")
Back to Journals » International Medical Case Reports Journal » Volume 7
Idiopathic CD4+ lymphocytopenia in Hispanic male: case report and literature review
Authors Said S, Alkhateeb HA, Cooper CJ, Rodriguez E, Trien R, Hernandez GT, Salameh HS
Received 10 March 2014
Accepted for publication 27 March 2014
Published 30 July 2014 Volume 2014:7 Pages 117—120
DOI https://doi.org/10.2147/IMCRJ.S63836
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Sarmad Said,1 Haider Alkhateeb,1 Chad J Cooper,1 Emmanuel Rodriguez,1 Remi Trien,1 German T Hernandez,1,2 Hasan S Salameh1,2
1Department of Internal Medicine, 2Department of Internal Medicine, Division of Nephrology and Hypertension, Paul L Foster School of Medicine, Texas Tech University Health Sciences Center at El Paso, El Paso, TX, USA
Introduction: Idiopathic cluster of differentiation 4 (CD4+) T-cell lymphocytopenia (ICL) is a rare non human immunodeficiency virus (HIV)-related syndrome with unclear natural history and prognosis that was first reported and defined in 1992. ICL has been observed in patients after the onset of an opportunistic infection without known immunosuppression.
Case presentation: A 20-year-old Hispanic male patient without significant past medical history presented with progressive shortness of breath and cough for 3 weeks. Chest computed tomography showed bilateral cavitary lesions in the upper lung lobes. The HIV rapid screening test as well as the sputum acid-fast bacilli test were both positive. The patient was started on antituberculosis therapy. The CD4 count was noticed to be low. However, the HIV Western blot test was negative, and the HIV viral load was within normal limit. Further radiologic studies, hemato-oncologic, and autoimmune workups were normal. The patient was discharged on the treatment for tuberculosis. Follow-up after 8 weeks revealed a persistent low CD4+ count, and the repeated HIV tests were negative.
Conclusion: The clinical features of ICL range from an asymptomatic condition to life-threatening complications that imitate the clinical course of HIV-infected patients. The differential diagnosis in adults comprises primarily HIV infection and other diseases or drug side effects. ICL is very rare and should be considered in the absence of any defined immunodeficiency or therapy associated with depressed levels of CD4+ T-cells. Early detection and recognition of the disease allow purposeful and systemic treatment approach and screening for the affected patients.
Keywords: CD4 count, immunodeficiency, lymphocytopenia, opportunistic infection
Introduction
Idiopathic cluster of differentiation 4 (CD4)+ lymphocytopenia (ICL) was first defined in 1992 by the US Centers for Disease Control and Prevention (CDC)1 as:
…a documented absolute CD4 T-lymphocyte count of <300 cells/mm3 or <20% of total T-cells on two separate time points at least six weeks apart without evidence of infection on HIV-1 or HIV-2 testing, and without immunodeficiency or therapy related to decrease of CD4 T-cells…
The CDC definition allows the inclusion of patients with pan-lymphocytopenia and normal CD4:CD8 ratio.
ICL is a very rare syndrome with an unknown etiology, not caused by human immunodeficiency virus (HIV)-1, HIV-2, human T-lymphotropic virus type I, or human T-lymphotropic virus type II, and not appearing to be caused by any transmissible agent.2 Sporadic ICL cases are reported from all over the world. There is no apparent predilection to one geographical area. ICL has been observed in patients after the onset of opportunistic infection without known immunocompromisation or immunosuppression.
We are reporting a case of a young Hispanic male who presented with progressive dry cough and shortness of breath, which turned out to be a case of ICL with pulmonary tuberculosis.
Case presentation
A 20-year-old Hispanic male with no past medical history presented to our institution complaining of generalized weakness for 4 weeks, progressive cough with yellowish sputum, as well as persistent shortness of breath for 3 weeks, and intermittent fever and chills for 3–5 days prior to admission. Family history was remarkable only for coronary artery disease in his father. He denied smoking, alcohol, and using illegal drugs. He had lived with his girlfriend for 2 years and stated to be monogamous over this period. He reported a total of two sexual partners in his life. He was born in Texas and had not traveled outside the US for the last 12 years. Physical examination revealed a cachectic and tachypneic patient in no acute distress with bilateral diffuse crackles and rhonchi. The initial laboratory workup was within normal limits. Computed tomography of the chest (Figure 1) showed diffuse tree-in-bud nodularity, bronchiectasis, and thick-walled cavities in the upper lobes and moderate right simple pleural effusion. The HIV-rapid enzyme-linked immunosorbent assay test was reactive. The acid-fast bacilli sputum test was positive, and the patient was started on combination treatment for tuberculosis. The patient improved clinically over the next 3 days. The HIV Western blot test was negative; the viral load was undetectable. The CD4+ and CD8 counts were low (124 and 185, respectively) with a low CD4:CD8 ratio of 0.7. Further workup including immunoglobulin A, immunoglobulin G, and immunoglobulin M levels, urine protein electrophoresis, and serum protein electrophoresis was unremarkable. The pleural fluid cytology revealed atypical lymphocytes consistent with the established diagnosis of pulmonary tuberculosis. Further imaging of the abdomen and pelvis showed no abnormalities.
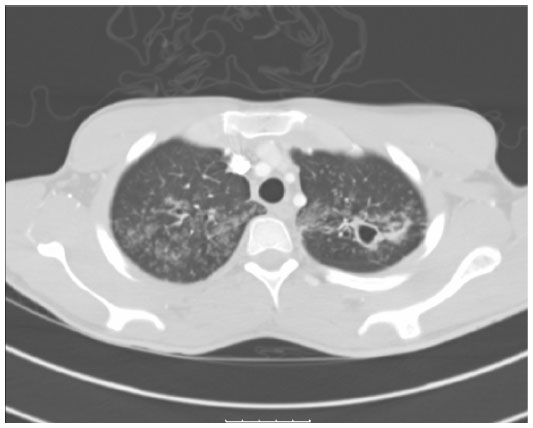 | Figure 1 CT of the chest (in lung window) showing multiple thick-walled cavities in both upper lobes with diffuse tree-in-bud nodularities. |
In addition, 8 weeks after discharge the patient was seen in clinic. He was taking his medication to treat tuberculosis and had regular health care follow-up. He was found to be completely asymptomatic with a CD4+ count of 188 cells/mm3. The repeated HIV test was negative, and the viral load was undetectable. After finishing his treatment for tuberculosis, he was evaluated in clinic, had a CD4+ count of 212 cells/mm3, and remained asymptomatic.
Discussion
With the increased incidence of acquired immunodeficiency syndrome and the related risk of opportunistic infections, the public attention was higher to investigate more about possible other associations. The CD4+ T-lymphocyte is reduced in HIV infection as well as in ICL, and the opportunistic infections that affect patients with ICL are similar to those in HIV-positive patients with low CD4+ T-lymphocyte counts.3
In our patient, the initial presentation with a false-positive HIV test might be attributed to the existing infection with tuberculosis. Several reports described this association over the last 20 years.4,5 However, HIV screening must be considered in the presence of tuberculosis or other opportunistic infections in a low-risk patient, as in our case.
In 1993, the CDC reviewed 23,179 cases in its acquired immunodeficiency syndrome reporting registry and discovered 47 cases that met the diagnostic criteria for ICL. There was no detectable bias of the age at diagnosis or in the sex of the patients (29 males and 18 females).6 No cases of ICL were identified among the 2,028 screened blood donors.7
The etiology of ICL is unknown and not clearly understood. A few observational studies have suggested the role of various immune defects and their association to CD4+ lymphocyte function. Apoptosis of CD4+ lymphocytes or the activation of CD4+ due to an unidentified pathogen stimulation could be factors that result in the development of ICL.8,9 Genetic predisposition may also be involved. However, very few familial cases have been reported who presented with ICL.10,11 The human leukocyte antigen-D (HLA-D) related CD4+ cells were discovered to be higher in patients with ICL compared to the control group.8 ICL-affected patients tend to have a progressive decline of CD4+ lymphocyte cell count; whereas, HIV-infected patients have a slower decline of CD4+ lymphocyte cell count. Several reports have demonstrated the expansion of γδ T-cells as a result of T-cell redistribution or concomitant infection.12,13
A differential diagnosis for CD4+ lymphocytopenia includes: autoimmune disorders; malignancy; infectious causes; medication-induced; or other primary immunodeficiency diseases or syndromes.14–18 The HIV infection is the most common differential diagnosis of low CD4+ lymphocytopenia. Bacterial, fungal, parasitic, and viral infections can depress the CD4+ count; however, the CD4:CD8 ratio remains unaffected.19,20 Cryptococcus is the most common cause of opportunistic infection in ICL, followed by Mycobacterium and herpes zoster virus. Kaposi’s sarcoma in ICL patients is associated with a severe reduction of CD4+ counts in combination with hypogammaglobulinemia or steroid therapy.21,22
Hematological malignancies have also been proposed to cause CD4+ lymphocytopenia. Ayoub et al described the association between non-Hodgkin’s lymphoma (mucosa-associated lymphatic tissue, large cell lymphoma, and Burkitt’s lymphoma) with lymphocytopenia and a normal CD4:CD8 ratio.23 Patients with ICL may have a predisposition for developing cervical neoplasias, depending on the human papillomavirus type.
According to several studies, ICL was present in patients with various autoimmune disorders.24–26 The risk of opportunistic infections in autoimmune patients with CD4+ lymphocytopenia is not known. Glück et al demonstrated in a cross-sectional study of 97 patients with autoimmune disease that the effect of immunosuppressive therapy is associated with higher infection rate and decrease of CD4+ T-lymphocyte cell counts.27 Cyclophosphamide caused a more profound decrease in lymphocytes and CD4+ cells (<250 cells/mL) compared to methotrexate or azathioprine.
The therapy of ICL includes treating the underlying cause. Prophylactic treatment for opportunistic infections is considered an experimental approach. A few reports have demonstrated an improved CD4+ T-lymphocyte cell count with antimycobacterial treatment in ICL patients with mycobacterial infections.28 ICL seems to persist despite treatment and remission in those with an autoimmune disorder.
Pneumococcal prophylaxis is recommended when the CD4+ T-cell count is <200 cells/uL.29 Lifetime prophylaxis is indicated for cryptococcosis and relapsing herpes infection. Females should be screened for cervical neoplasia every 6 months. Prophylaxis and treatment for hepatitis B and hepatitis C infection should be similar to the established recommendations for patients with HIV infection.
Conclusion
Clinical features of ICL range from being asymptomatic to life-threatening complications that are similar to the clinical course of patients with HIV infection. The simultaneous occurrence of opportunistic infections often permits the detection of new onset of ICL. Established guidelines for the treatment of idiopathic and non-HIV-related secondary CD4+ lymphocytopenia are not available, and current recommendations are controversial. This is mainly due to its rarity and unknown etiology or natural disease course. ICL should be considered in the absence of any immunodeficiency disorder or immunosuppressive therapy associated with decreased CD4+ T-lymphocyte cell count. Early consideration and recognition of the disease can allow for a more purposeful and systemic approach to screening and therapy.
Author contributions
SS was the primary physician in the team, the one who took care of the patient and arranged all required medical steps. He wrote the draft of the manuscript. CJC was the main physician who collected literature and reviewed the final version of the manuscript. ER took care of the patient as an outpatient and contributed in updating the team about the patient’s condition. He also reviewed the final version, edited it, and approved it. RT analyzed the manuscript and performed independent proofreading. GTH is a senior physician who analyzed the manuscript and performed independent proofreading. HSS is the senior physician of the team who controlled all the above-mentioned steps and made the final medical decisions. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work. No funding source has been obtained for this case report.
References
Centers for Disease Control (CDC). Unexplained CD4+ T-lymphocyte depletion in persons without evident HIV infection – United States. MMWR Morb Mortal Wkly Rep. 1992;41(30):541–545. | |
Fauci AS. CD4+ T-lymphocytopenia without HIV infection – no lights, no camera, just facts. N Engl J Med. 1993;328(6):429–431. | |
Walker UA, Warnatz K. Idiopathic CD4 lymphocytopenia. Curr Opin Rheumatol. 2006;18(4):389–395. | |
Ribeiro TT, Brites C, Moreira ED, et al. Serologic validation of HIV infection in a tropical area. J Acquir Immune Defic Syndr. 1993;6(3):319–322. | |
Werneck-Barroso E, Kritski AL, Vieira MA, et al. Tuberculosis as a cause of false-positive results in HIV screening EIA tests. Tuber Lung Dis. 1994;75(5):394–395. | |
Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. An investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328(6):373–379. | |
Busch MP, Valinsky JE, Paglieroni T, et al. Screening of blood donors for idiopathic CD4+ T-lymphocytopenia. Transfusion. 1994;34(3):192–197. | |
Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112(2):287–294. | |
Roger PM, Bernard-Pomier G, Counillon E, Breittmayer JP, Bernard A, Dellamonica P. Overexpression of Fas/CD95 and Fas-induced apoptosis in a patient with idiopathic CD4+ T lymphocytopenia. Clin Infect Dis. 1999;28(5):1012–1016. | |
Lin SJ, Chao HC, Yan DC, Kuo ML. Idiopathic CD4+ T lymphocytopenia in two siblings. Pediatr Hematol Oncol. 2001;18(2):153–156. | |
Freier S, Kerem E, Dranitzki Z, et al. Hereditary CD4+ T lymphocytopenia. Arch Dis Child. 1998;78(4):371–372. | |
Airò P, Caruso A, Stellini R, et al. Characterization of gammadelta T cells expressing CD158b, a killer cell inhibitory receptor, in a patient with chronic CD4(+) lymphocytopenia and disseminated Mycobacterium intracellulare infection. Clin Immunol. 2000;96(1):67–75. | |
Tassinari P, Deibis L, Bianco N, Echeverria de Pérez G. Lymphocyte subset diversity in idiopathic CD4+ T lymphocytopenia. Clin Diagn Lab Immunol. 1996;3(5):611–613. | |
Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32(6):1063–1064. | |
Petersen EJ, Rozenberg-Arska M, Dekker AW, Clevers HC, Verdonck LF. Allogeneic bone marrow transplantation can restore CD4+ T-lymphocyte count and immune function in idiopathic CD4+ T-lymphocytopenia. Bone Marrow Transplant. 1996;18(4):813–815. | |
Hequet O, Salles G, Espinousse D, et al. Multifocal progressive leukoencephalopathy occurring after refractory anemia and multiple infectious disorders consecutive to severe lymphopenia. Ann Hematol. 2002;81(6):340–342. | |
Kirtava Z, Blomberg J, Bredberg A, Henriksson G, Jacobsson L, Manthorpe R. CD4+ T-lymphocytopenia without HIV infection: increased prevalence among patients with primary Sjögren’s syndrome. Clin Exp Rheumatol. 1995;13(5):609–616. | |
Winfield JB, Winchester RJ, Kunkel HG. Association of cold-reactive antilymphocyte antibodies with lymphopenia in systemic lupus erythematosus. Arthritis Rheum. 1975;18(6):587–594. | |
Fantin B, Joly V, Elbim C, et al. Lymphocyte subset counts during the course of community-acquired pneumonia: evolution according to age, human immunodeficiency virus status, and etiologic microorganisms. Clin Infect Dis. 1996;22(6):1096–1098. | |
Laurence J. T-cell subsets in health, infectious disease, and idiopathic CD4+ T lymphocytopenia. Ann Intern Med. 1993;119(1):55–62. | |
Ben Rejeb A, Ebdelli N, Bouali MR, et al. [Primary digestive tract Kaposi sarcoma with idiopathic CD4+ lymphocytopenia, HIV negative, HHV8 positive]. Gastroenterol Clin Biol. 2001;25(6–7):707–710. French. | |
García-Silva J, Almagro M, Peña C, et al. CD4+ T-lymphocytopenia, Kaposi’s sarcoma, HHV-8 infection, severe seborrheic dermatitis, and onychomycosis in a homosexual man without HIV infection. Int J Dermatol. 1999;38(3):231–233. | |
Ayoub JP, Palmer JL, Huh Y, Cabanillas F, Younes A. Therapeutic and prognostic implications of peripheral blood lymphopenia in patients with Hodgkin’s disease. Leuk Lymphoma. 1999;34(5–6):519–527. | |
Henriksson G, Manthorpe R, Bredberg A. Antibodies to CD4 in primary Sjögren’s syndrome. Rheumatology (Oxford). 2000;39(2):142–147. | |
Smolen JS, Chused TM, Leiserson WM, Reeves JP, Alling D, Steinberg AD. Heterogeneity of immunoregulatory T-cell subsets in systemic lupus erythematosus. Correlation with clinical features. Am J Med. 1982;72(5):783–790. | |
Morimoto C, Reinherz EL, Distaso JA, Steinberg AD, Schlossman SF. Relationship between systemic lupus erythematosus T cell subsets, anti-T cell antibodies, and T cell functions. J Clin Invest. 1984;73(3):689–700. | |
Glück T, Kiefmann B, Grohmann M, Falk W, Straub RH, Schölmerich J. Immune status and risk for infection in patients receiving chronic immunosuppressive therapy. J Rheumatol. 2005;32(8):1473–1480. | |
Turett GS, Telzak EE. Normalization of CD4+ T-lymphocyte depletion in patients without HIV infection treated for tuberculosis. Chest. 1994;105(5):1335–1337. | |
Duncan RA, von Reyn CF, Alliegro GM, Toossi Z, Sugar AM, Levitz SM. Idiopathic CD4+ T-lymphocytopenia – four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328(6):393–398. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.