were expressed as mean ± standard deviation (SD). The significance of differences was determined by ANOVA analysis, followed by LSD test. A value of P<0.05 was considered statistically significant.
Results
Diabetes induction and immunization effect on serum anti-AGE level
Mice diabetic groups (group III as diabetic control and groups IV, V, and VI, which previously were immunized with AGE-BSA, KLH, and AGE-BSA-KLH, respectively) received STZ injection twice; the first injection was to induce moderate hyperglycemia and the second injection was to induce vascular complication, including nephropathy. Blood glucose level of diabetic groups (groups III, IV, V, VI) at 7 days after the first STZ injection revealed moderate hyperglycemia, yet did not reveal significant difference among these groups. Average values were 202 ± 31 mg/dL, 225 ± 21 mg/dL, 188 ± 3 mg/dL, and 198 ± 34 mg/dL, respectively, for groups III, IV, V, and VI compared with 147 ± 16 mg/dL and 127 ± 33 mg/dL for nondiabetic groups, I and II, respectively. Moreover, at 7 days after the last second STZ injection, blood glucose level of the diabetic groups increased, but did not reveal any significant differences among these groups. Average values were 385 ± 158 mg/dL, 361 ± 146 mg/dL, 272 ± 51 mg/dL, and 410 ± 136 mg/dL, respectively, for groups III, IV, V, and VI compared with 149 ± 9 mg/dL and 108 ± 44 mg/dL for nondiabetic groups, I and II, respectively. Compared with the nondiabetic groups (groups I and II), the diabetic control group (group III) showed significant difference at both measurements. Additionally, the nondiabetic AGE-BSA preimmunized group (group II) had lower glucose mean at both measurements compared with the normal control group (group I), yet did not reveal significant differences (P<0.05).
Serum anti-AGE levels in diabetic control mice (group III) were significantly higher compared with the normal control group (P<0.05) (Figure 1). This supported the evidence that hyperglycemia condition may stimulate the formation of autoantibody against AGE. Interestingly, compared with the nondiabetic AGE-BSA preimmunized group, the diabetic preimmunized groups for AGE-BSA and AGE-BSA-KLH (groups IV and VI) did not show any significant difference. This indicated that AGE immunization was successful in stimulating the anti-AGE antibody production in vivo.
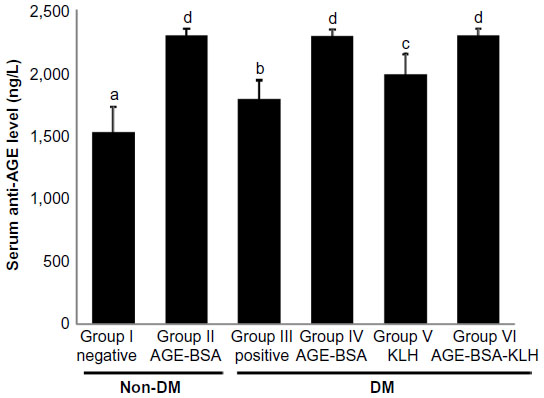 | Figure 1 Immunization effect on serum anti-AGE level. |
DN assessment
Proteinuria, glomerular hypertrophy, and mesangial expansion are included as major characteristics of DN. Significantly elevated proteinuria was shown in the diabetic control group compared with the normal group (P<0.05), whereas proteinuria levels were ameliorated in the diabetic groups preimmunized with AGE-BSA, KLH, or AGE-BSA-KLH (Figure 2A). There were no significant differences in the proteinuria level between the diabetic preimmunized groups compared with the normal control group.
 | Figure 2 Diabetic nephropathy assessments. |
The diabetic control group revealed significant increase of GV compared with the nondiabetic groups (Figure 2B). In contrast, GV in all diabetic preimmunized groups (groups IV, V, and VI) revealed no significant differences compared with the nondiabetic groups. Proportion of mesangial expansion area in the diabetic preimmunized groups also showed significant decrease compared with the diabetic control group (Figure 2C and D). For group II, there were no significant differences for proteinuria, GV, and mesangial expansion area compared with the normal control group (P<0.05).
This result indicated that immunizations of AGE-BSA, KLH, and AGE-BSA-KLH slowed down functional and pathological deterioration of DN in diabetic mice, and AGE immunization did not have any deterioration effect for functional renal normal mice.
Immunization of AGE-BSA, KLH, and AGE-BSA-KLH decreased serum AGE level and kidney AGE deposition
Chronic hyperglycemia induces AGE accumulation both in circulation and tissue. Trapping of circulating AGE can increase AGE deposition in the kidney that contributes to alter renal function and architecture. This study revealed that the diabetic control group showed significant increase both on the AGE level in serum (Figure 3A) and AGE deposition in kidney (Figure 3B and C) compared with the normal control group (P<0.05).
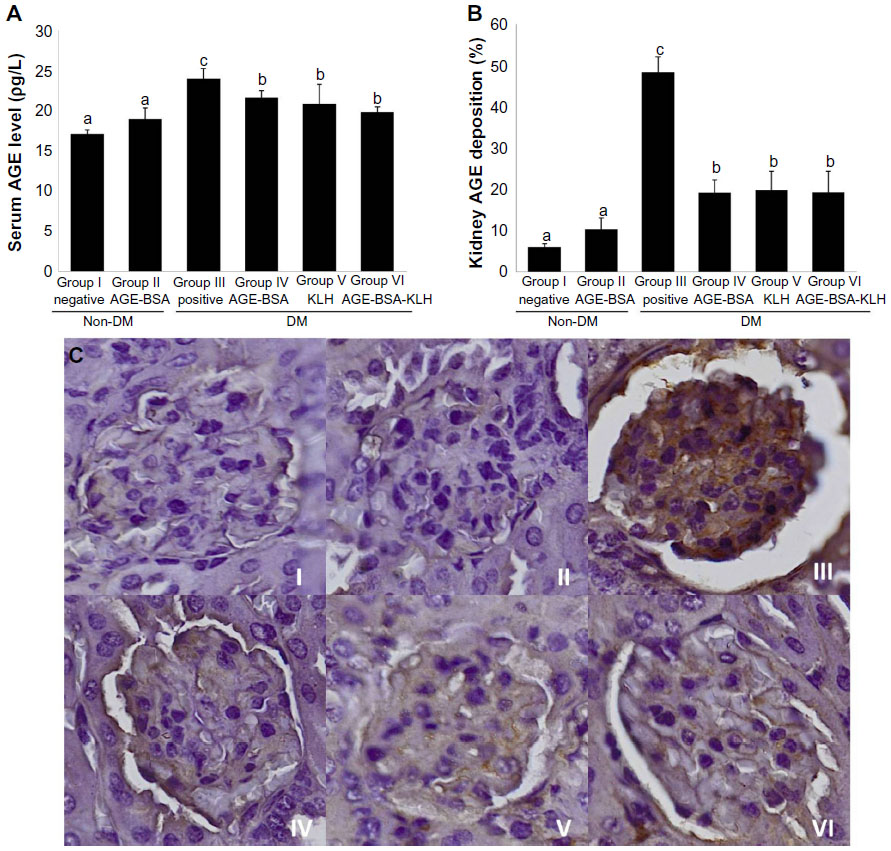 | Figure 3 Immunization of AGE-BSA, KLH, and AGE-BSA-KLH decreased serum AGE level and kidney AGE deposition. |
In contrast, the diabetic preimmunized groups (groups IV, V, and VI) revealed significant decrease in serum AGE level and kidney AGE deposition compared with the diabetic control group. Meanwhile, group II did not reveal any significant differences in the serum AGE level and kidney AGE deposition compared with the normal control group (P<0.05). This result indicated that immunization of AGE-BSA, KLH, and AGE-BSA-KLH decreased the serum AGE level and AGE kidney deposition in diabetic mice.
Immunization of AGE-BSA, KLH, and AGE-BSA-KLH maintained nephrin intensity
AGE accumulation can induce reduction in nephrin intensity as pivotal protein that composes slit diaphragm to prevent foot process effacement and further protein leakage. Diabetic control group revealed significant decrease in nephrin kidney intensity compared with normal group (P<0.05) (Figure 4). Interestingly, diabetic preimmunized groups (groups IV, V, and VI) revealed significant increase in nephrin intensity compared with both diabetic control and nondiabetic groups. Group II did not reveal any significant difference in nephrin intensity compared with normal control group (P<0.05). This result indicated that nephrin was upregulated in diabetic AGE-BSA, KLH, and AGE-BSA-KLH preimmunized mice.
 | Figure 4 Immunization of AGE-BSA, KLH, and AGE-BSA-KLH maintained nephrin intensity. |
Immunization of AGE-BSA, KLH, and AGE-BSA-KLH decreased kidney cells apoptosis
AGE accumulation induces apoptotic kidney cell death. Here, we examined caspase-3 as an early indicator for apoptosis. The diabetic control group revealed significant increase of kidney cell apoptosis compared with the normal control group (P<0.05) (Figure 5). In contrast, the diabetic preimmunized mice (groups IV, V, and VI) revealed significant decrease in kidney cell apoptosis compared with the diabetic control group. Meanwhile, group II did not reveal any significant differences in kidney cell apoptosis compared with the normal control group (P<0.05). This indicated that AGE-BSA, KLH, and AGE-BSA-KLH preimmunized in diabetic mice decreased the apoptosis of kidney cell death.
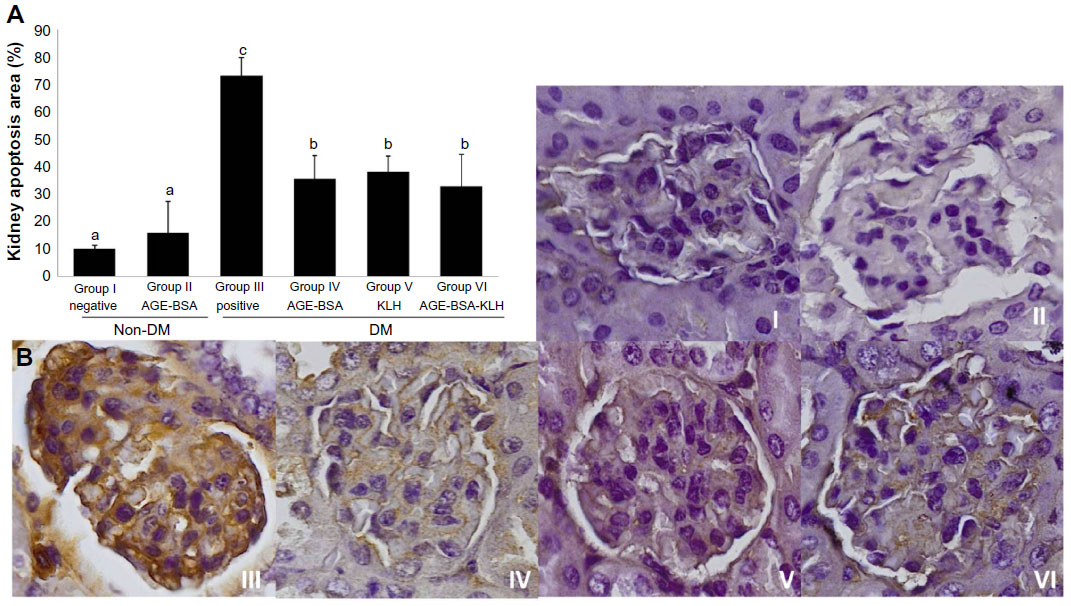 | Figure 5 Immunization of AGE-BSA, KLH, and AGE-BSA-KLH decreased kidney cells apoptosis. |
Discussion
Long-term hyperglycemia in diabetic condition triggers the protein glycation process, which results in increase of AGE. The previous studies have demonstrated that AGE formed from a wide variety of glycated proteins basically have antigenic properties.12 The existence of AGE in vivo is able to act as an immunogen that can trigger the formation of autoantibodies.18 It has been demonstrated that anti-AGE antibodies is found in the serum of patients with diabetes.12,19 Our study demonstrated that diabetic control group showed significant increase in anti-AGE levels compared with normal group (P<0.05). This supports the evidence that hyperglycemia in diabetic conditions triggers the formation of autoantibodies against AGE. Interestingly, diabetic AGE-BSA and diabetic AGE-BSA-KLH preimmunized groups did not show any significant differences compared with the nondiabetic AGE-BSA preimmunized group for anti-AGE antibody level (Figure 1). This finding is related to our hypothesis that immunization may be successful in inducing the production of protective anti-AGE antibody and may form immune complexes with antigen of AGE, thus inducing AGE clearance. We suggested that the initially diabetic preimmunized group produced higher anti-AGE antibody level compared with nondiabetic AGE-BSA preimmunized mice due to the antigen exposure yet would decrease due to immune complexes formation. This finding is supported by the previous research by Turk et al17 that there was inverse correlation between serum AGE level and AGE-IC), indicating that the serum level of AGE declines with the increasing presence of AGE-IC. The previous study also revealed that the specific antibody against AGE could be formed through the immunization of various types of glycated proteins by AGE.17 In line with this evidence, this study has demonstrated that both AGE-BSA and AGE-BSA-KLH immunization were able to increase the levels of serum anti-AGE. Our study showed significant increased levels of anti-AGE in all AGE-BSA and AGE-BSA-KLH preimmunized groups (groups II, IV, and VI) compared with the normal and diabetic control groups.
Furthermore, we examined the influence of AGE-BSA, KLH, and AGE-BSA-KLH immunization to DN pathogenesis. Assessed from the three hallmark characteristics of DN (proteinuria, glomerular hypertrophy represented by GV, and mesangial expansion), our results demonstrated that AGE-BSA, KLH, and AGE-BSA-KLH preimmunization were likely to succeed in ameliorating renal function and histopathology in diabetic mice. These also decreased the serum AGE level, kidney AGE deposition, and renal cells apoptosis. Interestingly, the diabetic immunized groups showed more upregulated nephrin expression than the normal group, and the diabetic control groups showed the lowest nephrin intensity compared with the other groups. This result study supports previous research, which stated that nephrin expression was transiently increased in the first 8 weeks of diabetes in the animal experimental model, followed by reduction in long-term studies in line with increase in proteinuria.20 Thus, AGE-BSA, KLH, or AGE-BSA-KLH immunization may delay the nephrin reduction in DN mice.
Kidney becomes the target and responsible for the increase of AGE. AGE deposition in the kidneys can be generated from the trapping of circulating AGE, local generation of new AGE on preexisting proteins, or decrease in AGE degradation and clearance. With increasing levels of anti-AGE induced by AGE-BSA and AGE-BSA-KLH immunization, it is expected to reduce AGE levels in circulation, thus further reducing the AGE deposition in kidney. In line with the previous study by Al-Farabi et al,13 this study also proved that immunization of AGE on prediabetic condition was able to reduce AGE level in the serum. Previous study by Turk et al17 and Al-Farabi et al13 demonstrated that anti-AGE antibody was able to bind to AGE to form immune complexes (AGE-IC). There was a correlation of decreased levels of serum AGE while serum AGE-IC increased.
AGE elimination pathway is through endocytosis by macrophages involves a wide range of receptors. Two-thirds are mediated by scavenging receptor and the third is mediated by FcγRII-B2, CD36, SR-BI, RAGE, and MARCO receptor,21–23 whereas the antibody–antigen complex between IgG and antigen will be recognized by Fcγ receptors on macrophages that will improve their phagocytosis ability.24 Mera et al25 proved that anti-Nε-[carboxyethyl]-lysine (anti-CEL) autoantibodies played role in CEL-modified protein uptake as one of lysine AGE derivatives by Kupffer cell for elimination of soluble AGE-IC in liver that mediated by scavenger receptor family (SR-A, SR-BI, LOX-1). Therefore, there is a possibility that the role of anti-AGE antibodies in lowering circulating AGE is through the formation of immune complex thus reducing the AGE deposition in kidney, and subsequently inhibits AGE related to pathogenesis in DN.
KLH is a glycoprotein conjugate that is known to have structural similarities with reactive carbonyl conjugate.26 KLH has proven to be immunogenic and is able to lead the production of reactive carbonyl anti-KLH antibodies in mice.27 Suspected anti-KLH will react with reactive carbonyl and lower AGE production, particularly from the glycated IgG that expresses L-chain.28 It has been demonstrated previously by Shcheglova et al14 that reactive immunization with KLH suppressed circulating AGE and reduced the progression of DN in diabetic rat. In vitro study proved that glycated protein from rat serum reacted faster with anti-KLH antibodies than normal IgG.14
Reactivity of Ig L-chains is suggested to have natural mechanism for the elimination of cytotoxic glycation products and that reactivity can be improved by specific reactive immunization against glycoprotein like previously demonstrated by KLH immunization.14 From this fact, it can be hypothesized that there is possibility of existence of anti-AGE antibodies in normal IgG and AGE-BSA or AGE-BSA-KLH immunization will increase the reactivity of the anti-AGE antibodies to bind and eliminate AGE in circulation. But this requires further research in vitro to test the difference of reactivity in glycated protein binding ability between anti-AGE antibodies induced by immunization compared with anti-AGE antibodies formed normally in the body.
The initial purpose of AGE-BSA conjugation with KLH was to increase the immunogenicity of AGE-BSA, thus further increasing the production of anti-AGE antibodies from immunization. AGE-BSA-KLH immunization will also induce anti-KLH antibodies production and anti-AGE antibodies production. Therefore, it can be hypothesized that the inhibitory effect against AGE related to pathogenesis was more potent in diabetic AGE-BSA-KLH preimmunization group than AGE-BSA or KLH preimmunization group alone. However, the result of this study showed similar inhibitory effect in DN progression among diabetic AGE-BSA, KLH, and AGE-BSA-KLH preimmunized groups. It can be due to this that immunization of AGE-BSA, KLH, or AGE-BSA-KLH are likely to increase IgG reactivity at a level similar to their natural ability to eliminate AGE in circulation, yet further study is needed.
In this study, as our focus is on AGE preimmunized effect, we also made a normal group with AGE-BSA preimmunization (group II). The purpose of this group is to examine the effect of immunization under normal conditions, whether they have any deteriorating effect or not. Interestingly, this group did not show any significant difference among all the indicators measured compared with the control normal group. This suggests that AGE-BSA immunization does not have any negative effect on renal function in normal conditions. Yet, it is possible that anti-AGE antibodies can form insoluble immune complexes when bound to AGE in tissues, and it is suspected that it could further damage the integrity of GBM and contribute to the progression of microvascular abnormalities if it accumulates in the glomeruli.17
Indeed, this study did not directly assess the presence of insoluble immune complex depositions in the kidney, and yet this study may weaken these allegations because immunization showed renoprotective effect. That effect of immunization was on decreasing of AGE deposition in kidney and on increasing circulating AGE elimination, which may further decrease the opportunity of anti-AGE to form an insoluble immune complex.
In conclusion, our results strengthen the evidence that the anti-AGE antibodies have a protective role against diabetic vascular complications, especially DN. AGE-BSA, KLH, and AGE-BSA-KLH immunization inhibit the progression of DN. This study provides a basis for the development of DN-based immunotherapy with AGE immunization as a potential candidate. Further in vitro studies are needed to prove inhibition mechanism of AGE signaling by anti-AGE and different reactivity between anti-AGE antibody, which is normally formed in the body compared with anti-AGE, which is induced by the immunization of various AGE antigen-modified proteins.
Acknowledgments
The authors thank Health Professional Education Quality (HPEQ) as a project from Directorate General of Higher Education, Ministry of Education and Culture of Indonesia, Republic of Indonesia for their financial support and also Brawijaya Biomedicine Laboratory, especially Kurnia and Heni Triwahyuni, for their excellent laboratory skill guide and advice as well.
Disclosure
The authors report no conflicts of interest in this work.
References
Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. J Intern Med. 2010;268:456–467. | |
World Health Organization (WHO). Diabetes Fact Sheet. Geneva: WHO; 2011. | |
Prodjosudjadi W. Incidence, prevalence, treatment and cost of end-stage renal disease in Indonesia. Ethn Dis. 2006;1:S2–S14. | |
Soegondo S, Prodjosudjadi W, Setiawati A. Prevalence and risk factors for microalbuminuria in a cross-sectional study of type-2 diabetic patients in Indonesia: a subset of DEMAND study. Med J Indones. 2009;18:124–130. | |
Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Natur Clin Prac Endocrinol Metab. 2008;4(8):444–452. | |
Ohshiro Y, Lee Y, King GL. Mechanism of diabetic nephropathy: role of protein kinase-C activation. ADS Stud Med. 2005;5(1A):S10–S19. | |
Goh S, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–1152. | |
Forbes JM, Cooper ME, Oldfield MD, Thomas MC. Role of advanced glycation end products in diabetic nephropathy. J Am Soc Nephrol. 2003;1:S254–S258. | |
Bohlender JM, Franke S, Stein G, Wolf G. Invited review advanced glycation end products and the kidney. Am J Physiol Renal Physiol. 2005;289:F645–F659. | |
Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3(2):101–108. | |
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. | |
Baydanoff S, Konova E, Ivanova N. Determination of anti-age antibodies in human serum. Glycoconj J. 1996;13:335–339. | |
Al-Farabi MJ, Ni’amita LF, Soeatmadji DW, Indra MR. Immunization with AGE-KLH prevent diabetic complication in mice. Indones Scholars J. 2013;1:28–32. | |
Shcheglova T, Makker S, Tramontano A. Reactive immunization suppresses advanced glycation and mitigates diabetic nephropathy. J Am Soc Nephrol. 2009;20:1012–1019. | |
Wu KK, Huan Y. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol. 2008;S40:5.47.1–5.47.14. | |
Rangan GK, Tesch GH. Methods in renal research quantification of renal pathology by image analysis. Nephrology. 2007;12:553–558. | |
Turk Z, Ljubic S, Turk N, Benko B. Detection of autoantibodies against advanced glycation end products and age- immune complexes in serum of patients with diabetes mellitus. Clin Chim Acta. 2001;303:105–115. | |
Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW. Nµ-(Carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry. 1995;34:10872–10878. | |
Shibayama R, Araki N, Ejima Y, et al. Study of autoantibodies against advanced glycation endproducts of the Maillard reaction. Diabetes. 1999;48:1842–1849. | |
Cooper ME, Mundel P, Boner G. Role of nephrin in renal disease including diabetic nephropathy. Semin Nephrol. 2002;22(5):393–398. | |
Stanton LW, White RT, Bryant CM, Protter AA, Endemann GA. Macrophage Fc receptor for IgG is also a receptor for oxidized low density lipoprotein. J Biol Chem. 1992;267(31):22446–22451. | |
Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem. 1993;268(16):11811–11816. | |
Smedsrod B, Melkko J, Araki N, Sano H, Horiuchi S. Advanced glycation end products are eliminated by scavenger-receptor-mediated endocytosis in hepatic sinusoidal Kupffer and endothelial cells. Biochem J. 1997;322(2):567–573. | |
Skogh T, Blomhoff R, Eskild W, Berg T. Hepatic uptake of circulating IgG immune complexes. Immunology. 1985;55:585–594. | |
Mera K, Nagai R, Takeo K, Izumi M, Maruyama T, Otagiri M. Anautoantibody against N(e)-(carboxyethyl)lysine (CEL): possible involvement in the removal of CEL-modified proteins by macrophages. Biochem Biophys Res Commun. 2011;407(2):420–425. | |
Wagner J, Lerner RA, Barbas CF. Efficient aldolase catalytic anti-bodies that use the enamine mechanism of natural enzymes. Science. 1995;270:1797–1800. | |
Armentano F, Knight T, Makker S, Tramontano A. Induction of covalent binding antibodies. Immunol Lett. 2006;103:51–57. | |
Gugliucci A, Bendayan M. Reaction of advanced glycation end products with renal tissue from normal and streptozotocin-induced diabetic rats. An ultrastructural study using colloidal gold cytochemistry. J Histochem Cytochem. 1995;43:591–600. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.