")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Identification of Two Novel Variants of the DMD Gene in Chinese Families with Duchenne Muscular Dystrophy
Authors Wu J, Ren L, Huang X, Hu L, Zhang L, Xie D, Li Z, Han N, Huang S
Received 8 April 2023
Accepted for publication 11 August 2023
Published 17 August 2023 Volume 2023:16 Pages 759—766
DOI https://doi.org/10.2147/PGPM.S416294
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Jiangfen Wu,1 Lingyan Ren,2 Xinyi Huang,3 Li Hu,2 Liangliang Zhang,2 Dan Xie,1 Zhimin Li,4 Naijian Han,4 Shengwen Huang1,2,5
1GuiZhou University Medical College, Guiyang, Guizhou, People’s Republic of China; 2Prenatal Diagnosis Center, Guizhou Provincial People’s Hospital, Guiyang, People’s Republic of China; 3School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, People’s Republic of China; 4Annoroad Gene Technology (Beijing) Co., Ltd, Beijing, People’s Republic of China; 5NHC Key Laboratory of Pulmonary Immunological Diseases, Guizhou Provincial, People’s Hospital, Guiyang, People’s Republic of China
Correspondence: Shengwen Huang, Guizhou Provincial People’s Hospital, Nanming District, Guiyang, People’s Republic of China, Tel +8615185134701, Fax +86-851-85937194, Email [email protected]
Background: Duchenne muscular dystrophy (DMD), an X-linked recessive neuromuscular disorder, is caused by pathogenic variants in the DMD gene encoding a large structural protein in muscle cells.
Methods: Two probands, a 6-year old boy and a 1-month old infant, respectively, were clinically diagnosed with DMD based on elevated levels of creatine kinase and creatine kinase isoenzyme. CNVplex and whole exome sequencing (WES) were performed for causal variants, and Sanger sequencing was used for verification.
Results: CNVplex found no large deletions or duplications in the DMD gene in both patients, but WES discovered a single-nucleotide deletion in exon 48 (NM_004006.2:c.6963del, p.Asp2322ThrfsTer16) in the proband of pedigree 1, and a nonsense mutation in exon 27 (NM_004006.2:c.3637A>T, p.K1213Ter) in the proband of pedigree 2.
Conclusion: The results of our study expand the mutation spectrum of DMD and enrich our understanding of the clinical characteristics of DMD. Genetic counseling was provided for the two families involved in this study.
Keywords: DMD gene, exon 48, exon 27, whole exome sequencing, Duchenne muscular dystrophy
Introduction
Duchenne muscular dystrophy (DMD, OMIM#310200) is a severe X-linked recessive, inherited neuromuscular disorder, characterized by rapidly progressive muscle weakness and muscle wasting throughout the body.1 DMD is more common in males than females, with an incidence rate of 1:5000 and 1:50,000,000, respectively.2 Female heterozygotes theoretically have 50% normal cells due to random X–inactivation and thus usually do not manifest muscle dysfunction. However, it has been reported that some female heterozygotes could develop cardiac dysfunction,3 muscle weakness or exercise intolerance,4 which could be attributed to the skewed X–inactivation. Patients with DMD generally show early symptoms at around 3–5 years of age, become wheelchair-dependent by the time of 10–12 years of age, and die of cardiac or respiratory failure at around 20 years of age.5
Genetically, DMD is mainly caused by mutations of the DMD gene. The DMD gene is known as the largest human gene with a length of 2.4 million base pairs and 79 exons. Hence, thousands of mutations in one or more exons have been found in patients with DMD, interfering with dystrophin production and function.6 Approximately 70–75% of mutations are large deletions/duplications covering single or multiple exons, and the rest are point mutations and small insertions/deletions.7 Therefore, in the majority of patients, multiplex ligation-dependent probe amplification (MLPA) or X-chromosome comparative genomic hybridization array is first used to screen large deletions or duplications. Patients who are negative need further parallel or targeted sequencing to screen small mutations.8,9
Herein, we discovered two novel variants from two families, including a single-base deletion in exon 48 (NM_004006.2:c.6963del) and a nonsense mutation in exon 27 (NM_004006.2:c.3637A>T) of the DMD gene by whole exome sequencing (WES). Both mutations are the first reported worldwide expanding the mutational spectrum of the DMD gene.
Materials and Methods
Ethical Compliance
The research was conducted in accordance with the Declaration of Helsinki. Gathering of relevant information, obtaining of participants’ consent, and protocols for this study were approved by the Medical Ethics Committee at Guizhou Provincial People’s Hospital (approval number 2022–05).
Subjects
Case 1
The proband (III-15) of pedigree 1 (Figure 1A) began walking unsteadily at age 4. By the time he came to our hospital on July 21, 2020 for genetic diagnosis at age 6, he could not stand up by himself when squatting, with waist muscle wasting, waddling gait, and frequent falls (Figures 2A and B). Creatine kinase (CK), creatine kinase isoenzyme (CKMB), lactate dehydrogenase (LDH) and α-hydroxybutyrate dehydrogenase (HBDH) levels were elevated to 13544U/L, 592U/L, 985U/L, and 713U/L. Computed tomography (CT) scan revealed a decreased density of soft tissue surrounding the posterior margin of the bilateral hip joints and the upper section of the bilateral femurs. Electromyography indicated myogenic impairment. All of the above findings correspond to manifestations of DMD. The proband has two brothers. III-12 is also a patient with DMD who is non-ambulant and spends most of his time in bed (Figure 2C and D), while III-13 is healthy without any symptoms of DMD by now.
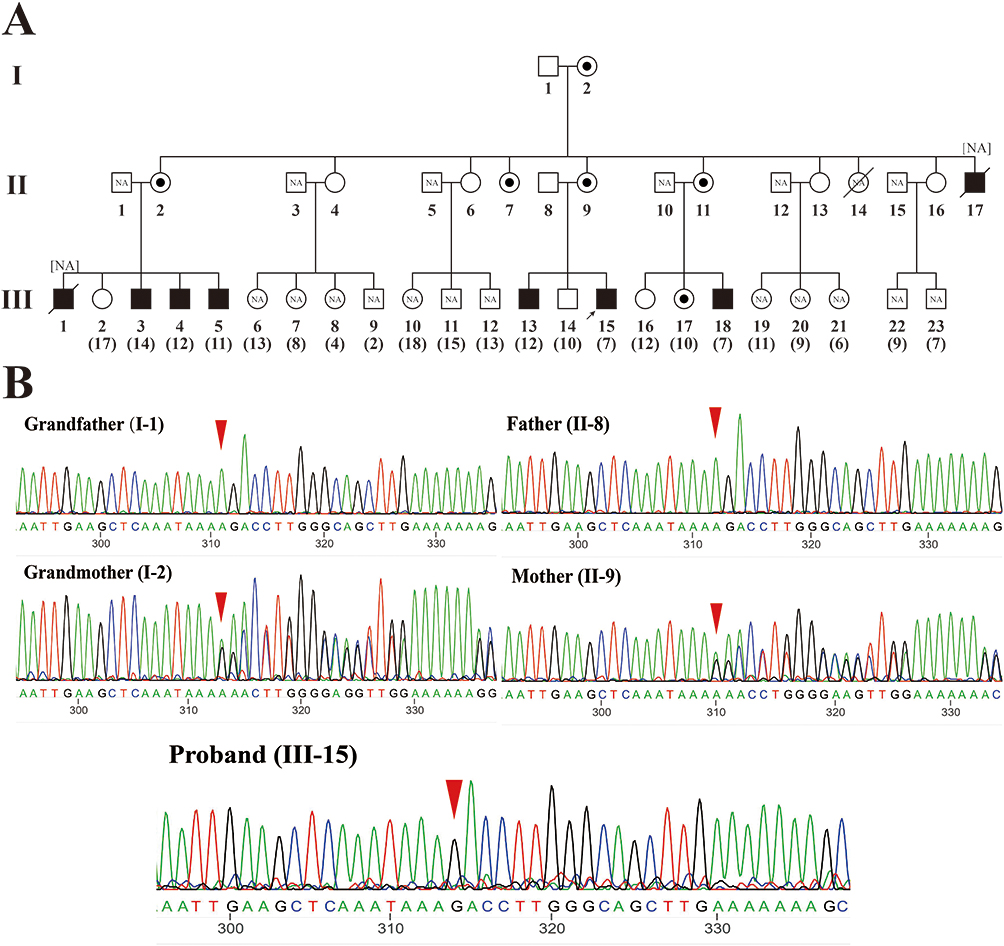 |
Figure 1 (A) Pedigree information of family 1 with DMD. The proband is marked with a black arrow. Black-filled squares represent patients. Slashes represent deceased family members. Circles with black dots represent female carriers. NA indicates the family members did not perform WES or Sanger sequencing. The numbers in parentheses represent the age of the family members. (B) Sanger sequencing results of the proband, his parents and grandparents. The red triangle indicates the mutation site (c.6963del). Both the mother and grandmother of the proband are mutant carriers. |
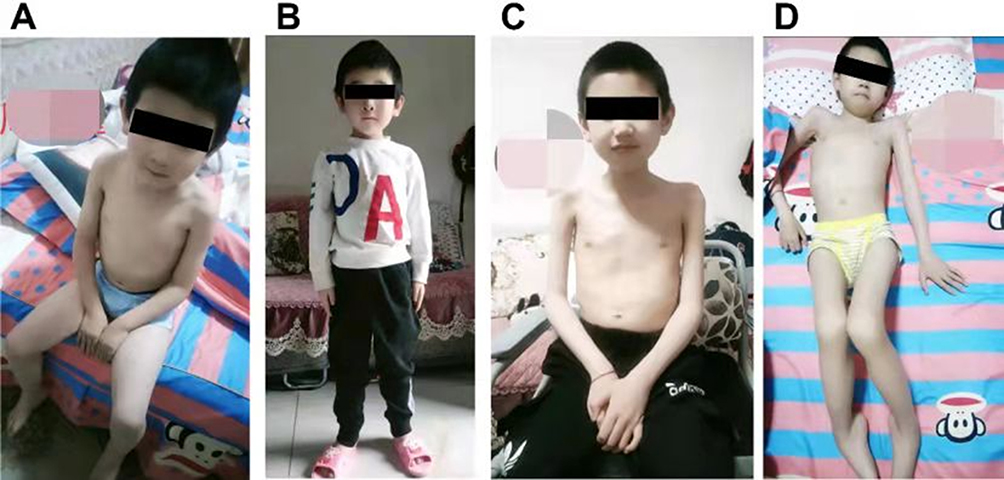 |
Figure 2 Clinical images of the proband III-15 (A and B) and his eldest brother (C and D) in pedigree 1. |
The proband’s mother (II-9) is asymptomatic and has seven sisters and one younger brother. All the sisters are asymptomatic except one (II-14) who died of an unknown reason at three months after birth. The younger brother (II-17), the first patient with DMD in the family, died at age 20. All the male patients in this family share very similar clinical manifestations such as walking instability and frequent falls as early as 3–5 years of age, high CK levels (9960–15520U/L), high CKMB levels (338–644U/L), extensive myogenic impairment as indicated by electromyography, and being fully non-ambulant at the age of 8–10.
Case 2
The 1-month old male proband (II-1) of pedigree 2 (Figure 3A) was born at a traditional Chinese medicine hospital in Songtao county, Guizhou province on January 14, 2022, who was then moved to the neonatal unit after birth for a “15-minute reduced muscle tone”. Serological test showed abnormal myocardial enzyme profile with elevated CK (19585 U/L) and CKMB (1174 U/L) (Table 1). Creatine phosphate was administered to nourish cardiomyocytes over the course of two weeks of hospitalization. The levels of CK and CKMB gradually decreased, but they are still higher than the upper limit of the reference ranges (Table 1). The infant was discharged from the hospital on January 26, 2022 because his parents decided to take him back to his hometown for Chinese Spring Festival. During this period, he did not receive any treatment. The infant returned to our pediatric department on February 16. Serological test showed CK and CKMB levels were elevated again (6237 U/L and 202 U/L). Additionally, elevated LDH (583U/L), HBDH (457U/L) were also detected. All of the above findings correspond to manifestations of DMD.
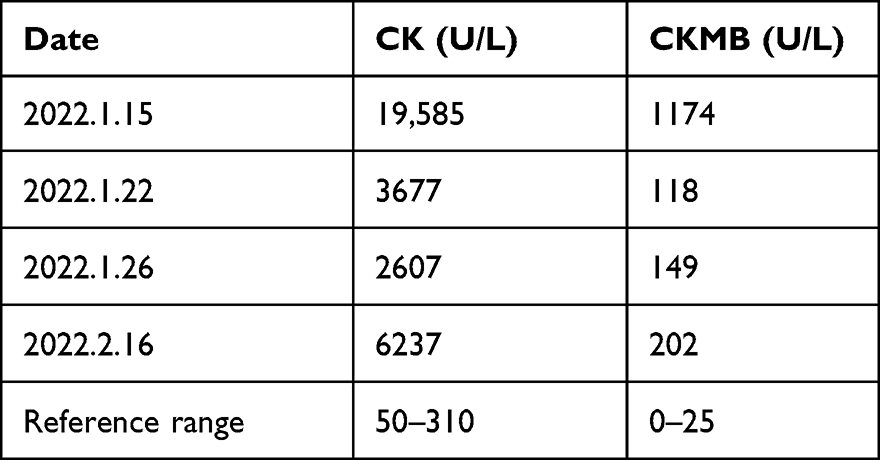 |
Table 1 Changes of CK and CKMB Levels of the Proband in Pedigree 2 (II-1) Before and After Hospitalization |
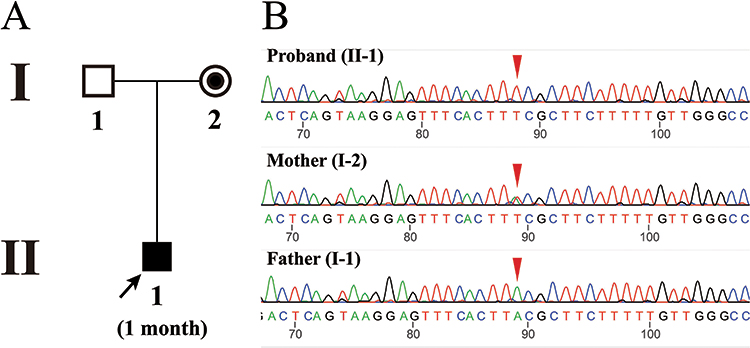 |
Figure 3 (A) Pedigree information of family 2 with DMD. The proband is marked with a black arrow. (B) Sanger sequencing results of the proband and his parents. The red triangle indicates the mutation site (c.3637A>T). |
DNA Sample Collection and Extraction
Genomic DNA was extracted by QIAamp DNA Blood Mini Kit (Qiagen, Germany) from the peripheral blood lymphocytes of participants. DNA quality was estimated by agarose gel electrophoresis and quantified by Qubit 2.0 fluorometer (ThermoFisher Scientific).
CNVplex
Large deletions or large duplications of the DMD gene were analyzed by CNVplex® (Genesky Biotechnologies), a high-throughput multiplex detection method for copy number variations similar to MLPA.10,11 79 target-specific probe pairs were designed for each exon of DMD (Table S1). Furthermore, 32 reference probes were chosen from various sub-chromosomal loci that had no reported copy number variations. The amplified products were detected using capillary electrophoresis on an ABI 3130xl Genetic Analyser (Applied Biosystems). Raw data were analyzed by GeneMapper 4.0 (Applied Biosystems) as previously described.11
Whole Exome Sequencing
The SureSelect Human All Exon V6 kit (Agilent Technologies) was used to prepare a library enriched with exons of all human genes. The library was sequenced on a NovaSeq 6000 platform (Illumina) with a paired-end 150bp strategy for an average depth of 100-fold. The reads were aligned to Genome Reference Consortium Human Build 37 (GRCh37). NextGENe V2.3.4 (SoftGenetics LLC) was used to find point mutations, which were then interpreted using InterVar software according to the ACMG/AMP guidelines.12,13
Sanger Sequencing
PCR primers for the candidate mutation were designed based on the reference sequence from GenBank (NM_004006.2) and synthesized by Sangon Biotech. The primers’ sequences for NM_004006.2:c.6963del were F: 5’–TGCAGTTGGCTATGCCTTTG–3’; R: 5’–CAAAAAGTTCCCTACCTTACG–3’. The primers’ sequences for NM_004006.2:c.3637A>T were F: 5’–AGAGCCACTGGTAGTTGGTG–3’; R: 5’– ACTGGGATGTTGTGAGAAAGAA–3’. The Ultra HiFidelity PCR Kit (TIANGEN) was used on an ABI 9700 thermal cycler (Applied Biosystems). PCR conditions were as follows: 94 °C 3 minutes; 94 °C 30 seconds, 55 °C 30 seconds, and 72 °C 60 seconds for 30 cycles; and 72 °C 5 minutes. Sanger sequencing was carried out on an ABI PRISM 3730 automatic sequencer (Applied Biosystems). The sequence chromatograms were compared and visualized by the TBtools software.14
Results
In both probands, CNVplex found no microdeletions or microduplications in the DMD gene (Figure S1). WES, on the other hand, discovered one nucleotide deletion in exon 48 (NM 004006.2:c.6963del) in the proband III-15 of pedigree 1 and one nucleotide substitution in exon 27 (NM 004006.2:c.3637A>T) in the proband II-1 of pedigree 2 (Figure S2). There were no other pathogenic variants of muscular dystrophy-related genes other than the DMD gene detected by WES. The c.6963del causes a frameshift and a premature stop codon (p. Asp2322ThrfsTer16). Sanger sequencing confirmed the mutations in the probands and their mothers (Figures 1B and 3B). Furthermore, the pedigree analysis of III-15 revealed that c.6963del are present in all male patients and their mothers, but absent in all healthy males and four females who had not given birth to patients (II-4, II-6, II-13, and II-16).
Based on four pieces of evidence from the ACMG guidelines, the c.6963del is categorized as a “pathogenic” mutation: (1) PVS1 evidence: the deletion resulting in protein truncation and loss of function; (2) PM2_supporting evidence: unreported previously in 1000 genome database, ExAC database, gnomeAD exon database, ClinVar database, and HGMD professional edition database; (3) PP1_strong evidence: the genotype of the mutation co-segregated with the DMD phenotype of the family; (4) PP4 evidence: the phenotypes of the patients were consistent with that of DMD. The c.3637A>T is also categorized as ‘pathogenic’ mutation, based on three pieces of evidence from the ACMG guidelines (11): (1) PVS1 evidence; (2) PM2_supporting evidence; and (3) PP4 evidence.
Discussion
In this study, we used WES to screen two probands with DMD for pathogenic factors, and found an A base deletion at the 6963rd site of the DMD gene (NM_004006.2:c.6963del) and an A to T substitution at the 3637th site (NM_004006.2:c.3637A>T). Because the probands’ parents refused muscle sampling, we could not perform any protein experiments to support the direct correlation between the mutations and DMD. However, there are several pieces of evidence that can reveal the two mutations as causative factors for DMD: 1. Whole exome sequencing did not detect any pathogenic mutations in other DMD-related genes. 2. The genotypes of the mutations co-segregated with the DMD phenotypes. 3. Both mutations could cause premature termination codons, so the transcripts would be subjected to nonsense-mediated mRNA decay (NMD).15 NMD is an RNA surveillance mechanism that detects the mRNAs harboring premature termination codons, and triggers the mRNA degradation to prevent the accumulation of truncated and potentially harmful proteins. Therefore, lack of dystrophins in muscles can lead to a severe phenotype of DMD but not BMD, which are consistent with the clinical manifestations of the probands and other males with DMD. Overall, we believe that these two variants are the pathogenic factors that cause two family members with DMD, respectively.
Exon 27 has a total of 25 “Pathogenic” mutations in the ClinVar database, including 15 nonsense mutations (Variant ID: 1322582, 1322366, 1322585, 11280, 1322368, 572550, 803887, 1322268, 1322701, 1322270, 575026, 287438, 1322372, 1072808, and 1322702), 7 frameshift mutations caused by small deletions (Variant ID: 585782, 963833, 803888, 526040, 1073854, 1322543, and 1072808), and 3 frameshift mutation caused by small insertions (Variant ID: 94601, 1409291, and 1428508). Exon 48 contrarily has fewer “Pathogenic” variants than exon 27, primarily due to fewer nonsense mutations. There are a total of 15 “pathogenic” mutations in exon 48, including 7 frameshift mutations (Variant ID: 501131, 803840, 803843, 455926, 455927, 409940, and 639718), 6 nonsense mutations (Variant ID: 640322, 803841, 803842, 197593, 11220, and 1322456), and 2 frameshift mutations (Variant ID: 94754 and 522612). We were particularly interested in one variant (NM 004006.2:c.6986del [p.lys2329SerfsTer9], Variant ID: 455927) that has been reported in several individuals,16,17 because it can result in a premature termination codon of the same length as the c.6963del mutation. This appears to be yet another piece of evidence supporting a loss of dystrophin function by the c.6963del mutation, though a functional study is still needed in the future.
At present, there is no curative treatment for patients with DMD and typical treatments such as corticosteroids and physical therapies only alleviate secondary symptoms of DMD.18 Several emerging therapies, focused on restoration of dystrophin production are being evaluated in clinical trials, some of which have received regulatory approval in the USA or Europe.19 For example, ataluren is an oral drug that can induce ribosomal readthrough of premature stop codons.20 Its safety and effectiveness led to conditional approval by the EMA.21 The c.3637A>T reported in our study causes a nonsense mutation, thus ataluren could be an effective therapy for this newborn. However, it is unfortunate that the proband’s parents chose not to pursue this therapy, primarily due to financial considerations.
Exon skipping is a quite promising treatment which aims to restore the disrupted open reading frame of the DMD transcripts in patients so that they can produce a Becker’s muscular dystrophy-like protein.22 It can bypass a defective exon from the pre-mRNA splicing machinery, restoring the reading frame and producing a partially functional, internally deleted dystrophin.23 Exon-skipping entails three experimental approaches: antisense oligonucleotides (ASOs), U7 snRNP, and CRISPR/Cas9 system.24 ASOs are short modified DNA or RNA homologs (usually 20–30 nucleotides) that can bind to their target exon prior to pre-mRNA splicing. This prevents the target exon from being recognized by the splicing machinery so that it will be spliced out when the mature mRNA is formed. Now only four ASOs that are designed for skipping exon 45 (Casimersen25), exon 51 (eteplirsen26) or exon 53 (golodirsen27 and viltolarsen28) have been granted conditional approval by the FDA. The mechanisms of U7 snRNP-based approach is very similar to ASOs. Adeno-associated virus vectors have been used to deliver modified U7 snRNP genes. A clinical trial of AAV-mediated RNA editing for exon 2 duplications is currently being tested.29 CRISPR/Cas9 system works at the DNA level by converting a DMD out-of-frame variant of genomic DNA in muscle cells into an in-frame variant. CRISPR also uses viral vectors to deliver the DNA editing machinery. Although no clinical trials in DMD have been conducted yet, a laboratory study using the CRISPR/Cas9 system successfully expressed a truncated, partially functional dystrophin with exon 48 skipping in HEK293 cells,30 providing a cellular basis for animal and clinical research and development in the future.
Conclusion
In summary, we report two rare family cases with DMD, confirming the critical role of genetic factors in DMD phenotypes. High throughput sequencing test, such as WES or even whole genome sequencing, is essential for accurate diagnosis, prognostic estimation, and personalized treatment in the era of precision medicine.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available due to the original design and validation of the project but are available from the corresponding author on reasonable request.
Acknowledgments
The authors appreciate all individuals participated in this study.
Funding
Guizhou Science and Technology Projects, Grant/Award Number: 20165670, 20192808 and 20205011; Guiyang Science and Technology Innovation Project, Grant/Award Number: 20161001; Science and Technology Foundation Project of Guizhou Provincial Health Commission, Grant/Award Number: gzwjkj2020-1-250.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 2004;5(9):872–876. doi:10.1038/sj.embor.7400221
2. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifiro G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. 2020;15(1):141. doi:10.1186/s13023-020-01430-8
3. Schade Van Westru SM, Hoogerwaard EM, Dekker L, et al. Cardiac abnormalities in a follow-up study on carriers of Duchenne and Becker muscular dystrophy. Neurology. 2011;77(1):62–66. doi:10.1212/WNL.0b013e318221ad14
4. Mercier S, Toutain A, Toussaint A, et al. Genetic and clinical specificity of 26 symptomatic carriers for dystrophinopathies at pediatric age. Eur J Hum Genet. 2013;21(8):855–863. doi:10.1038/ejhg.2012.269
5. Anaya-Segura MA, Garcia-Martinez FA, Montes-Almanza LA, et al. Non-invasive biomarkers for Duchenne muscular dystrophy and carrier detection. Molecules. 2015;20(6):11154–11172. doi:10.3390/molecules200611154
6. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7(1):13. doi:10.1038/s41572-021-00248-3
7. Wang L, Xu M, Li H, et al. Genotypes and Phenotypes of DMD Small Mutations in Chinese Patients with Dystrophinopathies. Front Genet. 2019;10:114. doi:10.3389/fgene.2019.00114
8. Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145–151. doi:10.1136/jmedgenet-2015-103387
9. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267. doi:10.1016/S1474-4422(18)30024-3
10. Zhang X, Xu Y, Liu D, et al. A modified multiplex ligation-dependent probe amplification method for the detection of 22q11.2 copy number variations in patients with congenital heart disease. BMC Genomics. 2015;16:364. doi:10.1186/s12864-015-1590-5
11. Liang Q, Qin H, Ding Q, et al. Molecular and clinical profile of VWD in a large cohort of Chinese population: application of next generation sequencing and CNVplex ® technique. Thromb Haemostasis. 2017;117(8):1534. doi:10.1160/TH16-10-0794
12. Li Q, Wang K. InterVar: clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100(2):267–280. doi:10.1016/j.ajhg.2017.01.004
13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
14. Chen C, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–1202. doi:10.1016/j.molp.2020.06.009
15. Lindeboom RG, Supek F, Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet. 2016;48(10):1112–1118. doi:10.1038/ng.3664
16. Okubo M, Minami N, Goto K, et al. Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. J Hum Genet. 2016;61(6):483–489. doi:10.1038/jhg.2016.7
17. Saad FA, Mostacciuolo ML, Trevisan CP, et al. Novel mutations and polymorphisms in the human dystrophin gene detected by double-strand conformation analysis. Hum Mutat. 1997;9(2):188–190. doi:10.1002/(SICI)1098-1004(1997)9:2<188::AID-HUMU15>3.0.CO;2-Z
18. Schreiber A, Brochard S, Rippert P, et al. Corticosteroids in Duchenne muscular dystrophy: impact on the motor function measure sensitivity to change and implications for clinical trials. Dev Med Child Neurol. 2018;60(2):185–191. doi:10.1111/dmcn.13590
19. Mercuri E, Bonnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019;394(10213):2025–2038. doi:10.1016/S0140-6736(19)32910-1
20. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487. doi:10.1002/mus.24332
21. Haas M, Vlcek V, Balabanov P, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscular Disord. 2015;25(1):5–13. doi:10.1016/j.nmd.2014.11.011
22. Niks EH, Aartsma-Rus A. Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert Opin Biol Th. 2017;17(2):225–236. doi:10.1080/14712598.2017.1271872
23. O’Brien A, Cohn RD. Genome Editing for Muscle Gene Therapy. In: Duan D, Mendell JR, editors. Muscle Gene Therapy. Cham: Springer International Publishing; 2019:275–287.
24. Takeda S, Clemens PR, Hoffman EP. Exon-Skipping in Duchenne Muscular Dystrophy. J Neuromuscular Dis. 2021;8(s2):S343–S358. doi:10.3233/JND-210682
25. Wagner KR, Kuntz NL, Koenig E, et al. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: a randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve. 2021;64(3):285–292. doi:10.1002/mus.27347
26. Alfano LN, Charleston JS, Connolly AM, et al. Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine. 2019;98(26):e15858. doi:10.1097/MD.0000000000015858
27. Frank DE, Schnell FJ, Akana C, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94(21):e2270–e2282. doi:10.1212/WNL.0000000000009233
28. Roshmi RR, Yokota T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drug Today. 2019;55(10):627–639. doi:10.1358/dot.2019.55.10.3045038
29. Gushchina LV, Frair EC, Rohan N, et al. Lack of Toxicity in Nonhuman Primates Receiving Clinically Relevant Doses of an AAV9.U7snRNA Vector Designed to Induce DMD Exon 2 Skipping. Hum Gene Ther. 2021;32(17–18):882–894. doi:10.1089/hum.2020.286
30. Dara M, Razban V, Talebzadeh M, Moradi S, Dianatpour M. Using CRISPR/Cas9 System to Knock out Exon 48 in DMD Gene. Avicenna J Med Biotechnol. 2021;13(2):54–57. doi:10.18502/ajmb.v13i2.5517
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.