Back to Archived Journals » Oncolytic Virotherapy » Volume 3
Immune cells: more than simple carriers for systemic delivery of oncolytic viruses
Authors Eisenstein S, Chen S, Pan P
Received 15 May 2014
Accepted for publication 8 August 2014
Published 6 November 2014 Volume 2014:3 Pages 83—91
DOI https://doi.org/10.2147/OV.S47143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Samuel Eisenstein,1 Shu-Hsia Chen,2 Ping-Ying Pan2
1Department of Surgery, 2Department of Oncological Sciences and Tisch Cancer Institute, The Icahn School of Medicine at Mount Sinai, New York, NY, USA
Abstract: Oncolytic virotherapy on its own has numerous drawbacks, including an inability of the virus to actively target tumor cells and systemic toxicities at the high doses necessary to effectively treat tumors. Addition of immune cell-based carriers of oncolytic viruses holds promise as a technique in which oncolytic virus can be delivered directly to tumors in smaller and less toxic doses. Interestingly, the cell carriers themselves have also demonstrated antitumor effects, which can be augmented further by tailoring the appropriate oncolytic virus to the appropriate cell type. This review discusses the multiple factors that go into devising an effective, cell-based delivery system for oncolytic viruses.
Keywords: oncolytic virus, cell carrier, immune cells, cancer therapy, myeloid-derived suppressor cells
Introduction
The promise of oncolytic virotherapy has far outpaced its practical efficacy. Since the first clinical trials of oncolytic viruses (OVs) and their modest results 15 years ago,1 efforts have been made to improve viral efficacy in the treatment of malignant tumors. While progress has been made in altering the virus particle itself, the very nature of OV, ie, being able to attack tumor cells but helpless against host immunity, has limited their use as conventional tumor therapies.
As has now been demonstrated, host immunity against an OV is rapid and strong. When exposed to an OV, there is a robust and rapid immune response mediated by complement, neutralizing antibodies, and cellular immunity. Viruses are rapidly cleared from the circulation within 30 minutes, taken up into cells, or complexed and cleared by the reticuloendothelial system.2 Very little of the virus itself ever reaches the tumor for which it was intended.
To improve delivery of OV, it is necessary to use a “smarter” carrier than the virus itself. A useful carrier is able to respond to and migrate toward tumor-secreted factors and protect and shed the virus appropriately. Cell-based carriers are the obvious choice. There are many cell types that, after viral infection, can migrate specifically toward tumors, and little engineering is required to achieve these goals. The earliest studies of carrier cell-mediated delivery of an OV employed tumor homing teratoma cell lines targeting the oncolytic herpes simplex virus to intraperitoneal ovarian cancer cells in a severe combined immunodeficiency (SCID) mouse model.3 However, the idea of injecting cancer cells into patients to cure their cancer is somewhat problematic, and it is unclear if these cells form new tumors after administration. The general applicability of this concept is also unclear, and ultimately the use of tumor cells as carriers for OVs, while still employed in some settings, is not the most promising frontier in the field of cell-based carriers for OVs.
In attempting to identify a vector that would effectively take up the virus, specifically traffic the virus to the tumor, protect the virus from host immunity, and unload the virus unaltered within the tumor, immune cells have emerged as the most promising option. It has long been demonstrated that a variety of host immune cells migrate to and accumulate within tumors.4–7 Initially it was felt that genetically modified immune cells could express oncolytic viral vectors which could then be activated when cells were activated at tumor sites. While this may still be a possibility, it requires a very complex cascade of events, which makes this process impractical. Instead, it was felt that it would be simpler to infect immune cells with OVs and adoptively transfer them into tumor-bearing hosts. Early studies were carried out with virally infected, antigen-specific T-cells and demonstrated that a virus could be passively coupled to the surface of an immune cell and brought to a tumor site, prolonging survival in tumor-bearing mice.8 Since these first experiments, simple T-cells are no longer often used for these purposes as the acquisition of tumor-specific T-cells is much more complex in humans than in mice and it has been demonstrated that virus bound on the surface of T-cells is readily neutralized in vivo.5
Over the last decade, new cell types have emerged as vehicles for the delivery of OVs. Dendritic cells,5,6 macrophages,7 peripheral blood mononuclear cells,9 lymphokine-activated killer cells,6 cytokine-induced killer (CIK) cells,2,10–12 and myeloid-derived suppressor cells (MDSCs)4,13 have all been shown to be effective in protecting OVs and delivering them directly to tumor sites. By employing these cells as OV carriers, there have been incremental improvements in tumor treatment in murine models. No work to date has been carried out in vivo in humans.
As we continue to study the synergistic effects used by immune cells to aid in OV therapy it has become clear that immune cells are more than just simple carriers that deliver their payload and have no further effect on tumor killing. In fact, there is a complex interplay between the virus, cell, and tumor, which is still being studied. This review discusses the many variables that go into selecting or developing a cell-based carrier for the delivery of an OV directly to tumors.
Viral selection
There are many OVs currently under study and in clinical trials in the USA and internationally (Table 1). The interplay between virus and cell can be critical in enhancing viral loading and migration, which is discussed later in this review. Ultimately, the synergy between the two agents is what will determine successful cell-based oncolytic virotherapy.
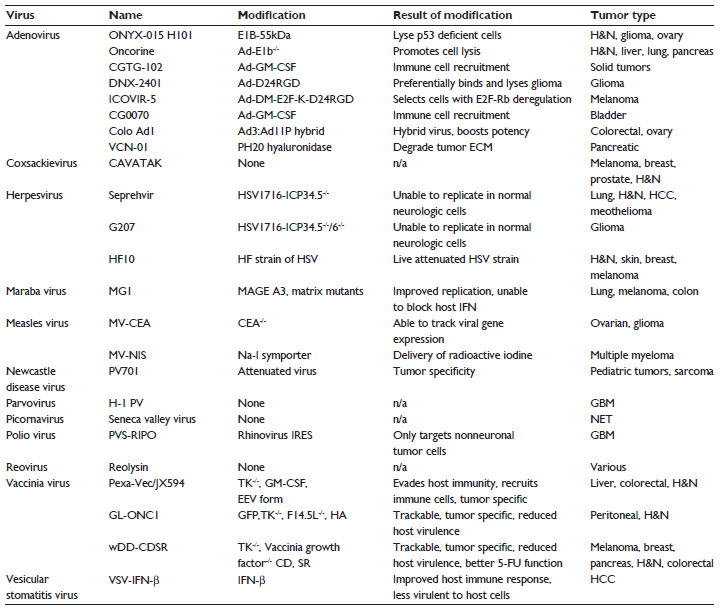 | Table 1 Oncolytic viruses that have undergone clinical trials, including any genetic modifications, the phenotypic results of those modifications, and the tumors which have been treated with the virus |
Numerous viruses have been engineered to improve their own homing to tumor cells. One of the original oncolytic adenoviruses, ONYX-015, was designed to replicate specifically in p53-deficient cells.14 Similarly, two separate oncolytic adenoviruses took advantage of the Δ24 mutation.15 DNX-2401 and ICOVIR-5 preferentially lyse tumor cells that have defective Rb gene function, such as in glioma cells. While the mutation does improve the therapeutic index of the OV by decreasing its ability to lyse normal cells, there is very little homing directly to tumor cells, thus limiting the administration route of this OV to intratumoral injections only. In fact, when examining the list of current and former OVs used in clinical trials (Table 1), it becomes apparent that not a single virus has the ability to directly home to and migrate toward a tumor-secreted factor. Because of this, many OVs had to be delivered intratumorally, which can prove difficult and often may require invasive surgery. Unfortunately, in delivering the virus directly to the tumor, the virus does not travel to sites of occult metastases unless it is administered in high doses systemically. This is likely the reason why the majority of the focus on OV modification has been on improving the safety profile of their effects on normal cells, because when they are delivered systemically, OVs enter tissues according to a concentration gradient and passive interactions.
Adding cell-based carriers to the system can ameliorate the need for built-in mechanisms of tumor targeting by the virus. The virus, in turn, can improve the ability of immune cells to target tumors. OVs, when infecting tumor cells, cause an inflammatory response. This inflammatory response promotes the migration of immune cells to the tumor microenvironment. The host immune response has been proposed by some to be the primary cause of tumor cell death in patients receiving OV therapy.16 This inflammatory response also promotes migration of exogenously delivered immune cells, and can improve the ability of immune cell-based carriers to migrate directly to tumor sites. This was demonstrated when oncolytic vaccinia virus was delivered by CIK cells. CIK cells target the NKG2D ligand, which is upregulated in virally infected tumor cells.2
Viruses are also being engineered to promote increased tumor-specific migration by cell carriers. Several OVs that secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) have been developed, from the adenovirus and vaccinia virus.16 When tumor cells are infected and GM-CSF is released, migration of monocytes and granulocytes to the tumor site is promoted, as well as increased production of these immune cells in the host. If GM-CSF-expressing viruses can be delivered by monocytic or granulocytic cells that respond to release of GM-CSF, viral infection of the tumor can promote tumor tropism of virally loaded immune cells. Similarly, survival in tumor-bearing mice was improved by loading CIK cells with adenovirus carrying the CCL5 gene, which has previously been shown to promote CIK migration to tumor sites17 and in our own experience has been shown to promote migration of monocytes and MDSCs to tumors (data not shown). CCL5 was secreted at the tumor site and improved tumor-specific targeting of CIK cells.10 Another interesting strategy involves use of OVs that express costimulatory molecules. Oncolytic adenovirus expressing T-cell costimulatory 4-1BB ligand has been shown to be effective in melanoma-bearing mice. 4-1BB ligand is a costimulatory molecule that, when released from OV-infected cells, primes the immune response by binding antigen-presenting cells. These antigen-presenting cells in turn stimulate a Th1 response, which promotes T-cell-based tumor killing.18,19
Another viral modification that may improve cell-based delivery is the addition of genes that are able to break down the peritumoral and intratumoral extracellular matrix. When a relaxin-expressing adenovirus was injected directly into melanomas, viral penetration was improved, tumor growth was inhibited, and overall survival was improved in an in vivo murine model.20 While this technique has yet to be employed in cell-based delivery methods, it is easy to see how it may aid in improving cell penetration and inhibiting tumor growth. Improvement in the virus to influence carrier cell efficacy is still in its infancy and there is much work that can still be done to promote this synergy.
Viral loading and protection
The mechanism used to load the virus not only affects the concentration of virus that will be delivered to the tumor, but also impacts the mechanisms by which the virus could potentially be neutralized prior to arriving within the tumor microenvironment. Virus can be either bound to the surface of its cell carrier or sequestered within cellular compartments. Surface-bound virus can be shed readily into the local environment and avoids the intracellular viral processing; however, the virus is exposed to neutralizing antibodies and complement. Conversely, intracellular virus can avoid the humoral immune system, but runs the risk of sequestration and neutralization or premature cell lysis, both of which may limit the amount of virus able to be released at the tumor site.
The first cells employed in the systemic delivery of OV were tumor-specific T-cells.8 It was observed that these cells, despite a lack of receptors on their surface for the retroviral envelope with which they were cultured, passively bound virus to their cell surface which they subsequently shed over the next 72–96 hours. These effects were further demonstrated using reovirus21 and vesicular stomatitis virus (VSV).4,22 Interestingly, while passive binding of virus to the cell surface appeared effective in naïve murine models, when preimmunized against viral antigens, this therapy lost its efficacy.22 Similarly, when cultured with human serum, reovirus bound to T-cells was neutralized and unable to lyse cells in an ex vivo murine model, likely due to pre-existing neutralizing antibodies and complement in the serum.5 In our experience, it has also proven difficult to obtain a high multiplicity of infection of virus to bind to the surface of certain cell types. MDSCs passively infected with VSV demonstrated less than one viral particle bound per cell.4 Thus, passive binding of the virus to the cell surface, although the method that has been used the longest, is likely to be the least effective way to join the virus to immune cells.
Internal packaging of the virus has also been demonstrated. Interestingly, a variety of cell types, including CIKs,10 dendritic cells,5 and likely even T-cells,23 have been shown to take up OV in this manner. While empirically it may seem that there is less likelihood of viral transfer once a cell has internalized virus, given that it is expected that normal cell-mediated immunity will make every attempt to clear the virus, this turns out to not always be true. Jennings et al demonstrated in vitro that murine dendritic cells could effectively take up oncolytic reovirus and release it to tumor cells.6 While no mechanism for this has been elucidated, dendritic cells similarly take up human immunodeficiency virus and transmit it to T-cells via exosomes.24
Viral internalization is an effective method for viral uptake in human dendritic cells, but it is difficult to extrapolate this to murine models because murine dendritic cells infected with viruses such as reovirus tend to allow the virus to replicate, rapidly leading to cell clearance by the immune system prior to the dendritic cell arriving at the tumor site. When selecting an immune cell as an OV carrier it is important to take this into consideration. The time taken for the cell to arrive at the tumor must be shorter than the time taken for the virus to replicate in and lyse the cell. For this reason, it is important to understand the kinetics of both cellular migration and viral replication. If the process can be timed correctly, the delivery cell can not only be used to bring the virus to the tumor, but it can also be used as a viral factory, increasing the local concentration of the virus within the tumor after the virus replicates and lyses the cell.
This phenomenon has been demonstrated in CIKs infected with oncolytic vaccinia virus. CIKs are a heterogeneous population of cells that demonstrate characteristics of both T-cells and natural killer cells. They have been shown to migrate to tumor sites within 72 hours after adoptive transfer in murine models.2 When infected with vaccinia virus, the virus undergoes an “eclipse period”. During this time the virus lays dormant and does not affect the CIK cell. Cell surface antigen expression, cell migration, and cell function remain unaffected during this time. Once the eclipse period ends after 48–72 hours, the virus replicates rapidly and lyses the CIK cell, leading to amplified viral release into the tumor microenvironment.2
One potential drawback of using CIKs is that only about 20% of cells actually take up virus under standard protocols,2 and while technically less demanding, passive culture of virus with immune cells may not be the optimal technique for binding the virus to its cell carrier for just this reason. When attempting to bind VSV to MDSCs, a population of heterogeneous immature immune cells that have been demonstrated to preferentially migrate to tumors and promote an immunosuppressive environment, we were not satisfied with the results obtained simply by culturing the virus with MDSCs. We found that less than one viral particle was being transferred by each MDSC. In an attempt to improve viral transfer by MDSCs, we explored several options: VSV was bound to a non-neutralizing antibody, which passively bound the MDSC Fc receptor (FcR); VSV was bound to an aluminum oxide nanoparticle, which we had previously demonstrated could be phagocytosed by MDSCs; and VSV was bound to a biotinylated non-neutralizing antibody which was in turn bound to an anti-Ly6C antibody that had streptavidin bound to its Fc segment. Interestingly, we demonstrated an 80-fold increase in viral binding for the VSV that was bound to the non-neutralizing antibody and in turn bound to the MDSC FcR. When we used this technique in survival studies, we found a significant increase in survival in mice with intrahepatic colon cancers.4
Tumor targeting
Once the virus has been loaded onto or into its cell carriers, the cells are adoptively transferred to their hosts. This is a time of great peril for both the virus and its cell carrier. The longer the migration, the greater the chance that either surface-bound virus will be shed or neutralized or the internalized virus will cause cell lysis before the cell reaches its target. Most murine studies demonstrate a peak concentration of immune cells within 48–72 hours of adoptive transfer, regardless of the type of cell used.2,4,8
Even if the virus is well protected and the cell is not lysed, it is important that the virally armed cell be able to home to tumor sites. Initially, this was ensured by selecting cells known to specifically migrate to tumor sites. T-cells, in and of themselves, do not specifically migrate to tumor sites. However, when T-cells are tumor-specific, as in the case of OT-1 T-cells that home to ovalbumin peptide (OVA)-expressing tumors, they will migrate specifically to tumor sites. While this has been demonstrated to be effective in murine models,8 it is not practical in humans because tumor-specific T-cells are much harder to isolate from cancer patients. However, T-cells do tend to migrate to lymphatic organs, so they could be useful in the treatment of occult lesions in patients with cancers that spread preferentially via lymphatics.
Numerous cell types have been employed to this end, including peripheral blood mononuclear cells,9 macrophages,7 dendritic cells,5 CIKs,2,10,12 lymphokine-activated killer cells,6 and MDSCs.4,25 All these cells demonstrate an ability to migrate to sites of inflammation due to a variety of chemokines that are released by either the tumor to attract immunosuppressant immune cells, such as T-regulatory cells and MDSCs, or by the host immune system to attract antitumor host immunity. Targeting inflammation is an attractive concept because, while various tumor epitopes may vary, inflammation is a hallmark of all tumors. This ensures that cells carrying OV can also target occult metastases, which are often more difficult to treat than the primary tumor. This inflammatory response can be further boosted using chemotherapy or radiation, and it has been demonstrated that macrophages bearing OV have a more robust antitumor response after these treatments.7
While many cell types do demonstrate tumor tropism, MDSCs, when compared with other cell types, demonstrated much more robust tumor tropism in a murine model of hepatic colon cancer metastases.4 Not only were 12 times as many MDSCs isolated from tumors when compared with other cell types, but far fewer MDSCs were isolated from other organs, particularly the spleen and liver, where CIKs were identified in large numbers. Interestingly, in the same study, MDSCs were also demonstrated to migrate specifically to the periphery of the tumor as well as perivascularly within the tumor on magnetic resonance imaging.
Another strategy for delivery of OVs, which has been used experimentally with moderate success, is to target the scaffolding in which the tumor grows. To this end, mesenchymal stem cells have been used to target the peritumoral stroma,26,27 endothelial progenitor cells have been used to target angiogenesis,28 and mesothelial cells have been used to target peritoneal cancers.29 These types of cells may prove very useful in not only attacking the tumor, but also in undermining the mechanisms via which tumors are established. They may also increase peritumoral inflammation, which can help trigger a host immune response and attract immune-based cell carriers to deliver virus as well. The downside of these cell types is that they may not penetrate the tumor as well as the immune cell carriers, as their actual target is the tissue surrounding the tumor, and they may not have as much specificity for occult metastases which have yet to completely establish their metastatic niche.
Work is just starting on altering both the cell and OV to promote tumor-specific homing. One novel approach was demonstrated with the extracellular enveloped viral form of the vaccinia virus. This form creates a membrane around itself with incorporated host proteins, protecting it from host neutralization. When murine CIKs were infected with this extracellular enveloped viral vaccinia virus, it was observed that some of the virus was able to leave the CIK, avoid host immune surveillance, and infect the tumor. Once the tumor was infected, the tumor microenvironment changed, becoming more proinflammatory, which ultimately led to more aggressive CIK migration to the tumor site. Once within the tumor microenvironment, the CIKs then lysed and released their viral payload, leading to a more vigorous antitumor response.10
Genetic modifications of both the virus and the carrier cell may enable improved tumor tropism. By engineering an OV to express cells attracting chemokines, repeated dosing of cell-based OV therapy could boost the effect of each sequential dose of OV that is injected subsequently.
Direct antitumor effect of immune cells
While immune cells were initially seen as simple carriers of virus to tumor sites, more recent efforts have focused on creating a synergy between the virus and the immune cell to enhance the antitumor effects of both. It has long been known that the host immune system performs tumor surveillance and eradication. By exploiting these innate mechanisms, it is possible to use cell carriers as an adjunct to the OV and promote a more vigorous antitumor response.
Much of the work in exploring this synergy has been done with CIK cells. It has been demonstrated that CIK cells identify their targets via the NKG2D receptor and its ligands, including the stress response ligands, MICA and MICB. These ligands have been shown to be upregulated in human tumors as a result of the various stresses imposed within the environment opposing tumor growth.30 Interestingly, viral infection is also a stressor, and can increase NKG2D ligand expression. When they encounter tumor cells, CIK cells act like natural killer cells and attempt to destroy the tumor. After infection with an OV and adoptive transfer into a tumor-bearing host, approximately 20% of CIK cells will lyse at the tumor site as a result of being infected, and the remaining 80% will attack the tumor.2 Activated CIK cells also release immunostimulatory mediators that prime the host immune response and help to create a memory response.2
CIK cells have also been shown to have increased antitumor efficacy when combined with interleukin (IL)-12 therapy in mouse models.31 IL-12 has been shown to have lethal toxicity after intravenous injection in Phase II trials.32 One recent study demonstrated that, by engineering an oncolytic adenovirus expressing human IL-12, it is possible to increase the therapeutic efficacy of OV-loaded CIK cells without the systemic toxicity of intravenous IL-12 injections. This ability to deliver toxic substances into the tumor microenvironment without disseminating them systemically is the hallmark of cell-based delivery of antitumor therapeutics.
Other recent work has investigated the use of dendritic cells as delivery vectors for OV. Dendritic cells have very little innate ability to kill tumor cells; however, they are very capable of migrating specifically to tumor sites. Lymphokine-activated killer cells are a heterogeneous group of natural killer cells and natural killer T-cells that have demonstrated an ability to target and kill tumor cells.33,34 Their efficacy is somewhat limited without a readily available source of IL-2; however, intravenous injection of IL-2 has significant toxicity, including increased vascular permeability and hypotension. Interestingly, lymphokine-activated killer cells can be combined with dendritic cells to deliver OV and synergistically promote tumor killing. Dendritic cells, when infected with an oncolytic reovirus, produce IL-2, which primes lymphokine-activated killers for killing. Dendritic cells also migrate to the tumor site and promote production of immunostimulatory cytokines such as interferon (IFN)-γ, tumor necrosis factor-α, IFN-α, and IL-12.6 By combining these two cell types, it becomes possible to overcome individual weaknesses and create more robust cancer therapy.
Similarly, a multifactorial antitumor effect was demonstrated when human peripheral blood mononuclear cells were infected with an oncolytic reovirus. The peripheral blood mononuclear cells delivered their OV to the tumor site effectively, and after infection with the virus, the natural killer cell fraction was activated preferentially and degranulated at tumor sites in an IFN-dependent manner. Inflammatory cytokines were also secreted after infection, further priming the immune response.1
Our own work with MDSCs has demonstrated an interesting synergy between cell carriers and OV. We have found that MDSCs demonstrate plasticity within their phenotype. While tumor-associated MDSCs typically demonstrate an immune suppressive, M2-like phenotype when the inhibitory paired immunoglobulin-like receptor B is blocked, murine MDSCs demonstrate an antitumor M1-like phenotype, secrete more inducible nitric oxide synthase and less arginase, and actively lyse tumor cells ex vivo.35 Another study demonstrated a similar effect by stimulating Toll-like receptor 3 with its agonist poly I:C.36 Evidence has emerged that a variety of pathogen-associated molecular patterns known to stimulate Toll-like receptors can stimulate M1-like differentiation of MDSCs. Subsequently, we have demonstrated that MDSCs develop this phenotype after infection with VSV, which synergistically aids in their targeted killing of tumor cells, prolonging survival in mice bearing intrahepatic colorectal cancer metastases.4
Delivery strategies
Peripheral administration of naked virus alone leads to widespread dissemination of the virus, both diminishing the dose that can be effectively administered directly to the tumor and increasing the amount of healthy tissue that can be exposed to the potentially harmful effects of the virus. Because of this, the cell-based delivery method has arisen; however, even by using the optimal virus and cell combination, there are ways to improve efficacy via delivery.
Naked OV, when delivered peripherally, promotes a robust antibody-mediated antiviral host memory response, limiting the ability to administer the virus multiple times. We have demonstrated that multiple administrations of OV loaded into MDSCs can improve survival.4 A single administration of VSV-loaded MDSCs improved survival in tumor-bearing mice; however, readministering VSV-loaded MDSCs every 3 days further improved survival for up to four doses. By loading OV onto MDSCs, the antiviral effect is attenuated. We are currently investigating this effect, and it is likely due to one of two phenomena. It is possible that the viral antigens are protected from either the MDSC loading itself or by the non-neutralizing antibody that we use to load the virus onto the cell. It may also be that the MDSC promotes a locally immune-tolerant environment, preventing immune priming against the virus.
Another technique that had been used to improve the efficacy of naked virus is to inject the virus either directly into the tumor or into the vascular supply feeding the tumor (ie, injection into the hepatic artery for liver tumors).37 While this strategy may improve the efficacy of OV, tumors are often in locations that require invasive procedures to reach them. Also, when relying on direct administration of OV into the tumor, the virus may miss any occult spread that could be reached using a delivery system that specifically targets tumor cells. One of the goals of adding a cell carrier for the OV is to eliminate the need for these invasive procedures. While most trials of cell-based OV delivery solely involve intravenous administration, there can be some advantage to adding intratumoral administration of OV to peripheral injection of immune cells. The inflammation induced by viral infection of the tumor can further attract both adoptively transferred immune cells laden with OV as well as promoting host immunity against tumor cells. In one series, adenovirus-expressing IL-12 was loaded onto CIK cells and injected directly into tumor cells. The virus-infected tumor cells were lysed, resulting in the release of IL-12, which has been shown to improve the homing and cell killing of CIK cells, augmenting the efficacy of CIK-based OV delivery.12
Induction of peritumoral inflammation, as previously mentioned, improves tumor targeting in most immune cell-based therapies. There has been a study demonstrating that chemotherapy and radiation followed 48 hours later by peripheral administration of macrophages armed with oncolytic adenovirus improved survival in mice with prostate cancer when compared with chemotherapy and radiation alone.25
Future directions
As previously mentioned, use of cell-based carriers of OV has yet to make an impact clinically. A trial is ongoing in the USA where measles virus encoding a thyroidal sodium-iodide symporter is being infected into mesenchymal stem cells and used to treat recurrent ovarian cancer by intraperitoneal injection. This trial is currently in Phase I/II according to the National Cancer Institute. No such trials are currently underway in Europe according to the European Organisation for Research and Treatment of Cancer.
There is much to be done before cell-based OV delivery is a viable therapy in humans with cancer. While many of the previously mentioned cell types can be amplified and harvested in mouse models, acquiring a sufficient number of cells to deliver an effective viral dose has proven difficult. We have observed that, in patients administered GM-CSF, a growth factor that promotes the production of granulocytes and monocytes, a large number of suppressive cells consistent with human MDSCs are released into the circulation and can be easily harvested and separated in large numbers from a unit of peripheral blood. At this time, it appears technically simpler to harvest these cells than to amplify them; however, with time and development of new techniques, this may become feasible.
Viral loading onto immune cells can certainly be enhanced. We have improved viral loading onto MDSCs by using a non-neutralizing antiviral antibody. While this worked well preliminarily, we feel that instead of conjugating OV to the surface of MDSCs, we may be able to conjugate virus to nanoparticles via this same non-neutralizing antibody, which can be phagocytosed by MDSCs and ultimately delivered to tumor sites. This would enable a significantly greater viral dose that can be delivered to the tumor tissue.
As viruses continue to be designed to be more tumor-specific and less toxic to normal cells, they also need to be modified to further enhance the synergy between cell and virus. As we home in on the optimal cell types via which to deliver OV, we can tailor a virus to secrete factors that can promote cellular migration to tumor sites, protecting the virus from host immunity and enhancing the cellular effects on tumor killing.
Currently, the most work appears to involve the adenovirus, which has been around the longest and has undergone the most extensive genetic modification, as well as vaccinia virus, which has been shown to be well suited as a cargo for cell-based carriers due to its eclipse phase. The most promising immune cell types employed to date include natural killer cell types such as the CIK cell due to the amount of studies that have been conducted to date, as well as the ability of the natural killer cell to synergistically kill tumor cells. MDSCs also demonstrate promise as they are able to take up virus in large quantities (with some modification), migrate to tumor sites robustly, and attack tumor cells in synergy with the virus.
Over a decade has passed since the initial work was performed with cell-based carriers of OVs, and the work can no longer be considered to be in its infancy. There is definitely a precedent for these clinical trials, given that both cell-based and viral therapies have been used to treat a variety of diseases. What is lacking is a connection between clinicians and scientists, which is necessary to bring these therapies to patients.
Conclusion
There is a growing body of literature demonstrating that immune cells are effective for delivering OVs directly to the tumor microenvironment as a result of their unique ability to take up virus and target tumor-induced inflammatory processes. In selecting appropriate carriers, it has become evident that cells such as natural killer cells and MDSCs are able to attack tumor cells directly when activated by either the virus or tumor, leading to a stronger, synergistic antitumor effect. The next frontier in engineering better cell-mediated OV therapy is to further enhance this synergy between cell and virus by altering both to improve tumor migration, viral uptake, and tumor killing.
Disclosure
The authors report no conflicts of interest in this work.
References
Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Therapy. Jan 2001;8(2):89–98. | |
Thorne SH, Contag CH. Integrating the biological characteristics of oncolytic viruses and immune cells can optimize therapeutic benefits of cell-based delivery. Gene therapy. May 2008;15(10):753–758. | |
Coukos G, Makrigiannakis A, Kang EH, et al. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. Jun 1999;5(6):1523–1537. | |
Eisenstein S, Coakley BA, Briley-Saebo K, et al. Myeloid-derived suppressor cells as a vehicle for tumor-specific oncolytic viral therapy. Cancer research. August 15, 2013;73(16):5003–5015. | |
Ilett EJ, Prestwich RJ, Kottke T, et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene therapy. May 2009;16(5):689–699. | |
Jennings VA, Ilett EJ, Scott KJ, et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. International Journal of Cancer. Journal International du Cancer. March 1, 2014;134(5):1091–1101. | |
Muthana M, Rodrigues S, Chen YY, et al. Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer Research. January 15, 2013;73(2):490–495. | |
Cole C, Qiao J, Kottke T, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nature medicine. Oct 2005;11(10):1073–1081. | |
Adair RA, Scott KJ, Fraser S, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. International Journal of Cancer. Journal International du Cancer. May 15, 2013;132(10):2327–2338. | |
Sampath P, Li J, Hou W, Chen H, Bartlett DL, Thorne SH. Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Molecular Therapy: the Journal of the American Society of Gene Therapy. Mar 2013;21(3):620–628. | |
Willmon C, Harrington K, Kottke T, Prestwich R, Melcher A, Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Molecular Therapy: the Journal of the American Society of Gene Therapy. Oct 2009;17(10):1667–1676. | |
Yang Z, Zhang Q, Xu K, et al. Combined therapy with cytokine-induced killer cells and oncolytic adenovirus expressing IL-12 induce enhanced antitumor activity in liver tumor model. PloS one. 2012;7(9):e44802. | |
Pan PY, Chen HM, Chen SH. Myeloid-derived suppressor cells as a Trojan horse: A cellular vehicle for the delivery of oncolytic viruses. Oncoimmunology. August 1, 2013;2(8):e25083. | |
Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nature Medicine. Jun 1997;3(6):639–645. | |
Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. January 6, 2000;19(1):2–12. | |
Bell J, McFadden G. Viruses for tumor therapy. Cell Host and Microbe. March 12, 2014;15(3):260–265. | |
Li J, O’Malley M, Urban J, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Molecular Therapy: the Journal of the American Society of Gene Therapy. Apr 2011;19(4):650–657. | |
Huang JH, Zhang SN, Choi KJ, et al. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Molecular Therapy: the Journal of the American Society of Gene Therapy. Feb 2010;18(2):264–274. | |
Kim HS, Kim-Schulze S, Kim DW, Kaufman HL. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Research. November 1, 2009;69(21):8516–8525. | |
Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. Journal of the National Cancer Institute. October 18, 2006;98(20):1482–1493. | |
Ilett EJ, Barcena M, Errington-Mais F, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. May 1, 2011;17(9):2767–2776. | |
Qiao J, Kottke T, Willmon C, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nature Medicine. Jan 2008;14(1):37–44. | |
Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Therapy. Feb 2007;14(4):324–333. | |
Wiley RD, Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. Proceedings of the National Academy of Sciences of the United States of America. January 17, 2006;103(3):738–743. | |
Liu G, Bi Y, Shen B, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Research. February 1, 2014;74(3):727–737. | |
Castleton A, Dey A, Beaton B, et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood. February 27, 2014;123(9):1327–1335. | |
Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. International journal of hematology. Jul 2007;86(1):8–16. | |
Jevremovic D, Gulati R, Hennig I, et al. Use of blood outgrowth endothelial cells as virus-producing vectors for gene delivery to tumors. American Journal of Physiology. Heart and Circulatory Physiology. Aug 2004;287(2):H494–H500. | |
Fujiwara S, Nawa A, Luo C, et al. Carrier cell-based delivery of replication-competent HSV-1 mutants enhances antitumor effect for ovarian cancer. Cancer Gene Therapy. Feb 2011;18(2):77–86. | |
Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. July 30, 1999;285(5428):727–729. | |
Helms MW, Prescher JA, Cao YA, Schaffert S, Contag CH. IL-12 enhances efficacy and shortens enrichment time in cytokine-induced killer cell immunotherapy. Cancer Immunology, Immunotherapy: CII. Sep 2010;59(9):1325–1334. | |
Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. October 1, 1997;90(7):2541–2548. | |
Steis RG, Urba WJ, VanderMolen LA, et al. Intraperitoneal lymphokine-activated killer-cell and interleukin-2 therapy for malignancies limited to the peritoneal cavity. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. Oct 1990;8(10):1618–1629. | |
Stewart JA, Belinson JL, Moore AL, et al. Phase I trial of intraperitoneal recombinant interleukin-2/lymphokine-activated killer cells in patients with ovarian cancer. Cancer research. October 1, 1990;50(19):6302–6310. | |
Ma G, Pan PY, Eisenstein S, et al. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity. March 25, 2011;34(3):385–395. | |
Shime H, Matsumoto M, Oshiumi H, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proceedings of the National Academy of Sciences of the United States of America. February 7, 2012;109(6):2066–2071. | |
Shinozaki K, Ebert O, Kournioti C, Tai YS, Woo SL. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Molecular Therapy: the Journal of the American Society of Gene Therapy. Mar 2004;9(3):368–376. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.