Back to Journals » OncoTargets and Therapy » Volume 13
Retracted Article: IL-6/STAT3 Signaling Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Through Targeting Cancer Stem Cells
Authors Li Y, Chen G, Han Z, Cheng H ![]() , Qiao L
, Qiao L ![]() , Li Y
, Li Y ![]()
Received 15 May 2020
Accepted for publication 4 September 2020
Published 30 September 2020 Volume 2020:13 Pages 9721—9730
DOI https://doi.org/10.2147/OTT.S262089
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
This paper has been retracted.
Yu Li, 1 Gang Chen, 1 Zhijian Han, 2 Huijuan Cheng, 2 Liang Qiao, 3 Yumin Li 1, 2
1General Surgery Department, Lanzhou University Second Hospital, Lanzhou, Gansu, People’s Republic of China; 2Key Laboratory of Digestive System Tumors of Gansu Province, Lanzhou University Second Hospital, Lanzhou, Gansu, People’s Republic of China; 3Storr Liver Unit at the Westmead Millennium Institute, The University of Sydney at Westmead Hospital, Sydney, NSW 2145, Australia
Correspondence: Yumin Li
Lanzhou University Second Hospital, Lanzhou 730030, People’s Republic of China
Tel +86-13893615421
Fax +86-0931-8458109
Email [email protected]
Liang Qiao
Storr Liver Unit at the Westmead Millennium Institute, The University of Sydney at Westmead Hospital, Westmead, Sydney, NSW 2145, Australia
Tel +86-17820019796
Email [email protected]
Background: Sorafenib is the standard first-line treatment for advanced hepatocellular carcinoma (HCC), even though acquired resistance to sorafenib has been found in many HCC patients, resulting in poor prognosis. Accumulating evidence demonstrates that liver cancer stem cells (LCSCs) contribute to sorafenib resistance in HCC. The inflammatory factor interleukin 6 (IL-6) plays a role in sorafenib resistance in HCC. However, the mechanism by which IL-6 in LCSCs is involved in the process of HCC sorafenib resistance remains elusive.
Methods: In this study, the sorafenib-resistant cell line PLC/PRF/5-R was generated by the concentration gradient method, and cell viability was determined by the CCK-8 assay. LCSCs were isolated from the PLC/PRF/5-R cell line by flow cytometry, and tumorigenesis was confirmed in nude mice. Blockade of IL-6 cells was achieved by lentiviral-mediated interference. The protein levels of stem cell markers (EpCAM, CD44), stemness markers (Oct3/4, β-catenin), and hepatocyte differentiation markers (glucose-6-phosphate, AFP) were measured by Western blotting analysis. Finally, a xenograft model was used to evaluate the function of IL-6 in the sorafenib resistance of HCC.
Results: The stable sorafenib-resistant HCC cell line PLC/PRF/5-R was established and showed significant epithelial–mesenchymal transition (EMT) characteristics; the isolated resistant LCSCs from PLC/PRF/5-R were more tumorigenic than the control LCSCs. We showed that IL-6, IL-6R, STAT3 and GP130 expression were dramatically increased in resistant LCSCs compared to control LCSCs. Downregulation of IL-6 expression with short hairpin RNA (shRNA) restored sorafenib sensitivity in resistant LCSCs, suggesting the critical roles of IL-6/STAT3 in inducing sorafenib resistance. Furthermore, a xenograft tumor model showed that IL-6 downregulation improved the antitumor effect of sorafenib.
Conclusion: LCSCs play an important role in sorafenib-resistant HCC, and inhibition of the IL-6/STAT3 signaling pathway improves the antitumor effects of sorafenib against HCC in vitro and in vivo. These findings demonstrate that IL-6 in LCSCs may function as a novel target for combating sorafenib resistance in HCC.
Keywords: IL-6/STAT3 signaling, cancer stem cells, hepatocellular carcinoma, drug resistance, sorafenib
Corrigendum for this paper has been published
Introduction
Hepatocellular carcinoma (HCC) is the sixth most diagnosed cancer and the fourth leading cause of cancer-related death worldwide.1 Surgical resection is the standard curative therapy for the early stage of HCC. Unfortunately, over 80% of patients with HCC are diagnosed during stages too advanced for surgery or liver transplantation.2,3 The recurrence rate of HCC is relatively high and is usually combined with metastasis. Chemotherapy, radiotherapy, radiofrequency ablation (RFA) and transcatheter arterial chemoembolization (TACE) might be alternative treatments, but their efficacy is limited. The response of HCC to chemotherapy diminishes quickly after the regimen is completed, usually due to acquired chemoresistance. Therefore, there is an urgent need to understand the mechanisms of chemoresistance that account for the primary or acquired refractoriness of HCC to anticancer drugs.
As an oral multikinase inhibitor of vascular endothelial growth factor (VEGF), platelet-derived growth factor receptor (PDGF-R), c-Kit and Raf kinase, sorafenib has been considered the first-line and standard treatment for advanced HCC.4 It has shown an overall survival advantage of 2–3 months in patients with advanced HCC. However, only 30% of patients with advanced HCC benefit from sorafenib, and acquired resistance often occurs within 6 months.5,6 No effective therapeutic options currently exist in patients who are resistant to sorafenib. Therefore, it is necessary to elucidate the resistance mechanism and discover effective strategies to improve therapeutic outcomes in HCC patients with sorafenib resistance. The reported mechanisms of sorafenib resistance in HCC mainly focus on changes in molecular targets, such as EGFR, c-Jun, and AKT, and the role of autophagy, apoptosis resistance, hypoxia-inducible factors and cancer stem cells (CSCs).7
CSCs have been proposed to be cancer-initiating cells that are responsible for recurrence, metastasis and chemotherapy resistance,8,9 including liver cancer sorafenib resistance.10,11 Liver cancer stem cells (LCSCs) are a small subpopulation of undifferentiated cells in HCC, and several LCSC markers, such as CD44, EpCAM, CD90, CD133, OV-6, CD24, CD13, CD47, ICAM-1 and DLK1 have been identified and characterized.12 Although LCSCs account for only a small part of the tumor, they are the most difficult part to eradicate with chemotherapy drugs, allowing eventual recurrence.13 Therefore, the study of CSCs is of great clinical significance in improving the therapeutic effect of chemotherapy drugs and overcoming drug resistance.
Interleukin-6 (IL-6) is a major driver of hepatocellular carcinogenesis and is crucial for the development of HCC.14 In the pathogenesis of cancer, elevated IL-6 serum levels cause excessive activation of JAK/STAT3 signaling, which is usually correlated with an increased risk of developing HCC and poor prognosis in patients.15,16 In a very recent study, hepatic stellate cells increased cancer cell viability, migration ability, and stemness and contributed to the tumor malignancy of HCC through the IL-6/STAT3 pathway.17 According to research, liver stem cells with activated STAT3 give rise to HCC due to aberrant IL-6 signaling.18
In the present study, we established a sorafenib-resistant HCC cell line (PLC/PRF/5-R) and isolated LCSCs from this cell line. The stemness and chemoresistance of LCSCs were confirmed both in vitro and in vivo. We also demonstrated that IL-6 was elevated in sorafenib-resistant LCSCs compared to control LCSCs. The expression of IL-6/STAT3 signaling conferred sorafenib resistance in LCSCs. These mechanisms might play an important role in the chemoresistance of HCC patients treated with sorafenib.
Materials and Methods
Cell Lines and Reagents
PLC/PRF/5 cell lines were obtained from the American Type Culture Collection (ATCC) and grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS (HyClone Laboratories Inc., Logan, UT, USA) and 5% antibiotics (penicillin and streptomycin) at 37°C under 5% CO2. Sorafenib was purchased from Bayer Corporation (Nexavar, Beijing, China). Sorafenib was dissolved in 100% dimethyl sulfoxide (DMSO), and the final concentration was 0.5 μM with DMSO 0.1% (v/v).
Antibodies and Constructs
All antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). β-Actin was purchased from Sigma-Aldrich (St Louis, MO, USA), and lentiviral vectors and plasmids expressing IL-6R-shRNA were purchased from Shanghai Gene-Pharma Co.
Generation of Sorafenib-Resistant HCC Cells (PLC/PRF/5-R)
First, we determined the IC50 of PLC/PRF/5 cells for sorafenib. After three days, the cells were incubated with CCK-8 reagent, and cell viability was determined. Next, we cultured PLC/PRF/5 cells in 6-well plates at 5×104 cells/well and incubated the cells with sorafenib concentrations just below the IC50. During the following weeks, we slowly increased the sorafenib dose by 0.5 μM every three days. Over approximately 6 months, we developed PLC/PRF/5 cell lines resistant to sorafenib. After 6 months of continuous stimulation with sorafenib, PLC/PRF/5-R cells developed. PLC/PRF/5-R was maintained by culturing them in the presence of 0.5 μM sorafenib.
Sorting and Grouping of LCSCs
PLC/PRF/5-R cells were resuspended in PBS in the logarithmic growth stage and incubated with FcR-blocking reagent (100 μL) at a cell density of 1×107 cells/100 μL. The cells were then mixed with 100 μL of EpCAM and CD44 antibodies and incubated at 4°C in the dark for 30 min. After centrifugation at 300 g for 10 min, cells were resuspended to create a single cell suspension using buffer solution, sorted by flow cytometry, and collected. After sorting, LCSCs were cultured in serum-free DMEM.
Cellular Viability Assay
Cells cultured in DMEM containing DMSO (0.01%) were used as negative controls, and the medium served as the blank control. The drug-resistant and nonresistant cell lines (100 μL, approximately 5000 cells) were injected into 96-well plates containing the indicated concentrations of sorafenib and cultured at 37°C under 5% CO2 for 24 hours. At the end of incubation, 10 μL of Cell Counting Kit-8 (CCK-8, Sigma, USA) solution was added to each well and cultured for another four hours. The absorbance was measured using a microplate reader (Thermo Scientific, USA) at 450 nm. The half-maximal inhibitory concentration (IC50) was then calculated from growth inhibition rates.
Cell Proliferation Assay
After selection, we plated cells in a 96-well plate and measured the optical density (OD) every 24 hours according to the manufacturer’s protocol. Before each detection, the CCK-8 reagent (10 μL) was added to each well, and an enzyme immunoassay instrument (Bio Rad Laboratories) was used for value readings.
Cell Apoptosis Assay
In accordance with the manufacturer’s protocol, apoptotic cells were analyzed using annexin V/PI double staining at 72 hours after sorting by flow cytometry. The cell suspension (100 μL) was incubated with 5 μL of annexin-V and 1 μL of propidium iodide at room temperature for 10 min. Total stained cells were analyzed using flow cytometry (BD-bio, San Diego, CA, USA).
Cell Transfection
Lentiviral vectors and plasmids expressing IL-6R-shRNA were purchased from Shanghai Gene-Pharma Co. (Shanghai, China). The shRNA was transfected into LCSCs using LipofectamineTM 2000 (Life Technologies) according to the manufacturer’s instructions. Transfection efficiency was determined by RT-qPCR and Western blotting after 24 hours.
Quantitative Real-Time Reverse Transcription PCR
Total RNA was isolated from each sample with TRIzol (Takara Biotechnology Co., Ltd., Dalian, China) and treated with DNase I (Takara Biotechnology Co., Ltd.) to remove residual DNA according to the manufacturer’s protocol. cDNA was reverse synthesized using a Prime-Script-RT reagent kit (Takara Biotechnology Co., Ltd.). SYBR Green qPCR was used to evaluate the mRNA levels of the indicated genes. The primers were synthesized by Takara Biotechnology Co. (Table 1). β-Actin was regarded as the internal control, and the relative transcriptional levels of the target genes were calculated by using the 2−ΔΔCT method. Each experiment was repeated three times.
 |
Table 1 Primers Sequences for RT-qPCR |
Western Blotting
Tissue and cells were lysed in RIPA lysis buffer (Beyotime, Shanghai, China) on ice for 30 min. The cytosolic fraction was collected after centrifugation at 12,000 g/min at 4°C for 15 min. The total protein was quantitatively assayed with a BCA Protein Assay Kit (Thermo Fisher, Shanghai, China). Protein (20 μg) was separated via 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, MA, USA) for Western blot analysis. The experiment was repeated independently three times.
Subcutaneous Xenograft Tumor Model
Nude mice were obtained from Shanghai Gene-Pharma Co. (Shanghai, China) and housed in a standard laboratory with free access to food and water. The study was approved by the Animal Care Committee of Lanzhou University Second Hospital and conducted following the ARRIVE guidelines. For the establishment of the subcutaneous xenograft tumor model, resistant LCSCs were resuspended to a density of 5×105/mL in PBS. Each mouse was subcutaneously injected with 100 μL of cells in the right dorsal area. When the tumor volume reached 150 mm3, the mice received sorafenib (100 mg/kg) or PBS orally once daily for 10 days. At the end of the experiment, mice were euthanized, and tumors were resected for testing gene or protein expression levels.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). The measurement data are presented as the mean±standard deviation (SD) of at least three independent experiments. Differences in mean values between two groups were analyzed by Student’s t-test (unpaired, two-tailed). P<0.05 was considered statistically significant.
Results
The Sorafenib-Resistant Hepatocellular Carcinoma Cell Line PLC/PRF/5-R Was Established and Exhibited EMT Characteristics
To study the mechanism of sorafenib resistance, PLC/PRF/5 cells were used to generate sorafenib-resistant HCC cells. PLC/PRF/5-R displayed a spindle shape and loose cell-cell contact, which is easy to peel off from the surface of a culture bottle (Figure 1A). The IC50 for the parental PLC/PRF/5 cell line was determined to be 5.464 μM, which is in accordance with previous reports,19 and in resistant cell lines, the IC50 (12.18 μM) showed a shift to a higher concentration compared to that in the parental line (Figure 1B). Furthermore, PLC/PRF/5-R cells showed typical EMT characteristics with downregulated E-cadherin and upregulated multidrug resistance gene (MDR) and vimentin compared with PLC/PRF/5 cells (Figure 1C-E). These results implied the successful establishment of sorafenib-resistant PLC/PRF/5 cells.
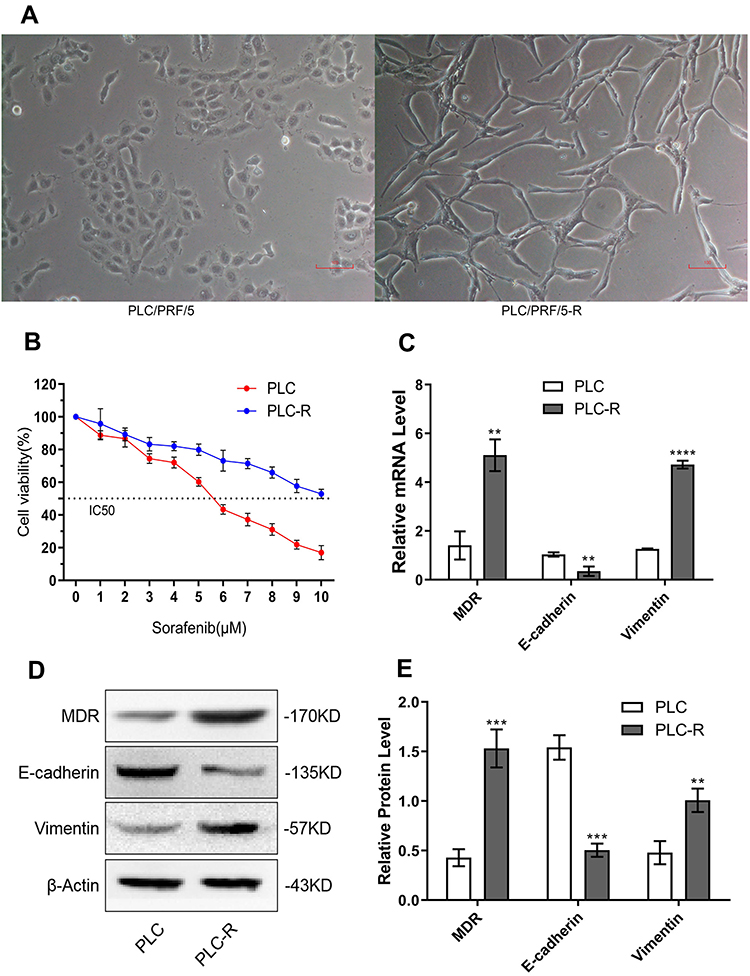 |
Figure 1 The sorafenib-resistant hepatocellular carcinoma cell line PLC/PRF/5-R was established. (A) Morphological characteristics of PLC/PRF/5 and PLC/PRF/5-R by microscopy (×100). (B) For PLC/PRF/5, the IC50 was approximately 5.464 μM; for the resistant cell lines, the IC50 shifted towards 12.18 μM. (C–E) MDR, E-cadherin and vimentin mRNA and protein expression levels in PLC/PRF/5 (PLC) and PLC/PRF/5-R (PLC-R) (**P<0.01, ***P<0.005, ****P<0.0001), PLC/PRF/5-R showed typical EMT characteristics. |
Isolation and Identification of PLC/PRF/5-R CSCs (Resistant LCSCs)
Since CD44 and EpCAM are common LCSC markers, flow cytometry was conducted to sort the cells that expressed CD44 and EpCAM positive cells or negative cells from PLC/PRF/5-R cells (Figure 2A and B). The selected positive (EpCAM+, CD44+) and negative (EpCAM−, CD44−) cells were tested using RT-qPCR. The results demonstrated that the mRNA levels of stem cell markers (EpCAM+, CD44+) in positive cells were higher than those in negative cells (Figure 2C).
 |
Figure 2 LCSC-R isolation and identification. (A) FACS plots demonstrating the isolation of EpCAM+ and CD44+ resistant LCSCs from PLC/PRF/5-R cells. (B) Cell morphology of resistant LCSCs by microscope (×100). (C) The sorted positive and negative cells were assessed using qPCR. (D, E) Cell proliferation and apoptosis assays were performed to compare the positive and negative cells, but there were no statistically significant differences. (F) Nude mouse xenograft experiment (R+: positive (EpCAM+, CD44+) resistant LCSCs, R-: negative (EpCAM−, CD44−)) resistant LCSCs, B+: positive (EpCAM+, CD44+) LCSCs, B-: negative (EpCAM−, CD44−) LCSCs (***P<0.005, ****P<0.0001). |
Subsequently, cell proliferation assays and apoptosis tests were carried out to evaluate the positive and negative cells, but there were no significant differences (Figure 2D and E). To further investigate the tumorigenesis of stem cells in vivo, both LCSCs and PLC/PRF/5-R cells were inoculated into nude mice to generate tumor xenografts. The results showed that the volume of LCSC-derived tumors was larger than that of PLC/PRF/5-R-derived tumors (Figure 2F), suggesting that LCSCs had more aggressive xenograft tumor growth.
The IL-6/STAT3 Signaling Pathway Proteins are Highly Expressed in Resistant LCSCs and are Pivotal for Maintaining the Stemness of Resistant LCSCs
The expression of IL-6, IL-6R, gp130 and STAT3 was higher in resistant LCSCs than in control cells (LCSCs) (Figure 3A). STAT3 protein expression in resistant LCSCs was enhanced compared with that in LCSCs (Figure 3B and C). The results demonstrated that the IL-6/STAT3 signaling pathway is significantly activated in resistant LCSCs.
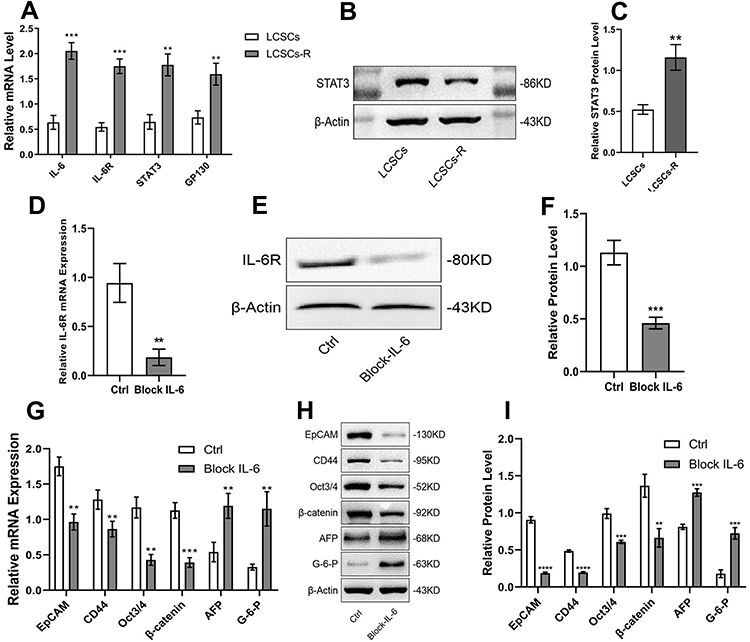 |
Figure 3 Blocking the IL-6/STAT3 signaling pathway in resistant LCSCs. (A) IL-6, IL-6R, STAT3 and GP130 mRNA levels in LCSCs and resistant LCSCs. (B, C) STAT3 protein expression in LCSCs and resistant LCSCs. (D–F) To confirm that IL-6 signaling was blocked in resistant LCSCs by qPCR and WB. (G–I) General stem cell markers (EpCAM and CD44), stemness markers (Oct3/4 and β-catenin), and hepatocyte differentiation markers (G-6-P and AFP) were detected using qPCR and WB (**P<0.01, ***P<0.005, ****P<0.0001), and there were significant differences. |
Blocking IL-6 with shRNA significantly downregulated IL-6R (Figure 3D-F), epithelial cell adhesion molecule (EpCAM) and key genes related to pluripotency (Oct3/4, β-catenin) and stemness (CD44). It also significantly upregulated hepatocyte differentiation markers (glucose-6-phosphate, AFP) (Figure 3G-I). These results illustrate that downregulation of IL-6/STAT3 diminished the expression of stemness-related genes of resistant LCSCs.
Sorafenib Combined with IL-6 Blocking Enhances Antitumor Effects in Xenograft Tumors
To further elucidate the role of IL-6 in LCSCs, a tumor xenograft model was generated in nude mice in vivo to examine tumorigenicity. The results showed that tumors in the shRNA IL-6 group grew slower and exhibited a smaller volume that those in the control group (Figure 4A). The sorafenib treatment combined with shRNA IL-6 group had a smaller tumor volume than the shRNA IL-6 group (Figure 4B). The tumor tissue IL-6 and STAT3 levels were measured by Western blotting (Figure 4C and D). LCSC markers (EpCAM, CD44), stemness markers (Oct3/4, β-catenin) and angiogenic factors (VEGF, VEGFR) were also examined in tumor tissue from nude mice. The results indicated that LCSC markers, stemness markers and angiogenic factors were downregulated in the shRNA IL-6 group (Figure 4E and F).
 |
Figure 4 Establishment of subcutaneous xenografts in nude mice using resistant LCSCs, which block the IL-6/STAT3 signaling pathway. (A) Tumor images of all subcutaneous xenografts in nude mice. Each indicated treatment group included 5 mouse tumors. (B) Growth curves of xenograft tumors from day 1 to day 11 in various treatment groups. (C, D) Analysis of IL-6R and STAT3 protein expression in xenograft tumors to confirm that the IL-6/STAT3 signaling pathway was blocked. (E, F) The LCSC markers (EpCAM and CD44), stemness markers (Oct3/4 and β-catenin) and angiogenic factors (VEGF and VEGFR) were assessed by WB (Sor-Block-IL-6): subcutaneous xenograft tumors in nude mice were established using resistant LCSCs, which blocked IL-6 signaling, treated with sorafenib, 100 mg/kg/d; sor: subcutaneous xenograft tumors in nude mice were established using resistant LCSCs, in which IL-6 signaling is NOT blocked, and sorafenib as the treatment, 100 mg/kg/d; PBS: subcutaneous xenograft tumors were established in nude mice using resistant LCSCs, in which IL-6 signaling is blocked, and PBS as the treatment (ns P>0.05, *P<0.05, **P<0.01, ***P<0.005). |
All the above results highlighted the important role of IL-6 in the maintenance of the stemness of LCSCs since IL-6 knockdown diminished the expression of stemness-related genes, suppressed the viability and tumorigenesis of LCSCs and sensitized PLC/PRF/5-R cells resistance to sorafenib.
Discussion
Sorafenib is the only standard clinical treatment against advanced HCC. One of the important reasons is the emergence of resistance during treatment. However, primary or acquired resistance to sorafenib often develops within 6 months, and only approximately 30% of HCC patients benefit from sorafenib due to treatment resistance. Based on the current situation of drug resistance to sorafenib, we established the sorafenib-resistant liver cancer cell line PLC/PRF/5-R. Our research confirmed that E-cadherin and vimentin levels in PLC/PRF/5-R cells are significantly different from those in control cells, which suggests that EMT promotes the drug resistance process.
The mechanism of sorafenib resistance is still elusive. However, increasing evidence suggests that CSCs may be one of the major factors in the drug resistance, metastasis and recurrence of cancer.20 In fact, drug-resistant cell populations have more CSCs,21,22 which are more resistant to conventional cancer therapies than the non-CSC subpopulation.23 CSCs mediate patients’ drug resistance through several different mechanisms. Studies of pancreatic cancer have revealed that there is a definite correlation between EMT-activated CSCs and tumor drug resistance.24 Similar to these findings, pretreatment of NSCLC cells with the gamma-secretase inhibitor GSI increased the sensitivity of anti-CD133+ non-small-cell lung cancer (NSCLC) stem cells to doxorubicin and paclitaxel.25 In the case of NSCLC, CD133+ CSCs may exhibit resistance by increasing PI3K/Akt and Bcl-2 expression.26 The expression of sonic hedgehog (SHH) and glioma-associated oncogene homolog 1 (GLI1), the most well-known signaling pathway molecules involved in drug resistance, is higher in enriched CD44+/Musashi-1+ gastric cancer stem cells, which consequently enhances drug resistance through high drug efflux pump activity.27 Research has reported that anti-CDK1 treatment can enhance sorafenib antitumor responses in a patient-derived tumor xenograft model by targeting CSCs, revealing the direct effects of CSCs on sorafenib resistance.28 Studies have shown that hypoxia can induce sorafenib resistance,29 and it was found that hypoxia may increase the HCC CSC population by altering androgen receptor/miR-520f-3p/SOX9 signaling.30 Combined sorafenib with silibinin significantly decreased the growth and induced the apoptosis of HCC cells with enhanced inhibition of the STAT3/ERK/ATK pathways by targeting CSCs.31 Recent research has reported that activation or overexpression of AMP-activated kinase (AMPK) decreases the characteristics of CSCs in HCC, restoring sensitivity to sorafenib.32 The above studies have shown the importance of CSCs in tumor drug resistance. Combining these reports, we sorted EpCAM+ and CD44+ subpopulations from PLC/PRF/5-R by flow cytometry according to common surface biomarkers of liver cancer stem cells. Interestingly, the observed differences between the positive and negative cells in the cell proliferation assay and apoptosis test study were not significant. However, the difference between the positive and negative cells in the xenograft tumor experiment was significant. This interesting result may be related to the stem cell characteristics of positive cells: the role of LCSCs in the sorafenib resistance of HCC is clear. We further investigated the role of the IL-6/STAT3 signaling pathway in resistant LCSCs.
The inflammatory cytokine IL-6 is known to be involved in the pathogenesis and progression of many types of cancer, including colorectal,33 ovarian,34 breast,35 prostate,36 lung,37 pancreatic,38 and head and neck cancers.39 In this context, IL-6 may be produced by the tumor cells themselves, in addition to infiltrating stromal cells of mesenchymal and hematological origin (macrophages and T cells).40 The dysregulated expression of IL-6 and downstream receptor signaling abnormalities is a common occurrence in cancer and is associated with poor clinical prognosis. Cancer-associated fibroblasts (CAFs) secrete high levels of IL-6, which promotes stem cell-like properties in HCC by activating Notch/STAT3 signaling.41 In this study, we found that the IL-6 signaling pathway in resistant LCSCs was obviously activated. Recent research also shows that IL-6 plays an important role in tumor development and the conversion of non-CSCs to CSCs.42,43 We demonstrated that the sorafenib resistance of LCSCs was dependent on IL-6 produced by HCC. The IL-6/STAT3 signaling cascade regulated the sorafenib resistance of LCSCs, and these effects were inhibited by the shRNA block of IL-6. Another study showed that IL-6 knockdown can increase the chemo-drug efficacy of cisplatin, inhibit tumor growth and reduce the potential for tumor recurrence and metastasis in laryngeal cancer via ALDH+ and CD44+ CSCs.44 Our research showed that the stemness properties of resistant LCSCs were weakened after blocking IL-6 signaling, and animal experiments also demonstrated that the expression of tumor angiogenesis-related factors that block IL-6 signaling is significantly reduced and tumor growth is inhibited. Thus, suppression of the IL-6/STAT3 signaling pathway by targeting CSCs in sorafenib-resistant HCC is probably a promising approach for advanced treatment and personalized management of patients.
Conclusion
In summary, our results demonstrate that LCSCs play a pivotal role in sorafenib-resistant HCC through the IL-6/STAT3 signaling pathway. Targeting IL-6 in LCSCs represents an effective therapeutic approach for overcoming acquired resistance to sorafenib in HCC patients.
Abbreviations
AFP, alpha fetoprotein; ANOVA, analysis of variance; CD44, cluster of differentiation 44; CSCs, cancer stem cells; EpCAM, epithelial cell adhesion molecule; G-6-P, glucose-6-phosphate; gp130, human glycoprotein 130; HCC, hepatocellular carcinoma; IL-6, interleukin 6; IL-6R, interleukin 6 receptor; JAK, Janus kinase; LCSCs, liver cancer stem cells; resistant LCSCs, sorafenib-resistant liver cancer stem cells; MDR, multidrug resistance gene; Oct3/4, octamer-binding transcription factor 3/4; PBS, phosphate-buffered saline; PDGF, platelet-derived growth factor; STAT3, signal transducer and activator of transcription 3; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Acknowledgments
The work was kindly supported by the National Natural Science Foundation of China (No. 81572437) and the Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (No. CY2017-MS05).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Johnson PJ, Berhane S, Kagebayashi C, et al. Assessment of liver function in patients with hepatocellular carcinoma: a new evidence-based approach-the ALBI grade. J Clin Oncol. 2015;33(6):550–558. doi:10.1200/JCO.2014.57.9151
3. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62(6):394–399. doi:10.3322/caac.21161
4. Kudo M. Systemic therapy for hepatocellular carcinoma: 2017 update. Oncology. 2017;93(Suppl 1):135–146. doi:10.1159/000481244
5. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
6. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a Phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi:10.1016/S1470-2045(08)70285-7
7. Niu L, Liu L, Yang S, Ren J, Lai P, Chen GG. New insights into sorafenib resistance in hepatocellular carcinoma: responsible mechanisms and promising strategies. Biochim Biophys Acta Rev Cancer. 2017;1868(2):564–570. doi:10.1016/j.bbcan.2017.10.002
8. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–728.
9. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi:10.1038/onc.2010.215
10. Shi DM, Bian XY, Qin CD, Wu WZ. miR-106b-5p promotes stem cell-like properties of hepatocellular carcinoma cells by targeting PTEN via PI3K/Akt pathway. Onco Targets Ther. 2018;11:571–585. doi:10.2147/OTT.S152611
11. Cheng -C-C, Chao W-T, Liao -C-C, et al. The roles of angiogenesis and cancer stem cells in sorafenib drug resistance in hepatocellular carcinoma. Onco Targets Ther. 2019;12:8217–8227. doi:10.2147/OTT.S217468
12. Cheng Z, Li X, Ding J. Characteristics of liver cancer stem cells and clinical correlations. Cancer Lett. 2016;379(2):230–238. doi:10.1016/j.canlet.2015.07.041
13. Xin HW, Ambe CM, Hari DM, et al. Label-retaining liver cancer cells are relatively resistant to sorafenib. Gut. 2013;62(12):1777–1786. doi:10.1136/gutjnl-2012-303261
14. Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–1415. doi:10.1016/j.jhep.2016.02.004
15. Chen Y, Wang J, Wang X, et al. STAT3, a poor survival predicator, is associated with lymph node metastasis from breast cancer. J Breast Cancer. 2013;16(1):40–49. doi:10.4048/jbc.2013.16.1.40
16. Macha MA, Matta A, Kaur J, et al. Prognostic significance of nuclear pSTAT3 in oral cancer. Head Neck. 2011;33(4):482–489. doi:10.1002/hed.21468
17. Iwahasi S, Rui F, Morine Y, et al. Hepatic stellate cells contribute to the tumor malignancy of hepatocellular carcinoma through the IL-6 pathway. Anticancer Res. 2020;40(2):743–749. doi:10.21873/anticanres.14005
18. Tang Y, Kitisin K, Jogunoori W, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105(7):2445–2450. doi:10.1073/pnas.0705395105
19. van Malenstein H, Dekervel J, Verslype C, et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013;329(1):74–83. doi:10.1016/j.canlet.2012.10.021
20. Hou Y, Zhu Q, Li Z, et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017;8(3):e2659. doi:10.1038/cddis.2017.53
21. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911–1918. doi:10.1172/JCI66024
22. Tovar V, Cornella H, Moeini A, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut. 2017;66(3):530–540. doi:10.1136/gutjnl-2015-309501
23. Peng Y, He G, Tang D, et al. Lovastatin inhibits cancer stem cells and sensitizes to chemo- and photodynamic therapy in nasopharyngeal carcinoma. J Cancer. 2017;8(9):1655–1664. doi:10.7150/jca.19100
24. Meidhof S, Brabletz S, Lehmann W, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7(6):831–847. doi:10.15252/emmm.201404396
25. Liu YP, Yang CJ, Huang MS, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;73(1):406–416. doi:10.1158/0008-5472.CAN-12-1733
26. Sarvi S, Mackinnon AC, Avlonitis N, et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014;74(5):1554–1565. doi:10.1158/0008-5472.CAN-13-1541
27. Xu M, Gong A, Yang H, et al. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett. 2015;369(1):124–133. doi:10.1016/j.canlet.2015.08.005
28. Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8(14):3737–3750. doi:10.7150/thno.25487
29. Méndez-Blanco C, Fondevila F, Fernández-Palanca P, et al. Stabilization of hypoxia-inducible factors and bnip3 promoter methylation contribute to acquired sorafenib resistance in human hepatocarcinoma cells. Cancers (Basel). 2019;11:12. doi:10.3390/cancers11121984
30. Xiao Y, Sun Y, Liu G, et al. Androgen receptor (AR)/miR-520f-3p/SOX9 signaling is involved in altering hepatocellular carcinoma (HCC) cell sensitivity to the Sorafenib therapy under hypoxia via increasing cancer stem cells phenotype. Cancer Lett. 2019;444:175–187. doi:10.1016/j.canlet.2018.11.004
31. Mao J, Yang H, Cui T, et al. Combined treatment with sorafenib and silibinin synergistically targets both HCC cells and cancer stem cells by enhanced inhibition of the phosphorylation of STAT3/ERK/AKT. Eur J Pharmacol. 2018;832:39–49. doi:10.1016/j.ejphar.2018.05.027
32. Bort A, Sánchez BG, Mateos-Gómez PA, Vara-Ciruelos D, Rodríguez-Henche N, Díaz-Laviada I. Targeting AMP-activated kinase impacts hepatocellular cancer stem cells induced by long-term treatment with sorafenib. Mol Oncol. 2019;13(5):1311–1331. doi:10.1002/1878-0261.12488
33. Waldner MJ, Foersch S, Neurath MF. Interleukin-6–a key regulator of colorectal cancer development. Int J Biol Sci. 2012;8(9):1248–1253. doi:10.7150/ijbs.4614
34. Macciò A, Madeddu C. The role of interleukin-6 in the evolution of ovarian cancer: clinical and prognostic implications–a review. J Mol Med (Berl). 2013;91(12):1355–1368. doi:10.1007/s00109-013-1080-7
35. Dethlefsen C, Højfeldt G, Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat. 2013;138(3):657–664. doi:10.1007/s10549-013-2488-z
36. Culig Z, Puhr M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol. 2012;360(1–2):52–58.
37. Chang CH, Hsiao CF, Yeh YM, et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer. 2013;132(9):1977–1985. doi:10.1002/ijc.27892
38. Miura T, Mitsunaga S, Ikeda M, et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas. 2015;44(5):756–763. doi:10.1097/MPA.0000000000000335
39. Mojtahedi Z, Khademi B, Hashemi SB, et al. Serum interleukine-6 concentration, but not interleukine-18, is associated with head and neck squamous cell carcinoma progression. Pathol Oncol Res. 2011;17(1):7–10. doi:10.1007/s12253-010-9261-y
40. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70(Suppl 1):i104–108. doi:10.1136/ard.2010.140145
41. Xiong S, Wang R, Chen Q, et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018;8(2):302–316.
42. Wan S, Zhao E, Kryczek I, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393–1404. doi:10.1053/j.gastro.2014.08.039
43. Wang CQ, Sun HT, Gao XM, et al. Interleukin-6 enhances cancer stemness and promotes metastasis of hepatocellular carcinoma via up-regulating osteopontin expression. Am J Cancer Res. 2016;6(9):1873–1889.
44. Fu Q, Liu P, Sun X, et al. Ribonucleic acid interference knockdown of IL-6 enhances the efficacy of cisplatin in laryngeal cancer stem cells by down-regulating the IL-6/STAT3/HIF1 pathway. Cancer Cell Int. 2017;17:79. doi:10.1186/s12935-017-0448-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.