Back to Journals » International Medical Case Reports Journal » Volume 18
Idiopathic Acute Exudative Polymorphous Vitelliform Maculopathy: A Case Report
Authors Faria Pereira A ![]() , Tavares-Ferreira J, Santos-Silva R, Oliveira-Ferreira C
, Tavares-Ferreira J, Santos-Silva R, Oliveira-Ferreira C
Received 20 November 2024
Accepted for publication 31 January 2025
Published 17 February 2025 Volume 2025:18 Pages 255—263
DOI https://doi.org/10.2147/IMCRJ.S507569
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Ana Faria Pereira,1 João Tavares-Ferreira,1 Renato Santos-Silva,1,2 Cláudia Oliveira-Ferreira1,3
1Department of Ophthalmology, Unidade Local de Saúde de São João, Porto, Portugal; 2Department of Sense Organs, Faculty of Medicine, University of Porto, Porto, Portugal; 3Department of Surgery and Physiology, Faculty of Medicine, University of Porto, Porto, Portugal
Correspondence: Ana Faria Pereira, Department of Ophthalmology of Local Health Unit of São João, Avenida Prof. Hernâni Monteiro, Porto, 4202-451, Portugal, Email [email protected]
Purpose: Acute exudative polymorphous vitelliform maculopathy (AEPVM) is a rare retinal disorder first described by Gass in 1988. Characterized by multifocal yellow-white lesions at the level of the retinal pigment epithelium (RPE) and associated with serous retinal detachments, AEPVM typically presents with acute visual disturbances and, in some cases, headaches. Despite its rarity, with fewer than 40 cases reported in the literature, its clinical course and etiology remain poorly understood. Patients generally experience gradual recovery of vision, though retinal abnormalities may persist. Various hypotheses suggest infectious, inflammatory, autoimmune, or even paraneoplastic mechanisms, but definitive causes remain elusive. In this report, we describe the clinical course and multimodal imaging findings of a patient diagnosed with AEPVM, contributing to the limited understanding of this condition’s progression and management.
Case Presentation: We report the case of a 40-year-old Caucasian female presenting with blurred vision for two weeks, with a best corrected visual acuity of 20/20 in both eyes, preceded by flu-like symptoms. Initial clinical evaluation, including fundus examination and spectral-domain optical coherence tomography (SD-OCT), revealed multiple small serous retinal detachments bilaterally. Multimodal imaging (fundus autofluorescence, fluorescein angiography, and indocyanine green angiography) appeared normal initially. A comprehensive systemic workup excluded autoimmune, infectious, and neoplastic etiologies. Upon follow-up, yellowish retinal lesions and hyperautofluorescence emerged, leading to the diagnosis of idiopathic AEPVM. The patient continues to be monitored for visual recovery and potential complications.
Conclusion: AEPVM is a rare macular disorder that requires comprehensive multimodal evaluation to establish an accurate diagnosis. Ruling out autoimmune, infectious, and especially neoplastic causes, including paraneoplastic syndromes, is critical for confirming its idiopathic nature. While visual recovery is common, the risk of recurrence and complications such as choroidal neovascularization necessitates vigilant long-term monitoring.
Keywords: acute exudative polymorphous vitelliform maculopathy, serous retinal detachment, multimodal imaging, fundus autofluorescence, fluorescein angiography, indocyanine green angiography, vitelliform lesions
Introduction
Acute exudative polymorphous vitelliform maculopathy (AEPVM) is a rare retinal disorder first described by Gass in 1988, characterized by the sudden onset of visual disturbances accompanied by bilateral serous neurosensory retinal detachments and multifocal, yellow-white subretinal lesions at the level of the retinal pigment epithelium (RPE). These lesions often evolve into polymorphous subretinal deposits resembling the vitelliform lesions seen in Best disease. While the disease primarily affects the posterior pole, small bleb-like lesions may scatter across the retina, creating a honeycomb pattern.1 Fewer than 40 cases of idiopathic AEPVM have been documented to date, with patients typically presenting symptoms such as acute vision loss, blurred vision, and occasionally headaches.2,3 The etiology of AEPVM remains unclear, though several potential mechanisms have been proposed. Some cases have been linked to an infectious prodrome, such as a flu-like illness or upper respiratory infection, suggesting an immune-mediated or inflammatory basis. Response to corticosteroids and the presence of autoantibodies in some patients support an inflammatory or autoimmune component.4 Other cases have been associated with paraneoplastic syndromes but further research is needed to fully understand its pathogenesis.5 Despite its often benign course, with gradual visual recovery over several months to years, residual electrophysiologic abnormalities and photoreceptor damage may persist, resulting in incomplete visual restoration.6,7 In this report, we describe the case of a patient diagnosed with idiopathic AEPVM, detailing the clinical presentation, multimodal imaging findings, comprehensive systemic screening and disease progression, contributing to the limited literature on this rare condition and its diagnostic and therapeutic challenges.
Case Presentation
We present the case of a 40-year-old Caucasian female nurse who presented with complaints of blurred vision that had developed over the preceding two weeks. The patient denied experiencing other symptoms, including pain, ocular redness or scotoma. Notably, she reported flu-like illness one week prior to the onset of her visual symptoms, during which she self-initiated treatment with oral prednisolone (20 mg/day, during 3 days) and bilastine (10 mg/day, during 3 days).
Her medical history is significant for vitiligo and hypertension, which has been well-controlled with telmisartan (40mg/day) since diagnosis, 3 years earlier. The patient has no known allergies and no relevant family medical history. Her visual acuity was assessed at 20/20 with a refractive error of −0.25 in both eyes at baseline.
Biomicroscopy of the anterior segment was unremarkable, and intraocular pressure (IOP) was recorded at 14 mmHg in the right eye (OD) and left eye (OS). Pupillary reflexes were normal, with equal and reactive pupils. Extraocular movements were intact.
Fundoscopic examination under mydriasis revealed the absence of the foveal reflex, along with a suspected subfoveal serous retinal detachment and multiple small serous retinal detachments in the posterior pole, but no other abnormalities. The retinal vessels appeared normal without signs of vasculitis, and no vitreous opacities or chorioretinal lesions were identified (Figure 1).
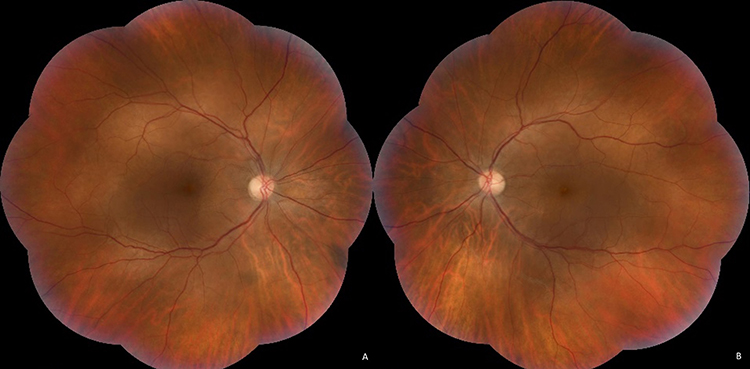 |
Figure 1 Color fundus photographs of OD (A) and OS (B) show the absence of the foveal reflex, with evidence of a suspected subfoveal serous retinal detachment, along with multiple small serous retinal detachments in the posterior pole. Abbreviations: OD, right eye; OS, left eye. |
Infrared and spectral-domain optical coherence tomography (SD-OCT) in OD and OS revealed bilateral, multiple, bleb-like small serous retinal detachments in the macula and along the vascular arcades, with varying sizes. A large confluent detachment was present in both eyes, temporal to the fovea (Figure 2). Fundus autofluorescence (FAF) appeared normal, showing no significant alterations at this stage. Fluorescein angiography (FA) and indocyanine green angiography (ICG) presented a normal aspect without any leakage or pooling. (Figure 3).
 |
Figure 2 Infrared and macular SD-OCT of OD (A) and OS (B) and peripapillary SD-OCT of OD (C) and OS (D) revealing multiple small serous retinal detachments. Abbreviations: OD, right eye; OS, left eye; SD-OCT, Spectral-Domain Optic Coherence Tomography. |
 |
Figure 3 FAF of OD (A) and OS (B), WF-FA and ICG of OD (C and D respectively) and WF-FA and ICG of OS (E and F respectively) with no abnormalities. Abbreviations: WF-FA, wide field fluorescein angiography; FAF, fundus autofluorescence; ICG, indocyanine green angiography; OD, right eye; OS, left eye; SD-OCT, spectral-domain optic coherence tomography. |
A survey for possible infectious causes was carried out and the patient lived in an apartment in an urban area with good sanitary conditions but had contact with a dog and cows/goats once a week, cleaning them. There is no history of international travel within the past five years, nor any recent hair coloring. There was no recent tick bite, no contact with patients with known pulmonary tuberculosis, nor any recent work accident, such as a needle stick. No recent or past consumption of intravenous drugs and no recent or previous use of topiramate or any MEK inhibitor. No recent or past risky sexual relations and no consumption of unpasteurized milk and its derivatives or consumption of undercooked meat.
Regarding vaccination, the patient’s immunization status is in accordance with the national vaccination schedule, including the COVID-19 vaccination. Two initial doses of the Pfizer-BioNTech COVID-19 vaccine were administered at the beginning of 2021, followed by a booster dose later in the same year. An annual booster has been received since then, with the most recent dose administered 6 months prior to the onset of symptoms.
A comprehensive laboratory workup was conducted to investigate potential infectious, autoimmune, and systemic etiologies for the serous retinal detachments. Hematological evaluation, including a complete blood count and platelet levels, returned within normal limits. Liver function tests, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), and gamma-glutamyl transferase (GGT), as well as kidney function markers (creatinine and urea), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) were all normal. Electrolyte levels were also within normal ranges, and thyroid function tests, including thyroid-stimulating hormone (TSH) and free thyroxine (T4), revealed no abnormalities. Angiotensin-converting enzyme (ACE) levels and ionized calcium were likewise normal.
To explore autoimmune causes, tests for antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies (ANCA), anti-double-stranded DNA (anti-dsDNA), anti-extractable nuclear antigen (anti-ENA), anti–Sjögren’s-syndrome-related antigens A and B (anti-SSA and anti-SSB) and anti-Smith antibodies (anti-SM) were all negative. Rheumatoid factor testing was also negative. Syphilis serology (TPPA) was unremarkable.
Infectious serologies showed that IgG antibodies for cytomegalovirus (CMV), herpes simplex virus 1 (HSV1), Epstein-Barr virus (EBV), and parvovirus were positive, while IgM antibodies for these infections were negative, suggesting past infections. Serologies for herpes simplex virus 2 (HSV2), varicella-zoster virus, Toxoplasma gondii, Borrelia, Coxsackie B virus, Coxiella burnetii, and Rickettsia were negative for both IgG and IgM antibodies. Bartonella DNA/PCR testing was negative, as was the Wright reaction for brucellosis. Serological tests for HIV, hepatitis B, and hepatitis C were also negative. Tuberculosis screening with an interferon-gamma release assay (IGRA) and a Mantoux test yielded negative results, with 0 mm of induration. Urine test revealed no hematuria or proteinuria.
Additional imaging and systemic evaluations were conducted. A brain and orbit magnetic resonance imaging (MRI) and thoraco-abdominopelvic computerized tomography (CT) scan were both normal. Further assessments, including upper gastrointestinal endoscopy, colonoscopy, breast CT, breast ultrasound, cervical cytology, transvaginal ultrasound, and dermatological screening, revealed no significant abnormalities.
Three months after the onset of the visual symptoms, in a follow-up consultation, her best corrected visual acuity (BCVA) remained 20/20 in OD but decreased to 20/25 in OS. Biomicroscopy of the anterior segment was unremarkable, and IOP was 14 mmHg in both eyes. Fundoscopic examination revealed multifocal yellowish subretinal polymorphous material scattered throughout the macula and seemed to gravitate along the lower margin of the serous retinal detachment, forming a curvilinear meniscus along temporal inferior vascular arcade (Figure 4). The previously observed bilateral multiple small serous retinal detachments have coalesced into a single larger detachment, while an accumulation of hyperreflective material in the subretinal space is observed, which translates the fundoscopic vitelliform deposits, as seen on SD-OCT (Figure 4). Additionally, FAF demonstrated the characteristic hyperautofluorescence of the polymorphous deposits, that corresponded to the yellow spots observed during fundoscopic examination. The same lesions showed hyperfluorescence on FA in all phases of the angiogram and ICG showed mild hypofluorescence in all phases of the angiogram (Figure 5).
 |
Figure 4 Color fundus photographs of OD (A) and OS (B) revealing multifocal subretinal yellowish lesions dispersed throughout the superior macula, the foveal area and along the lower margin of the serous retinal detachment, forming a curvilinear meniscus along temporal inferior vascular arcade. Macular SD-OCT of OD (C) and OS (D) revealing a single serous retinal detachment, remarkable thickening of the photoreceptor layer and a preserved integrity of external limiting membrane and ellipsoid. Accumulation of hyperreflective material in the subretinal space is observed in OD (E) and OS (F), representing the fundoscopic vitelliform deposits. Abbreviations: OD, right eye; OS, left eye; SD-OCT, spectral-domain optic coherence tomography. |
 |
Figure 5 FAF of OD (A) and OS (B), early phase FA of OD (C) and OS (D) and late phase FA of OD (E) and OS (F) demonstrated hyperfluorescent areas dispersed throughout the superior macula, the foveal area and along the temporal inferior vascular arcade. Early phase ICG of OD (G) and OS (H) and late phase ICG of OD (I) and OS (J) showed hypofluorescent areas in the foveal area and inferior macula. Abbreviations: FAF, fundus autofluorescence; FA, fluorescein angiography; ICG, indocyanine green angiography; OD, right eye; OS, left eye. |
In both eyes, full field electroretinogram was normal, as electrooculogram (Arden ratio was 2.39 in OD and 2.51 in OS) and pattern visually evoked potentials. Pattern Electroretinogram was normal in OD but showed a mild macular dysfunction in OS (reduction of P50-wave amplitude).
Based on the clinical presentation, multimodal imaging findings, evolving retinal changes, and comprehensive systemic screening to rule out autoimmune, infectious, and neoplastic etiologies, a diagnosis of idiopathic AEPVM was established. The patient is being monitored for visual recovery and the potential need for further interventions.
Discussion
AEPVM is a rare macular disorder. Establishing an accurate diagnosis for patients with AEPVM requires a comprehensive multimodal evaluation, extended follow-up periods, and a high level of clinical suspicion.8–10 Confirming its idiopathic nature necessitates thorough systemic screening to exclude autoimmune, infectious, neoplastic and, in selected cases, genetic etiologies.11,12
Despite the increasing documentation of the clinical characteristics of AEPVM in the literature, its underlying pathogenesis remains poorly understood. Recent studies propose that the autofluorescent material observed in AEPVM arises not solely from lipofuscin but also from non-digestible remnants of phagocytized photoreceptor outer segments, which accumulate due to inadequate apposition between the retina and the RPE. Some researchers have suggested that variations in the chemical composition within the subretinal space develop as the disease progresses. This theory posits that an initial transudate, stemming from RPE dysfunction, eventually becomes enriched with lipofuscin and debris from shedded photoreceptor outer segments. FAF findings support this idea, as they show a gradual increase in autofluorescence correlating with the accumulation of lipofuscin and the indigestible components of the outer segments. In the presented case, the characteristic vitelliform lesions of AEPVM that accumulate at the level of the RPE only became visible after 3 months of monitoring.12,13
Literature reports suggest that a prodromal influenza-like illness or upper respiratory tract infection may precede the condition, leading many to hypothesize an inflammatory or autoimmune origin. However, the exclusion of a paraneoplastic component is of critical importance, as there are also documented cases linked to malignancies such as cutaneous melanoma, breast, testicular, and lung carcinoma. Given the potential association with cancer, it is imperative to thoroughly rule out any underlying neoplastic processes when diagnosing AEPVM, which was a key consideration in this case.4,12
Currently, there is no consensus regarding the efficacy of early systemic or intraocular treatments, such as steroids or intraocular antivascular endothelial growth factor (anti-VEGF) agents, in altering the clinical progression of the disease.14
Regardless of the treatment approach, patients often experience gradual visual recovery over time, with the disease typically following a benign course. While electrophysiological abnormalities may persist, the condition usually resolves spontaneously, and most patients experience significant visual improvement, making the prognosis generally favorable.7,12
In a case series review by Barbazetto et al, symptoms and clinical findings of idiopathic AEPVM were shown to persist for several months to years. Half of their subjects had prodromal symptoms, such as our case, and one of their patients developed choroidal neovascularization (CNV). They concluded that younger patients who present with similar symptoms, especially those in their teens, should have genetic testing to rule out a genetic basis for the disease, especially a bestrophinopathy and that autofluorescent imaging remains one of the key diagnostic tests. They also identified a patient who developed recurrence of the condition 2 years post complete resolution, a phenomenon which had not been previously described, underscoring the importance of vigilant follow-up for these patients.12
Baddar et al reported two cases of AEPVM occurring subsequent to COVID-19 vaccination. The authors postulated that the vaccine might induce an immune response, potentially via autoantibodies directed against the SARS-CoV-2 virus glycoproteins, which could cross-react with retinal pigment epithelial cells. While this hypothesis is of significant interest, it is currently not possible to definitively establish a causal relationship between the COVID-19 vaccination and the onset of AEPVM. Given the widespread nature of COVID-19 vaccination, the high incidence of vaccinated individuals precludes the ability to conclusively attribute AEPVM to the vaccination. Consequently, further research is warranted to explore this potential association and elucidate the underlying mechanisms involved.15
We acknowledge that the absence of long-term outcome data is a limitation of the case, and further studies with extended follow-up periods would provide valuable insights into the natural progression and potential complications of AEPVM.
Conclusion
AEPVM is a rare macular disorder that requires comprehensive multimodal evaluation to establish an accurate diagnosis. Ruling out autoimmune, infectious, and especially neoplastic causes, including paraneoplastic syndromes, is critical for confirming its idiopathic nature. While visual recovery is common, the risk of recurrence and complications such as CNV necessitates vigilant long-term monitoring.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed.
Statements of Ethics
Written informed consent was obtained from the patient for the publication of this paper. Approval to publish the case details was not required in our institution.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was not supported by any sponsor or funder.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Gass JD, Chuang EL, Granek H. Acute exudative polymorphous vitelliform maculopathy. Trans Am Ophthalmol Soc. 1988;86:354–366.
2. Torres-Costa S, Penas S, Carneiro A, et al. Idiopathic acute exudative polymorphous vitelliform maculopathy: insight into imaging features and outcomes. Case Rep Ophthalmol Med. 2020;2020:7254038. doi:10.1155/2020/7254038
3. Massaro D, Pece A, Pichi F, et al. A case of acute exudative polymorphous vitelliform maculopathy: follow-up and wide-field spectral-domain optical coherence tomography. Eur J Ophthalmol. 2015;25(5):e91–94. doi:10.5301/ejo.5000616
4. Koreen L, He SX, Johnson MW, Hackel RE, Khan NW, Heckenlively JR. Anti-retinal pigment epithelium antibodies in acute exudative polymorphous vitelliform maculopathy: a new hypothesis about disease pathogenesis. Arch Ophthalmol. 2011;129(1):23–29. doi:10.1001/archophthalmol.2010.316
5. Grajewski RS, Schuler-Thurner B, Mauch C, et al. Ocular diseases in metastatic cutaneous melanoma: review of 108 consecutive patients in two German tertiary centers. Graefes Arch Clin Exp Ophthalmol. 2014;252(4):679–685. doi:10.1007/s00417-013-2563-5
6. Murtagh P, Treacy M, Stephenson K, Dooley I. Acute exudative polymorphous vitelliform maculopathy syndrome; natural history and evolution of fundal and OCT images over time. BMJ Case Rep. 2018;2018.
7. Kozma P, Locke KG, Wang YZ, Birch DG, Edwards AO. Persistent cone dysfunction in acute exudative polymorphous vitelliform maculopathy. Retina. 2007;27(1):109–113. doi:10.1097/01.iae.0000226537.95346.ca
8. Cruz-Villegas V, Villate N, Knighton RW, Rubsamen P, Davis JL. Optical coherence tomographic findings in acute exudative polymorphous vitelliform maculopathy. Am J Ophthalmol. 2003;136(4):760–763. doi:10.1016/S0002-9394(03)00426-4
9. Vianna RN, Muralha A, Muralha L. Indocyanine-green angiography in acute idiopathic exudative polymorphous vitelliform maculopathy. Retina. 2003;23(4):538–541. doi:10.1097/00006982-200308000-00016
10. Astakhov YS, Astakhov SY, Lisochkina AB, Nechiporenko PA. Eight-year multimodal follow-up of recurrent idiopathic acute exudative polymorphous vitelliform maculopathy. J Fr Ophtalmol. 2020;43(6):500–516. doi:10.1016/j.jfo.2020.01.004
11. Chan CK, Gass JD, Lin SG. Acute exudative polymorphous vitelliform maculopathy syndrome. Retina. 2003;23(4):453–462. doi:10.1097/00006982-200308000-00002
12. Barbazetto I, Dansingani KK, Dolz-Marco R, et al. Idiopathic acute exudative polymorphous vitelliform maculopathy: clinical spectrum and multimodal imaging characteristics. Ophthalmology. 2018;125(1):75–88. doi:10.1016/j.ophtha.2017.07.020
13. Vaclavik V, Ooi KG, Bird AC, Robson AG, Holder GE, Webster AR. Autofluorescence findings in acute exudative polymorphous vitelliform maculopathy. Arch Ophthalmol. 2007;125(2):274–277. doi:10.1001/archopht.125.2.274
14. Scupola A, Abed E, Sammarco MG, et al. Intravitreal dexamethasone implant for acute exudative polymorphous vitelliform maculopathy. Eur J Ophthalmol. 2014;24(5):803–807. doi:10.5301/ejo.5000477
15. Baddar D, Fayed AE, Tawfik CA, Bassily S, Gergess MM, El-Agha MH. COVID-19 vaccine-induced acute exudative polymorphous vitelliform maculopathy: case reports. Retin Cases Brief Rep. 2024;18(1):66–70. doi:10.1097/ICB.0000000000001319
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Multimodal Imaging of Yellow-White Transformation in Chorioretinal Hemorrhages: Structural and Biochemical Insights
Tanaka T, Muraoka Y, Kogo T, Hama Y, Akiyama Y, Kadomoto S, Hata M, Ooto S, Tsujikawa A
Clinical Ophthalmology 2026, 20:571106
Published Date: 21 March 2026