Back to Journals » Journal of Asthma and Allergy » Volume 14
Imbalanced Coagulation in the Airway of Type-2 High Asthma with Comorbid Obesity
Authors Womble JT ![]() , McQuade VL, Ihrie MD, Ingram JL
, McQuade VL, Ihrie MD, Ingram JL ![]()
Received 29 April 2021
Accepted for publication 20 July 2021
Published 10 August 2021 Volume 2021:14 Pages 967—980
DOI https://doi.org/10.2147/JAA.S318017
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Amrita Dosanjh
Jack T Womble, Victoria L McQuade, Mark D Ihrie, Jennifer L Ingram
Division of Pulmonary, Allergy, and Critical Care Medicine, Duke University Medical Center, Durham, NC, 27710, USA
Correspondence: Jennifer L Ingram
Division of Pulmonary, Allergy, and Critical Care Medicine, Duke University Medical Center, Box 2641, DUMC Research Drive, Durham, NC, 27710, USA
Tel +1 919 668-1439
Fax +1 919 668-0494
Email [email protected]
Abstract: Asthma is a common, chronic airway inflammatory disease marked by airway hyperresponsiveness, inflammation, and remodeling. Asthma incidence has increased rapidly in the past few decades and recent multicenter analyses have revealed several unique asthma endotypes. Of these, type-2 high asthma with comorbid obesity presents a unique clinical challenge marked by increased resistance to standard therapies and exacerbated disease development. The extrinsic coagulation pathway plays a significant role in both type-2 high asthma and obesity. The type-2 high asthma airway is marked by increased procoagulant potential, which is readily activated following damage to airway tissue. In this review, we summarize the current understanding of the role the extrinsic coagulation pathway plays in the airway of type-2 high asthma with comorbid obesity. We propose that asthma control is worsened in obesity as a result of a systemic and local airway shift towards a procoagulant and anti-fibrinolytic environment. Lastly, we hypothesize bariatric surgery as a treatment for improved asthma management in type-2 high asthma with comorbid obesity, facilitated by normalization of systemic procoagulant and pro-inflammatory mediators. A better understanding of attenuated coagulation parameters in the airway following bariatric surgery will advance our knowledge of biomolecular pathways driving asthma pathobiology in patients with obesity.
Keywords: asthma, obesity, coagulation, bariatric surgery, extrinsic pathway, inflammation
Introduction
Asthma is characterized by airway hyperresponsiveness (AHR), remodeling, and inflammation resulting in moderate-to-severe respiratory symptoms.1 Numerous cluster analyses have identified asthma as a heterogeneous disease consisting of several manifestations, with most patients categorized into two main endotypes: Type-2 high and Type-2 low, with varying phenotypic presentations.1–4 These endotypes are based on a predominance (high) or a scarcity (low) of eosinophilic and type 2 cytokine (interleukin (IL)-13, IL-4, and IL-5)-driven airway inflammation.
Type-2 high asthma further comprises two phenotypes. The first is typically characterized as early-onset (<12 years of age), allergen-sensitized asthma concurrent with elevated sputum eosinophilia (>1%-3%)5 and serum immunoglobulin E (IgE).4–6 The second Type-2 high asthma phenotype is typically characterized as late-onset (>12 years of age), non-allergic, eosinophilic asthma that does not display elevated serum IgE levels.7 Both Type-2 high asthma phenotypes are more responsive to inhaled and oral corticosteroids when compared to the Type-2 low endotype, with the early-onset allergic phenotype relatively the most sensitive to corticosteroid therapy, and management of the late-onset non-atopic phenotype more reliant on oral corticosteroids.7 Type-2 high asthma with comorbid obesity is a significant healthcare problem due to changes in therapeutic success. Compared to lean Type-2 high asthma patients, patients with obesity are more likely to be hospitalized, use oral steroids, be admitted to the intensive care unit (ICU),8 and experience greater asthma severity through augmented small airway dysfuction9 and bronchial hyperresponsiveness.8 These trends strongly suggest that the development of obesity significantly impacts pulmonary function; however, no longitudinal studies have investigated the effect of weight gain on Type-2 high asthma pathophysiology. The increased resistance to corticosteroid therapy and limited alternative therapies for Type-2 high asthma with comorbid obesity10,11 places this population at increased risk of poor clinical outcomes.
A key feature present in both obesity and asthma is an imbalance in coagulation. Activation of the coagulation cascade occurs via two mechanisms: the intrinsic and extrinsic pathways. The intrinsic pathway begins with vascular injury, resulting in the exposure of endothelial collagen to plasma, causing activation of factor XII.12 The extrinsic pathway also begins as a result of tissue damage; whereby plasma leaks into the extravascular space. There, coagulation factor VII (FVII) complexes with tissue factor (TF), which is presented by various cell types,12 activating downstream zymogens, the inactive precursor of an enzyme which lacks catalytic capability. TF is expressed in perivascular cells,13 adventitial tissue,14 and epithelial cells,15 and is in greatest concentration in vital organs such as the brain, lungs, and kidney, allowing for rapid activation of the coagulation cascade following vascular injury.15,16
The two coagulation pathways converge as the common pathway with the activation of factor X (FX), which becomes factor X active (FXa). Downstream processes result in the conversion of prothrombin to thrombin, which cleaves fibrinogen into fibrin fragments, the main component in clot structure. Once the clot has stabilized and tissue repair occurs, fibrinolysis—the breakdown of fibrin clots into fibrin degradation products (FDPs)—is initiated by activation of tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA). tPA/uPA cleave the zymogen plasminogen, forming plasmin, the enzyme responsible for fibrinolysis. Figure 1 illustrates extrinsic pathway activation in the airway following injury to the airway epithelium.
In disease states, fluctuation in clotting and fibrinolytic factors results in improper coagulation. In pathologies such as Type-2 high asthma with comorbid obesity, a significant shift in this equilibrium exists, favoring the procoagulant state through increased production of coagulation factors.17 Additionally, these pathologies are characterized by increased anti-fibrinolytic mediators, thus prolonging clot time.17,18
While coagulation has been studied separately in obesity and asthma, to date, no studies specifically evaluate how imbalanced coagulation within obesity impacts airway pathology in the Type-2 high asthma phenotype. Given the shift in equilibrium toward clot formation and hypofibrinolysis in both obesity and asthma, it is imperative to better understand coagulation in this setting. These studies would help advance our knowledge of asthma pathophysiology and promote therapeutic development for Type-2 high asthma with comorbid obesity.
Bariatric surgery is a promising therapy for improving asthma with comorbid obesity.19–21 Studies evaluating both asthma19,20 and coagulation22,23 report improvement of coagulant and inflammatory parameters following weight loss through conventional diet and exercise regimens or bariatric surgery, but minimal literature is available regarding the mechanisms by which attenuated coagulation improves Type-2 high asthma symptoms, lung function, airway remodeling, and quality of life for patients within this population.
The goal of this review is to present current knowledge of the extrinsic coagulation pathway and regulatory effects of the coagulation cascade on asthma pathobiology in Type-2 high asthma patients with comorbid obesity. The size and complexity of the coagulation cascades narrows the topic of discussion toward the extrinsic pathway due to its potential therapeutic targets. The intrinsic pathway and numerous autogenic loops in coagulation may also serve as potential targets to regulate asthma exacerbations and other pathologic features, but the breadth of regulators is beyond the scope of this review.
Given the procoagulant and hypo-fibrinolytic environment in both obesity and Type-2 high asthma, the coagulation cascade may serve as a link between the influences of increased adiposity and diminished pulmonary function or AHR in asthma. In an attempt to combat the challenges associated with the development of pharmacologic therapies for improper coagulation, we propose bariatric surgery as a possible therapy, with potential for return to normal coagulation processes and a decrease in inflammatory and procoagulant mediators following surgery and/or weight loss.
Coagulation Imbalance in Type-2 High Asthma
Pro-Coagulation in Type-2 High Asthma
Type-2 high asthma is a well-characterized endotype, and the airways of patients with Type-2 high asthma show shifts in coagulation profiles resulting in a propensity towards coagulation and with decreased fibrinolysis. Recent evidence supports that the type-2 cytokine, IL-13, increases both the expression and release of TF from human airway epithelial cells,24 contributing to the procoagulant environment in the airway. Additionally, induced sputum obtained from severe eosinophilic asthma patients exhibits significantly elevated soluble TF compared to sputum from healthy controls.25 Brims et al noted that this group of severe asthma patients who used inhaled corticosteroids (ICS) had increased expression of plasminogen activator inhibitor-1 (PAI-1)—an anti-fibrinolytic mediator that interferes with activation of plasminogen by inhibiting tPA and uPA—, thrombin activatable fibrinolysis inhibitor (TAFI) which protects fibrin clots from lysis, and fibrin deposition, suggesting that ICS use contributes to airway coagulation and fibrosis in severe eosinophilic asthma.25 Furthermore, in an ovalbumin-mouse model of allergic asthma, FVIIa deficiency resulted in improved asthma response seen through decreased lung eosinophilia and AHR.26 Additionally, a study by Ollivier et al found that TF-FVIIa augmented vascular endothelial growth factor (VEGF) production by human airway fibroblasts,27 promoting angiogenesis, a hallmark feature of airway remodeling in asthma.28–30
Downstream activation of FX is also predictive of impaired airway physiology. In ovalbumin-allergen challenged mice, FX expression is significantly elevated compared to controls, and mice treated with fondaparinux (FPX), an inhibitor of FX, experience attenuated airway mucin production, AHR, airway thickening, and airway fibrosis.31 Additionally, FXa increases airway fibroblast proliferation and procollagen synthesis, contributing to airway stiffening and reduced lung compliance.32
Progressing through the coagulation cascade, further zymogen activation contributes to worsening asthma. Asthma is associated with enhanced thrombin activation,17 contributing to increased fibrinogen cleavage while also binding with protease-activated receptors (PARs), driving inflammation.33 Platelet accumulation and fibrin deposition are further known to exacerbate asthma. In asthma, platelet accumulation in the airway contributes to bronchial hyperresponsiveness, bronchoconstriction, airway inflammation, and airway remodeling.34 Furthermore, FDPs contribute to the inflammatory and infiltration profiles characteristic of asthma.35,36 More specifically, the fibrin E fragment increases leukocyte invasion/migration and IL-1β and IL-6 production, while fibrin D dimers augment IL-1β, IL-6, and PAI-1 release by peripheral blood monocytes.35,37 During tissue repair, under homeostatic conditions, fibrinolysis facilitates clot breakdown and FDP clearance, potentially decreasing pro-inflammatory cytokine production and immune system activation.
Hypofibrinolysis in Type-2 High Asthma
Augmentation of a procoagulant environment in Type-2 high asthma is also met with hypofibrinolysis. The major direct inhibitor of fibrinolysis is PAI-1, which inhibits both tPA and uPA, prolonging clot presence at the injury site. Type-2 high asthma is associated with increased production of PAI-1, which is produced by numerous cell types in the lung, including macrophages, mast cells, fibroblasts, and bronchial epithelial cells36,38 and is strongly associated with impaired lung function.39 Furthermore, sputum from asthma patients25 and bronchoalveolar lavage (BAL) fluid from ovalbumin-sensitized rodents both exhibited a marked increase in PAI-140–42 relative to healthy and saline-treated controls. Administration of aerosolized tPA reduces AHR in ovalbumin-challenged mice,40 and aerosolized uPA in this same model results in improved AHR and subepithelial fibrosis.43
Activated protein C (APC)—a collateral cascade initiated following thrombin activation that decreases the extent of clot formation—is reduced in Type-2 high asthma44,45 and is suggestive as a potential therapy. In a randomized human trial, recombinant human APC administration did not alter the procoagulant state in the airway but did decrease neutrophil influx and degranulation while leaving eosinophil influx unaltered.46 However, inhalation of APC before ovalbumin-challenge in mice resulted in a strong attenuation of allergic inflammation with decreased eosinophil influx, IL-4, IL-5, and IL-13 and IgE levels in BAL fluid, and diminished AHR.47 These findings suggest that APC plays an anti-inflammatory role through modulation of dendritic cell and neutrophil trafficking in asthma,46,48 and these effects are independent of the functions of APC in the coagulation pathway. Therefore, reduced levels of APC in Type 2-high asthma reflect increased inflammation in the airway, and administration of APC inhibits inflammatory cell migration, suggesting a role as an anti-inflammatory therapeutic agent for asthma.
Current asthma therapies target asthma symptoms and exacerbations, but few, if any, specifically address the mediators of the coagulation pathway. This discrepancy may, in part, be due to the complexity of the coagulation cascade and difficulties faced when regulating its mediators. In a study conducted by Peyvandi et al, deficiency in fibrinogen, FX, and FXIII associated strongly with severe bleeding episodes.49 Additional studies attempted to knockout TF in mice, but this approach resulted in a significant increase in embryonic death due to severe hemorrhagic events and poor vascular development.50,51 Therefore, therapeutic strategies attempting to target mediators of the coagulation cascade must carefully consider the detrimental effects of even mildly altering expression and activity of these factors.
Type-2 high asthma with comorbid obesity characteristically involves distal airways52 and presents as a severe asthma phenotype.1–4,6 Recent findings from a case study of status asthmaticus found extensive fibrin deposition in distal airways of difficult-to-treat steroid-resistant asthma; these findings were also seen in a Type-2 high asthma mouse model conducted by the same group.40 In addition to increased fibrin deposition, Wagers et al noted an impaired fibrinolytic system within the mouse model.40 Given that obesity is an important risk factor for increased asthma severity8 and that impaired coagulation presents in both obesity and Type-2 high asthma alone, Type-2 high asthma patients with comorbid obesity are at elevated risk for rapid disease progression. The interactions of airway mediators with the mediators of the coagulation cascade and their downstream effects on asthma pathobiology are listed in Table 1.
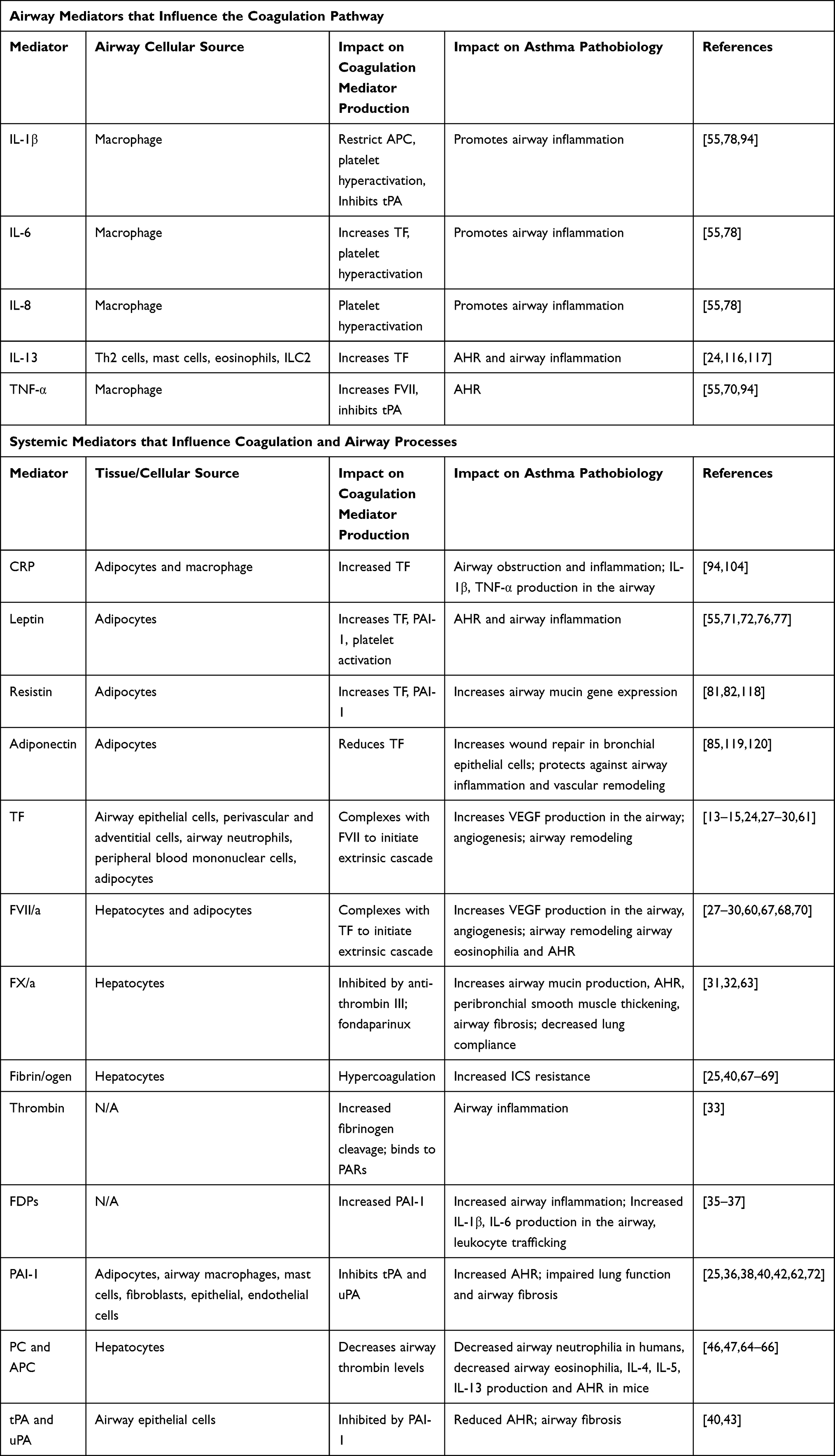 |
Table 1 Cytokine, Adipokines, and Coagulation Mediators Contributing to Asthma Pathobiology |
Coagulation in Obesity
Obesity is typically defined in the literature by body mass index (BMI ≥ 30 kg/m2 for adults) or waist circumference (WC ≥ 40 inches for adult males or ≥ 35 inches for adult females).53 Obesity contributes to chronic low-grade systemic inflammation due to an increase in pro-inflammatory adipokines and cytokines.54 Pro-inflammatory mediators increased in obesity include IL-1β, IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α), among others.55 This inflammatory profile associated with obesity is attributed to polarization from anti-inflammatory M2 to pro-inflammatory M1 macrophages within adipose tissue,56–58 which results in increased secretion of Type 1 and Type 17 cytokines.The impact of systemic inflammation in obesity is a significant topic of interest due to its contribution to numerous comorbidities such as type II diabetes, stroke, pulmonary embolism, and asthma, among others.59
Obesity promotes a systemic and local procoagulant and anti-fibrinolytic environment,60 which accounts for the increased risk of cardiovascular diseases,59 as depicted in Figure 2. This shift favoring coagulation with increased adiposity is due, in part, to increased adipose production of TF61 and PAI-1.62 These mediators also increase in systemic circulation due to Type 1 and Type 17 cytokine interaction with various organ systems. Mice on high-fat diet, a model of obesity, experience a significant increase in TF expression in numerous vital organs, including the brain, lung, and kidneys, with the largest increase seen in the brain and lungs.61
Aside from augmented coagulation and inhibition of fibrinolysis, obesity is also associated with a decrease of the anti-coagulation mediator, anti-thrombin III,63 which inhibits FX and prothrombin activation. Obesity is characterized by increased APC,46,47,64–66 despite the persistent procoagulant state, and these levels are attenuated following weight loss.64 The mechanism driving the diminished efficacy of APC in obesity may be related to thrombin generation.64
A majority of coagulation factor synthesis occurs in the liver, and numerous factors increase in obesity, including FVII and fibrinogen.67–69 Adipocytes exposed to TNF-α, which is elevated in obesity, also contribute to increased synthesis of FVII70 which complexes with TF initiating the extrinsic coagulation cascade. Given these findings, the augmented presence of FVII, supplied both by adipocytes70 and hepatocytes,60,67,68 increases the coagulation potential within obesity.
A major adipokine elevated in obesity is leptin; a satiety hormone that synapses in the lateral geniculate nucleus of the thalamus and inhibits hunger. Leptin signaling becomes imbalanced during obesity, leading to leptin hyposensitivity, unregulated caloric intake, and adipogenesis. Leptin is a significant contributor to chronic low-grade systemic inflammation54 and influences mediators within the coagulation pathway, such as TF expression, in neutrophils and peripheral blood mononuclear cells71 and PAI-1 production by vascular endothelial cells.72 Leptin also interacts with type-2 cytokines to promote mucin and eotaxin secretion by airway cells and eosinophil chemotaxis in the airway,73–75 suggesting a role for leptin in augmenting airway remodeling and inflammation in Type-2 high asthma with comorbid obesity.
Obesity not only contributes to augmented susceptibility to pro-coagulation through increased production of procoagulant and anti-fibrinolytic factors, but also through enhanced platelet hyperreactivity.69 The effect of leptin on platelet activation is still under debate, but platelets cultured in physiological equivalent levels of leptin, adenosine diphosphate (ADP), and thrombin seen in obesity resulted in increased aggregation, while this did not occur in levels comparable to those observed in lean patients.76,77 Additionally, IL-1β, IL-6, and IL-8 were all found to promote hypercoagulability.78 More specifically, IL-1β impaired anti-coagulation processes restricting activation of PC; IL-6 increased TF, priming for coagulation initiation; and addition of all three cytokines to whole blood produced the most pronounced platelet hyper-activation.78
Additional adipokines impacted in obesity include augmented resistin (pro-inflammatory) and decreased adiponectin (anti-inflammatory and atherogenetic).79 The impact of resistin and adiponectin on coagulation factor synthesis is under debate. An in vitro study conducted by Ayer et al found no significant effect of increasing doses of resistin on TF production by peripheral blood mononuclear cells.80 However, Calabró et al observed augmented TF expression in human coronary artery endothelial cells when treated with resistin.81 Additionally, resistin gene knockdown in 3T3-L1 adipocytes resulted in decreased PAI-1 gene and protein production.82 A mouse study conducted by Kato et al found no significant difference between platelet count or levels of coagulation parameters between wild type and adiponectin knockout mice.83 Furthermore, the addition of adiponectin to human platelets had no impact on platelet activation/aggregation.84 Despite these reports, adiponectin appears to inhibit macrophage TF expression, possibly providing a protective role for the initiation of the extrinsic pathway.85
To date, no studies have reported the impact of diet and exercise on coagulation profiles specifically in Type-2 high asthma or in those patients with asthma and comorbid obesity. However, a clinical trial found that a low sodium diet in asthma patients had no impact on systemic inflammation or coagulation profiles following 6 weeks.86 Additionally, a study conducted by Liang et al noted that improvements in physical activity, physical fitness, and/or weight loss were associated with decreased procoagulation parameter production, including plasma levels of PAI-1.87 Thus, weight reduction through diet and exercise may have some beneficial effects on the imbalance of coagulation/fibrinolysis pathways in obesity, but these effects on asthma pathology remain unclear.
Bariatric Surgery, Comorbid Obesity and Allergic Asthma and Coagulation
The lack of effective pharmacologic treatments targeting progression and exacerbation of asthma in comorbid obesity places this population at risk for poor clinical outcome and quality of life. As such, the development of new therapies is needed to address the challenges faced within this subset of patients. Interestingly, bariatric surgery independently improves features of improper coagulation19–23 in patients with obesity; however, the impact of attenuated coagulation within the airway and in asthma patients following bariatric surgery has yet to be determined.
The implication of obesity on asthma pathophysiology is multifactorial, complicating disease management, treatment, and progression, which is attenuated following bariatric surgery and weight loss. In a study of patients with comorbid obesity and asthma, those undergoing bariatric surgery experienced a significant increase in asthma control, reduced medication usage, and improved quality of life.88,89 Of note, those with normal IgE levels experienced significant AHR improvement, which was not found in subjects with high IgE levels88—a feature of the Type-2 high allergic asthma phenotype. However, this finding is not surprising. Augmented AHR is attributed to increased adiposity through reduced lung and chest wall compliance largely due to compression,90–92 which is corrected following weight loss.93 Despite this, all patients experienced a significant improvement in controlling asthma exacerbations.88 Lack of change in AHR in patients with high IgE suggests the pre-existing allergic asthma-associated airway pathology is still present following weight loss from bariatric surgery, contributing to the lack of significant effect found by Dixon et al. These findings provide insight into the role that increased adiposity plays in complicating asthma management, which is corrected following weight loss; further studies are needed to better understand the metabolic and cellular signaling mechanisms whereby bariatric surgery impacts features of asthma exacerbations and management.
In addition to improved asthma control, bariatric surgery corrects inflammatory cytokine and adipokine profiles. Circulating levels of leptin, TNF-α, IL-6 and C-reactive protein (CRP), an indirect inhibitor of tPA through augmentation of IL-1β and TNF-α,94 significantly decrease 3-to-12 months following bariatric surgery,95–100 with a concomitant increase seen in circulating adiponectin levels.95–97 Serum levels of Type-2 cytokines IL-13 and chitinase-3-like protein 1 (YKL-40) are also shown to decrease following bariatric surgery.101,102 Furthermore, the airways following bariatric surgery show a significant decrease in mast cells 12 months post-operative,98 an important cell type for mediating allergic responses and initiating asthma pathobiology.103
The impact of bariatric surgery on parameters of coagulation has also been identified and are shown in Table 2. As previously mentioned, bariatric surgery reduces CRP levels. CRP is known to increase TF expression by monocytes,104 thereby increasing pro-thrombotic risk. Independent of the impact of CRP on TF production, weight loss alone results in a significant decrease in TF.65 Serum levels of the co-initiator of the extrinsic coagulation cascade, FVII, also significantly decrease 12 months after bariatric surgery,99 further reducing the pro-thrombotic potential. Cugno et al further found a significant decrease in serum CRP, PAI-1, and fibrinogen 3 months post-operative, with the lowest levels achieved at 12 months.99
 |
Table 2 The Impact of Bariatric Surgery on Mediator Production |
The increased circulating fibrinogen in patients with obesity contributes to rapid clot formation through augmented hypercoagulability and increased resistance to fibrinolysis, which significantly improves following bariatric surgery and weight loss.18,23 Furthermore, Stolberg et al found a significant decrease in systemic levels of thrombin, fibrinogen, and PAI-1 antigens following Roux-en-Y gastric bypass (RYGB) at 6- and 24-months post-operative, leading to improved clot lysis.22 Similar results were noted by Ay et al who showed significant decreases in PAI-1, TF, and prothrombin fragments 2 years after gastric banding and RYGB,65 suggesting that bariatric surgery patients may experience sustained reductions in levels of pro-coagulant mediators for several years after surgery.
Bariatric surgery induces improvement in coagulation through normalized anti-coagulant and anti-fibrinolytic mediators.22,63–66,97,99,100,105 Batist et al reported increased systemic anti-thrombin III associated with bariatric surgery-stimulated weight loss.63,105 Moreover, circulating PC/APC levels decreased 1 year following bariatric surgery due to reduced thrombomodulin and thrombin potential.64–66 Bariatric surgery also promoted a significant reduction in systemic PAI-1 as early as 3-months post-operative,22,65,97,99,100 reducing the inhibition of tPA and uPA, leading to improved fibrinolysis.
Indeed, epigenetic mechanisms may provide a link between bariatric surgery and improved coagulation profiles in patients with obesity. DNA methylation presents as a possible molecular response following bariatric surgery and may be involved in reversing some of the detrimental effects of obesity. Compared to lean individuals, adults with obesity exhibit global hypomethylation, with associated increased expression of proinflammatory genes.106 A study conducted by Nicoletti et al found that surgically-induced weight loss resulted in decreased methylation of IL6, while an increase in IL6 methylation was observed following weight loss through restricted diet.107 These authors further demonstrated that baseline SERPINE1 (the gene encoding PAI-1) methylation was significantly associated with percentage of weight loss, suggesting that baseline methylation markers of coagulation pathway-associated genes may predict future weight loss in patients.107 These results are intriguing and point to the potential of epigenomics studies to offer greater insight into the intersection of obesity and asthma pathobiology.
It should be noted that the attenuated coagulation factors observed following bariatric surgery were seen in systemic circulation. To date, there are no studies investigating how these coagulation parameter profiles change in the airway following bariatric surgery. Despite this knowledge gap, attenuated circulating levels of procoagulant/anti-fibrinolytic mediators are suspected to improve asthma outcomes, which are worsened through an imbalance in the coagulation cascade in Type-2 high asthma patients with comorbid obesity. In asthma, there is an increase in vascularity,108,109 vascular permeability,110–112 and plasma exudation,40,113,114 thereby readily exposing extravascular tissues to the circulating mediators of the coagulation cascade.
Conclusion
The increased prevalence of obesity worldwide poses new challenges within health care. More specifically, previously understood and controlled pathologies are being complicated by increased adiposity at an alarming rate, as seen in comorbid obesity and asthma. Impaired coagulation in Type-2 high asthma with comorbid obesity presents as a unique pathologic mechanism; advancing our understanding of this pathway will allow for better therapeutic development for this growing population of patients.
At present, the lack of treatments for Type-2 high asthma with comorbid obesity places these patients at increased risk for hospitalization, ICU admission, poor symptom control, and quality of life.8 As presented herein, an imbalance in the extrinsic coagulation cascade resulting from increased adiposity may play a significant role in impaired management of asthma exacerbations. Increased levels of TF and FVII in the airway and systemic circulation respectively, primes the pathway for rapid activation. Increased presence and synthesis of downstream mediators FX and fibrinogen, among others, not only aids in rapid clot formation within the airway, but also produces collateral effects driving AHR, remodeling, and inflammation. Additionally, augmented production of anti-fibrinolytic mediators prolongs clot presence and impairs responsiveness to ICS. Given the complexity and heterogeneity of asthma and the general influence of obesity on cardio/pulmonary vascular pathobiology, these pathways may be critical in other phenotypic presentations of asthma, including Type-2 low asthma with obesity, an area in need of much greater understanding.
Currently, available asthma therapies do not target mediators within the coagulation cascade. This circumstance is due, in part, to the challenges and complications faced when modulating systemic levels of mediators of this complex pathway. Interestingly, inhaled heparin, an antioxidant and anti-coagulant, has been identified as a novel therapy to enhance asthma management through decreased airway obstruction, improved airway inflammation, and augmented delivery of other inhaled therapies.115 Despite heparin’s potential benefit in asthma management, it does not address the underlying shift towards a procoagulant environment in the airway.
Obesity is characterized by a significant pathway modification favoring procoagulant and anti-fibrinolytic mediators, both in the airway and in systemic compartments. The use of concurrent ICS and heparin addresses the symptomatic responses of asthma but cannot address the shift in inflammatory and pro-coagulation profiles. However, bariatric surgery addresses these imbalances on a systemic scale leading to improved responsiveness to therapies.
We hypothesize that bariatric surgery, unlike other pharmacological interventions, provides significant improvements in asthma control as seen through improved AHR, lung compliance, and normalized coagulant and inflammatory parameters. Future research will identify specific metabolic and cellular mechanisms whereby bariatric surgery influences airway coagulation and physiology and reveal potential applications to clinical care. A better understanding of bariatric surgery-induced metabolic and cellular mechanisms regulating mediators of the coagulation pathway, whether those mechanisms be weight loss-dependent or -independent, holds promise for the development of potential therapies to benefit all patients with Type-2 high asthma, not just those with comorbid obesity.
 |
Figure 1 Extrinsic Coagulation Cascade in Response to Allergen Exposure-Induced Airway Damage Allergen exposure and/or tissue damage initiates the extrinsic coagulation pathway in the extravascular compartments of the airway. Downstream activation via antigen presenting cells (APCs) promotes clinical features of asthma pathobiology, including AHR, airway inflammation, and fibrosis. In Type 2-high asthma with comorbid obesity, the pathway becomes imbalanced, favoring pro-coagulation (black) and anti-fibrinolysis (blue). The above figure is used to exemplify the coagulation pathway. Created with BioRender.com. |
 |
Figure 2 Coagulation and Inflammatory Mediators Promoting Asthma Pathophysiology Increased adiposity promotes pro-inflammatory cytokine and adipokine production leading to chronic low-grade systemic inflammation. These mediators augment procoagulant factor production, contributing to increased asthma pathogenesis in Type-2 high asthma with comorbid obesity. Additionally, the impact of IL-6, TNF-α, IL-1β, monocyte chemoattractant protein-1 (MPC-1), and free fatty acids (FFAs) leads to insulin resistance, hepatic steatosis, and non-alcoholic fatty liver disease.69 In response to upregulated mediators, the airway experiences increased leukocyte infiltration, fibrosis, narrowing, mucin production, angiogenesis and hyperresponsiveness. The figure only includes coagulation mediators of the extrinsic and common pathway; the intrinsic coagulation mediators are beyond the scope of this review. Red arrows indicates contribution to worsening asthma and green arrows indicate improvement in asthma. Created with BioRender.com. |
Acknowledgments
The authors thank Dr. Eric Monson from the Duke University Center for Data and Visualization Sciences with his guidance on the figures.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
Dr. Ingram’s research is supported by NIH grants 5R01-HL130234 and 1R21AI146761-01.
Disclosure
Jennifer L Ingram reports grants from National Institutes of Health, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178(3):218–224. doi:10.1164/rccm.200711-1754OC
2. Lefaudeux D, De Meulder B, Loza MJ, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol. 2017;139(6):1797–1807. doi:10.1016/j.jaci.2016.08.048
3. Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–323. doi:10.1164/rccm.200906-0896OC
4. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. 2019;56(2):219–233. doi:10.1007/s12016-018-8712-1
5. Walford HH, Doherty TA. Diagnosis and management of eosinophilic asthma: a US perspective. J Asthma Allergy. 2014;7:53–65.
6. Trivedi M, Denton E. Asthma in children and adults-what are the differences and what can they tell us about asthma? Front Pediatr. 2019;7:256. doi:10.3389/fped.2019.00256
7. Gerday S, et al. Asthmatics with concordant eosinophilic disease classified according to their serum IgE status. Respir Med Res. 2021;79:100797.
8. Holguin F, Bleecker ER, Busse WW, et al. Obesity and asthma: an association modified by age of asthma onset. J Allergy Clin Immunol. 2011;127(6):1486–93e2. doi:10.1016/j.jaci.2011.03.036
9. Abdo M, Trinkmann F, Kirsten A-M, et al. Small airway dysfunction links asthma severity with physical activity and symptom control. J Allergy Clin Immunol Pract. 2021. doi:10.1016/j.jaip.2021.04.035
10. Wang M, Gao P, Wu X, et al. Impaired anti-inflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir Res. 2016;17(1):153. doi:10.1186/s12931-016-0462-0
11. Hinks TS, Levine SJ, Brusselle GG. Treatment options in type-2 low asthma. Eur Respir J. 2020;57:1.
12. Chaudhry R, Usama SM, Babiker HM. Physiology, Coagulation Pathways. Treasure Island (FL): StatPearls; 2020.
13. Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134(5):1087–1097.
14. Wilcox JN, Smith KM, Schwartz SM, et al. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. 1989;86(8):2839–2843. doi:10.1073/pnas.86.8.2839
15. Fleck RA, Rao LVM, Rapaport SI, et al. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59(2):421–437. doi:10.1016/0049-3848(90)90148-6
16. Smith SA, Travers RJ, Morrissey JH. How it all starts: initiation of the clotting cascade. Crit Rev Biochem Mol Biol. 2015;50(4):326–336. doi:10.3109/10409238.2015.1050550
17. Bazan-Socha S, Mastalerz L, Cybulska A, et al. Asthma is associated with enhanced thrombin formation and impaired fibrinolysis. Clin Exp Allergy. 2016;46(7):932–944. doi:10.1111/cea.12734
18. Samuels J, Lawson PJ, Morton AP, et al. Prospective assessment of fibrinolysis in morbid obesity: tissue plasminogen activator resistance improves after bariatric surgery. Surg Obes Relat Dis. 2019;15(7):1153–1159. doi:10.1016/j.soard.2019.03.048
19. Boulet L-P, Turcotte H, Martin J, et al. Effect of bariatric surgery on airway response and lung function in obese subjects with asthma. Respir Med. 2012;106(5):651–660. doi:10.1016/j.rmed.2011.12.012
20. Chaaban TA. Bariatric surgery: a potential cure for asthma? Eur Respir Rev. 2019;28(152):190003. doi:10.1183/16000617.0003-2019
21. Chapman DG, Irvin CG, Kaminsky DA, et al. Influence of distinct asthma phenotypes on lung function following weight loss in the obese. Respirology. 2014;19(8):1170–1177. doi:10.1111/resp.12368
22. Stolberg CR, Mundbjerg LH, Funch-Jensen P, et al. Effects of gastric bypass followed by a randomized study of physical training on markers of coagulation activation, fibrin clot properties, and fibrinolysis. Surg Obes Relat Dis. 2018;14(7):918–926. doi:10.1016/j.soard.2018.03.022
23. Ditschuneit HH, Flechtner-Mors M, Adler G. Fibrinogen in obesity before and after weight reduction. Obes Res. 1995;3(1):43–48. doi:10.1002/j.1550-8528.1995.tb00119.x
24. Mitchel JA, Antoniak S, Lee J-H, et al. IL-13 augments compressive stress-induced tissue factor expression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2016;54(4):524–531. doi:10.1165/rcmb.2015-0252OC
25. Brims FJ, Chauhan AJ, Higgins B, et al. Coagulation factors in the airways in moderate and severe asthma and the effect of inhaled steroids. Thorax. 2009;64(12):1037–1043. doi:10.1136/thx.2009.114439
26. Shinagawa K, Ploplis VA, Castellino FJ. A severe deficiency of coagulation factor VIIa results in attenuation of the asthmatic response in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296(5):L763–L770. doi:10.1152/ajplung.90638.2008
27. Ollivier V, Bentolila S, Chabbat J, et al. Tissue factor-dependent vascular endothelial growth factor production by human fibroblasts in response to activated factor VII. Blood. 1998;91(8):2698–2703. doi:10.1182/blood.V91.8.2698.2698_2698_2703
28. Chetta A, Zanini A, Olivieri D. Therapeutic approach to vascular remodelling in asthma. Pulm Pharmacol Ther. 2007;20(1):1–8. doi:10.1016/j.pupt.2005.10.009
29. Walters EH, Reid D, Soltani A, et al. Angiogenesis: a potentially critical part of remodelling in chronic airway diseases? Pharmacol Ther. 2008;118(1):128–137. doi:10.1016/j.pharmthera.2008.01.007
30. Walters EH, Soltani A, Reid DW, et al. Vascular remodelling in asthma. Curr Opin Allergy Clin Immunol. 2008;8(1):39–43. doi:10.1097/ACI.0b013e3282f42696
31. Shinagawa K, Martin JA, Ploplis VA, et al. Coagulation factor Xa modulates airway remodeling in a murine model of asthma. Am J Respir Crit Care Med. 2007;175(2):136–143. doi:10.1164/rccm.200608-1097OC
32. Blancbrude OP, Archer F, Leoni P, et al. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res. 2005;304(1):16–27. doi:10.1016/j.yexcr.2004.10.021
33. Reed CE, Kita H. The role of protease activation of inflammation in allergic respiratory diseases. J Allergy Clin Immunol. 2004;114(5):
34. Kornerup KN, Page CP. The role of platelets in the pathophysiology of asthma. Platelets. 2007;18(5):319–328. doi:10.1080/09537100701230436
35. Jennewein C, Tran N, Paulus P, et al. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med. 2011;17(5–6):568–573. doi:10.2119/molmed.2010.00146
36. Levi M, Schultz MJ, Rijneveld AW, et al. Bronchoalveolar coagulation and fibrinolysis in endotoxemia and pneumonia. Crit Care Med. 2003;31(4 Suppl):S238–42. doi:10.1097/01.CCM.0000057849.53689.65
37. Robson SC, Shephard EG, Kirsch RE. Fibrin degradation product D-dimer induces the synthesis and release of biologically active IL-1 beta, IL-6 and plasminogen activator inhibitors from monocytes in vitro. Br J Haematol. 1994;86(2):322–326. doi:10.1111/j.1365-2141.1994.tb04733.x
38. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38(2 Suppl):S26–34. doi:10.1097/CCM.0b013e3181c98d21
39. Cho S, Kang J, Lyttle C, et al. Association of elevated plasminogen activator inhibitor 1 levels with diminished lung function in patients with asthma. Ann Allergy Asthma Immunol. 2011;106(5):371–377. doi:10.1016/j.anai.2010.12.021
40. Wagers SS, Norton RJ, Rinaldi LM, et al. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 2004;114(1):104–111. doi:10.1172/JCI200419569
41. Oh CK, Ariue B, Alban RF, et al. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem Biophys Res Commun. 2002;294(5):1155–1160. doi:10.1016/S0006-291X(02)00577-6
42. Kucharewicz I, Mogielnicki A, Kasacka I, et al. Plasmin system regulation in an ovalbumin-induced rat model of asthma. Int Arch Allergy Immunol. 2008;147(3):190–196. doi:10.1159/000142041
43. Kuramoto E, Nishiuma T, Kobayashi K, et al. Inhalation of urokinase-type plasminogen activator reduces airway remodeling in a murine asthma model. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L337–L346. doi:10.1152/ajplung.90434.2008
44. Schouten M, Van de pol MA, Levi M, et al. Early activation of coagulation after allergen challenge in patients with allergic asthma. J Thromb Haemost. 2009;7(9):1592–1594. doi:10.1111/j.1538-7836.2009.03523.x
45. Hataji O, Taguchi O, Gabazza EC, et al. Activation of protein C pathway in the airways. Lung. 2002;180(1):47–59. doi:10.1007/s004080000080
46. de Boer JD, Berger M, Majoor CJ, et al. Activated protein C inhibits neutrophil migration in allergic asthma: a randomised trial. Eur Respir J. 2015;46(6):1636–1644. doi:10.1183/13993003.00459-2015
47. Yuda H, Adachi Y, Taguchi O, et al. Activated protein C inhibits bronchial hyperresponsiveness and Th2 cytokine expression in mice. Blood. 2004;103(6):2196–2204. doi:10.1182/blood-2003-06-1980
48. Matsumoto T, Matsushima Y, Toda M, et al. Activated protein C modulates the proinflammatory activity of dendritic cells. J Asthma Allergy. 2015;8:29–37.
49. Peyvandi F, Palla R, Menegatti M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10(4):615–621. doi:10.1111/j.1538-7836.2012.04653.x
50. Carmeliet P, Mackman N, Moons L, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383(6595):73–75. doi:10.1038/383073a0
51. Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24(6):1015–1022. doi:10.1161/01.ATV.0000130465.23430.74
52. Marin G, Gamez AS, Molinari N, et al. Distal airway impairment in obese normoreactive women. Biomed Res Int. 2013;2013:707856. doi:10.1155/2013/707856
53. Guidelines (2013) for managing overweight and obesity in adults. Preface to the Expert Panel Report (comprehensive version which includes systematic evidence review, evidence statements, and recommendations). Obesity (Silver Spring). 2014;22 Suppl 2:S40.
54. Bullo M, García-Lorda P, Megias I, et al. Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res. 2003;11(4):525–531. doi:10.1038/oby.2003.74
55. Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:139239. doi:10.1155/2013/139239
56. Molofsky AB, Nussbaum JC, Liang H-E, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–549. doi:10.1084/jem.20121964
57. Miethe S, Karsonova A, Karaulov A, et al. Obesity and asthma. J Allergy Clin Immunol. 2020;146(4):685–693. doi:10.1016/j.jaci.2020.08.011
58. Samad F, Ruf W. Inflammation, obesity, and thrombosis. Blood. 2013;122(20):3415–3422. doi:10.1182/blood-2013-05-427708
59. Guh DP, Zhang W, Bansback N, et al. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9(1):88. doi:10.1186/1471-2458-9-88
60. Vilahur G, Ben-Aicha S, Badimon L. New insights into the role of adipose tissue in thrombosis. Cardiovasc Res. 2017;113(9):1046–1054. doi:10.1093/cvr/cvx086
61. Samad F, Pandey M, Loskutoff DJ. Regulation of tissue factor gene expression in obesity. Blood. 2001;98(12):3353–3358. doi:10.1182/blood.V98.12.3353
62. Eriksson P, Reynisdottir S, Lönnqvist F, et al. Adipose tissue secretion of plasminogen activator inhibitor-1 in non-obese and obese individuals. Diabetologia. 1998;41(1):65–71. doi:10.1007/s001250050868
63. Batist G, Bothe A, Bern M, et al. Low antithrombin III in morbid obesity: return to normal with weight reduction. JPEN J Parenter Enteral Nutr. 1983;7(5):447–449. doi:10.1177/0148607183007005447
64. Sola E, Navarro S, Medina P, et al. Activated protein C levels in obesity and weight loss influence. Thromb Res. 2009;123(5):697–700. doi:10.1016/j.thromres.2008.07.017
65. Ay L, Kopp H-P, Brix J-M, et al. Thrombin generation in morbid obesity: significant reduction after weight loss. J Thromb Haemost. 2010;8(4):759–765. doi:10.1111/j.1538-7836.2010.03766.x
66. Rega-Kaun G, Kaun C, Ebenbauer B, et al. Bariatric surgery in morbidly obese individuals affects plasma levels of protein C and thrombomodulin. J Thromb Thrombolysis. 2019;47(1):51–56. doi:10.1007/s11239-018-1744-9
67. Mertens I, Van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obes Rev. 2002;3(2):85–101. doi:10.1046/j.1467-789X.2002.00056.x
68. Heinz S, Braspenning J. Measurement of blood coagulation factor synthesis in cultures of human hepatocytes. Methods Mol Biol. 2015;1250:309–316.
69. Faber DR, de Groot PG, Visseren FL. Role of adipose tissue in haemostasis, coagulation and fibrinolysis. Obes Rev. 2009;10(5):554–563. doi:10.1111/j.1467-789X.2009.00593.x
70. Takahashi N, Yoshizaki T, Hiranaka N, et al. The production of coagulation factor VII by adipocytes is enhanced by tumor necrosis factor-α or isoproterenol. Int J Obes (Lond). 2015;39(5):747–754. doi:10.1038/ijo.2014.208
71. Rafail S, Ritis K, Schaefer K, et al. Leptin induces the expression of functional tissue factor in human neutrophils and peripheral blood mononuclear cells through JAK2-dependent mechanisms and TNFalpha involvement. Thromb Res. 2008;122(3):366–375. doi:10.1016/j.thromres.2007.12.018
72. Singh P, Peterson TE, Barber KR, et al. Leptin upregulates the expression of plasminogen activator inhibitor-1 in human vascular endothelial cells. Biochem Biophys Res Commun. 2010;392(1):47–52. doi:10.1016/j.bbrc.2009.12.158
73. Hao W, Wang J, Zhang Y, et al. Leptin positively regulates MUC5AC production and secretion induced by interleukin-13 in human bronchial epithelial cells. Biochem Biophys Res Commun. 2017;493(2):979–984. doi:10.1016/j.bbrc.2017.09.106
74. Watanabe K, Suzukawa M, Arakawa S, et al. Leptin enhances cytokine/chemokine production by normal lung fibroblasts by binding to leptin receptor. Allergol Int. 2019;68:S3–S8. doi:10.1016/j.alit.2019.04.002
75. Kato H, Ueki S, Kamada R, et al. Leptin has a priming effect on eotaxin-induced human eosinophil chemotaxis. Int Arch Allergy Immunol. 2011;155(4):335–344. doi:10.1159/000321195
76. Dellas C, Schäfer K, Rohm I, et al. Absence of leptin resistance in platelets from morbidly obese individuals may contribute to the increased thrombosis risk in obesity. Thromb Haemost. 2008;100(12):1123–1129. doi:10.1160/TH08-05-0314
77. Wallace AM, McMahon AD, Packard CJ, et al. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation. 2001;104(25):3052–3056. doi:10.1161/hc5001.101061
78. Bester J, Pretorius E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes platelets and clot viscoelasticity. Sci Rep. 2016;6:32188.
79. Muc M, Todo-Bom A, Mota-Pinto A, et al. Leptin and resistin in overweight patients with and without asthma. Allergol Immunopathol (Madr). 2014;42(5):415–421. doi:10.1016/j.aller.2013.03.004
80. Ayer JG, Song C, Steinbeck K, et al. Increased tissue factor activity in monocytes from obese young adults. Clin Exp Pharmacol Physiol. 2010;37(11):1049–1054. doi:10.1111/j.1440-1681.2010.05430.x
81. Calabro P, Cirillo P, Limongelli G, et al. Tissue factor is induced by resistin in human coronary artery endothelial cells by the NF-ĸB-dependent pathway. J Vasc Res. 2011;48(1):59–66. doi:10.1159/000318775
82. Ikeda Y, Tsuchiya H, Hama S, et al. Resistin regulates the expression of plasminogen activator inhibitor-1 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2014;448(2):129–133. doi:10.1016/j.bbrc.2014.03.076
83. Kato H, Kashiwagi H, Shiraga M, et al. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol. 2006;26(1):224–230. doi:10.1161/01.ATV.0000194076.84568.81
84. Elbatarny HS, Netherton SJ, Ovens JD, et al. Adiponectin, ghrelin, and leptin differentially influence human platelet and human vascular endothelial cell functions: implication in obesity-associated cardiovascular diseases. Eur J Pharmacol. 2007;558(1–3):7–13. doi:10.1016/j.ejphar.2006.11.052
85. Okamoto Y, Ishii S, Croce K, et al. Adiponectin inhibits macrophage tissue factor, a key trigger of thrombosis in disrupted atherosclerotic plaques. Atherosclerosis. 2013;226(2):373–377. doi:10.1016/j.atherosclerosis.2012.12.012
86. Forrester DL, Britton J, Lewis SA, et al. Impact of adopting low sodium diet on biomarkers of inflammation and coagulation: a randomised controlled trial. J Nephrol. 2010;23(1):49–54.
87. Liang G, Huang X, Hirsch J, et al. Modest gains after an 8-week exercise program correlate with reductions in non-traditional markers of cardiovascular risk. Front Cardiovasc Med. 2021;8:669110. doi:10.3389/fcvm.2021.669110
88. Dixon AE, Pratley RE, Forgione PM, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128(3):
89. Guerron AD, Ortega CB, Lee H-J, et al. Asthma medication usage is significantly reduced following bariatric surgery. Surg Endosc. 2019;33(6):1967–1975. doi:10.1007/s00464-018-6500-x
90. Pelosi P, Croci M, Ravagnan I, et al. The effects of body mass on lung volumes, respiratory mechanics, and gas exchange during general anesthesia. Anesth Analg. 1998;87(3):654–660.
91. Hedenstierna G, Santesson J. Breathing mechanics, dead space and gas exchange in the extremely obese, breathing spontaneously and during anaesthesia with intermittent positive pressure ventilation. Acta Anaesthesiol Scand. 1976;20(3):248–254. doi:10.1111/j.1399-6576.1976.tb05036.x
92. Sharp JT, Henry JP, Sweany SK, et al. The total work of breathing in normal and obese men. J Clin Invest. 1964;43(4):728–739. doi:10.1172/JCI104957
93. Dixon AE, Peters U. The effect of obesity on lung function. Expert Rev Respir Med. 2018;12(9):755–767. doi:10.1080/17476348.2018.1506331
94. Singh U, Devaraj S, Jialal I. C-reactive protein decreases tissue plasminogen activator activity in human aortic endothelial cells: evidence that C-reactive protein is a procoagulant. Arterioscler Thromb Vasc Biol. 2005;25(10):2216–2221. doi:10.1161/01.ATV.0000183718.62409.ea
95. Yadav R, Hama S, Liu Y, et al. Effect of Roux-en-Y bariatric surgery on lipoproteins, insulin resistance, and systemic and vascular inflammation in obesity and diabetes. Front Immunol. 2017;8:1512. doi:10.3389/fimmu.2017.01512
96. Shih K-C, Janckila AJ, Lee W-J, et al. Effects of bariatric weight loss surgery on glucose metabolism, inflammatory cytokines, and serum tartrate-resistant acid phosphatase 5a in obese Chinese adults. Clin Chim Acta. 2016;453:197–202. doi:10.1016/j.cca.2015.11.004
97. Netto BDM, Bettini SC, Clemente APG, et al. Roux-en-Y gastric bypass decreases pro-inflammatory and thrombotic biomarkers in individuals with extreme obesity. Obes Surg. 2015;25(6):1010–1018. doi:10.1007/s11695-014-1484-7
98. van Huisstede A, Rudolphus A, Castro Cabezas M, et al. Effect of bariatric surgery on asthma control, lung function and bronchial and systemic inflammation in morbidly obese subjects with asthma. Thorax. 2015;70(7):659–667. doi:10.1136/thoraxjnl-2014-206712
99. Cugno M, Castelli R, Mari D, et al. Inflammatory and prothrombotic parameters in normotensive non-diabetic obese women: effect of weight loss obtained by gastric banding. Intern Emerg Med. 2012;7(3):237–242. doi:10.1007/s11739-011-0522-x
100. Pedersen NB, Stolberg CR, Mundbjerg LH, et al. Reductions in plasmin inhibitor and fibrinogen predict the improved fibrin clot lysis 6 months after obesity surgery. Clin Obes. 2020;10(6):e12397. doi:10.1111/cob.12397
101. Nestvold TK, Nielsen EW, Ludviksen JK, et al. Lifestyle changes followed by bariatric surgery lower inflammatory markers and the cardiovascular risk factors C3 and C4. Metab Syndr Relat Disord. 2015;13(1):29–35. doi:10.1089/met.2014.0099
102. Hempen M, Kopp H-P, Elhenicky M, et al. YKL-40 is elevated in morbidly obese patients and declines after weight loss. Obes Surg. 2009;19(11):1557–1563. doi:10.1007/s11695-009-9917-4
103. Bradding P, Arthur G. Mast cells in asthma–state of the art. Clin Exp Allergy. 2016;46(2):194–263.
104. Jialal I, Devaraj S, Venugopal SK. C-reactive protein: risk marker or mediator in atherothrombosis? Hypertension. 2004;44(1):6–11. doi:10.1161/01.HYP.0000130484.20501.df
105. Pardina E, Ferrer R, Rivero J, et al. Alterations in the common pathway of coagulation during weight loss induced by gastric bypass in severely obese patients. Obesity (Silver Spring). 2012;20(5):1048–1056. doi:10.1038/oby.2011.361
106. Ali MM, Naquiallah D, Qureshi M. et al. DNA methylation profile of genes involved in inflammation and autoimmunity correlates with vascular function in morbidly obese adults. Epigenetics;2021. 1–17. doi:10.1080/15592294.2021.1876285
107. Nicoletti CF, Nonino CB, de Oliveira BAP, et al. DNA methylation and hydroxymethylation levels in relation to two weight loss strategies: energy-restricted diet or bariatric surgery. Obes Surg. 2016;26(3):603–611. doi:10.1007/s11695-015-1802-8
108. Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med. 1997;156(1):229–233. doi:10.1164/ajrccm.156.1.9607066
109. Salvato G. Quantitative and morphological analysis of the vascular bed in bronchial biopsy specimens from asthmatic and non-asthmatic subjects. Thorax. 2001;56(12):902–906. doi:10.1136/thorax.56.12.902
110. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611.
111. Kanazawa H, Hirata K, Yoshikawa J. Involvement of vascular endothelial growth factor in exercise induced bronchoconstriction in asthmatic patients. Thorax. 2002;57(10):885–888. doi:10.1136/thorax.57.10.885
112. Asai K, Kanazawa H, Kamoi H, et al. Increased levels of vascular endothelial growth factor in induced sputum in asthmatic patients. Clin Exp Allergy. 2003;33(5):595–599. doi:10.1046/j.1365-2222.2003.01576.x
113. Moreno RH, Hogg JC, Pare PD. Mechanics of airway narrowing. Am Rev Respir Dis. 1986;133(6):1171–1180.
114. Goldie RG, Pedersen KE. Mechanisms of increased airway microvascular permeability: role in airway inflammation and obstruction. Clin Exp Pharmacol Physiol. 1995;22(6–7):387–396. doi:10.1111/j.1440-1681.1995.tb02028.x
115. Shute JK, Puxeddu E, Calzetta L. Therapeutic use of heparin and derivatives beyond anticoagulation in patients with bronchial asthma or COPD. Curr Opin Pharmacol. 2018;40:39–45. doi:10.1016/j.coph.2018.01.006
116. Schmid-Grendelmeier P, Altznauer F, Fischer B, et al. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J Immunol. 2002;169(2):1021–1027. doi:10.4049/jimmunol.169.2.1021
117. Maggi L, Mazzoni A, Capone M. et al. The dual function of ILC2: from host protection to pathogenic players in type 2 asthma. Mol Aspects Med;2021. 100981. doi:10.1016/j.mam.2021.100981
118. Kwak S, Kim Y-D, Na HG, et al. Resistin upregulates MUC5AC/B mucin gene expression in human airway epithelial cells. Biochem Biophys Res Commun. 2018;499(3):655–661. doi:10.1016/j.bbrc.2018.03.206
119. Medoff BD, Okamoto Y, Leyton P, et al. Adiponectin deficiency increases allergic airway inflammation and pulmonary vascular remodeling. Am J Respir Cell Mol Biol. 2009;41(4):397–406. doi:10.1165/rcmb.2008-0415OC
120. Zhu XL, Qin XQ, Xiang Y, et al. Adipokine adiponectin is a potential protector to human bronchial epithelial cell for regulating proliferation, wound repair and apoptosis: comparison with leptin and resistin. Peptides. 2013;40:34–41. doi:10.1016/j.peptides.2012.11.017
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.