Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Identification of two novel mutations, PSEN1 E280K and PRNP G127S, in a Malaysian family
Authors Ch’ng G, An SSA ![]() , Bae SO, Bagyinszky E, Kim S
, Bae SO, Bagyinszky E, Kim S ![]()
Received 9 April 2015
Accepted for publication 18 May 2015
Published 8 September 2015 Volume 2015:11 Pages 2315—2322
DOI https://doi.org/10.2147/NDT.S86334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Gaik-Siew Ch’ng,1,* Seong Soo A An,2,* Sun Oh Bae,2 Eva Bagyinszky,2 SangYun Kim3,4
1Department of Genetics, Kuala Lumpur Hospital, Malaysia; 2Department of Bionano Technology, Gachon University, 3Department of Neurology, Seoul National University College of Medicine, 4Seoul National University Bundang Hospital, South Korea
*These authors contributed equally to this work
Abstract: Alzheimer’s disease (AD) is the most common form of dementia, which can be categorized into two main forms: early onset AD and late onset AD. The genetic background of early onset AD is well understood, and three genes, the APP, PSEN1, and PSEN2 have been identified as causative genes. In the current study, we tested three siblings from Malaysia who were diagnosed with early onset dementia, as well as their available family members. The family history was positive as their deceased father was similarly affected. Patients were tested for mutations in APP, PSEN1, PSEN2, and PRNP. A novel variant, E280K, was discovered in exon 8 of PSEN1 in the three siblings. In silico analyses with SIFT, SNAP, and PolyPhen2 prediction tools and three-dimensional modeling were performed, and the results suggested that the mutation is probably a pathogenic variant. Two additional pathogenic mutations were previously been described for codon 280, E280A, and E280G, which could support the importance of the E280 residue in the PS1 protein contributing to the pathogenic nature of E280K. Additional ten family members were screened for the E280K mutation, and all of them were negative. Six of them presented with a variety of neuropsychiatric symptoms, including learning disabilities, epilepsy, and schizophrenia, while four family members were asymptomatic. A novel PRNP G127S mutation was found in a step-niece of the three siblings harboring the PSEN1 E280K mutation. In silico predictions for PRNP G127S mutation suggested that this might be possibly a damaging variant. Additional studies to characterize PRNP G127S would be necessary to further understand the effects of this mutation.
Keywords: Alzheimer’s disease, presenilin-1, prion, mutation
Introduction
Alzheimer’s disease (AD) is a genetically heterogeneous disorder, and several genes were associated with the disease. AD could be defined as early onset AD (EOAD) and under 65 years of age and late onset AD, which could onset under and over 65 years of age, respectively. The genetic background of EOAD has been well defined. At least three causative genes have been reported for EOAD: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2; http://www.alzforum.org/mutations).1 The causative mutations in APP for EOAD were found in exons 16 and 17, which were near the cleavage sites of beta and gamma secretases. PSEN1 is a part of gamma secretase, where the majority of causative mutations for EOAD were reported.1,2 Currently, more than 200 mutations were found in PSEN1, which may be involved in AD or in other disease phenotypes, such as frontotemporal dementia (FTD), Pick’s disease, parkinsonism, spastic paraparesis, or myoclonus. PSEN2 is also involved in gamma secretase cleavage. Initially, only a few mutations were described in PSEN2 for AD, but recent studies have discovered additional pathogenic or probable pathogenic variants causing other disease phenotypes such as FTD, dementia with Lewy bodies, and atypical dementias. In EOAD, the majority of mutations revealed a positive family history in which the disease inheritance followed a Mendelian pattern. However, several EOAD cases were described without any family history, suggesting de novo cases for EOAD.2,3
At least 30 pathogenic mutations have been described for the prion gene (PRNP; http://www.federationofscientists.org/pmpanels/TSE/Priprogene.asp), which could also be involved in different disease phenotypes, such as Creutzfeldt–Jakob disease, familial fatal insomnia, Gerstmann–Straussler–Scheinker disease, or kuru.4 Pathological similarities were found between AD and prion disease, as neurodegenerative diseases may be based on the aggregation of abnormal proteins. The main signs of AD, cerebral deposits of amyloid plaques, and aggregated tau fibrils could also be present in prion diseases. There are no specific clinical or biochemical diagnostic markers for AD and prion diseases. The diagnosis of dementia is difficult, particularly in patients in the early presymptomatic stage in the absence of any significant neurological signs. Postmortem histopathological analysis could also be important for the confirmation of clinical diagnosis.3 Since AD and prion diseases have many similarities, patients with early onset dementia should be screened for mutations in both PRNP and the major causative genes of EOAD.5,6
In this manuscript, we reported the cases of three Malaysian siblings who were diagnosed with EOAD. They had a positive family history of the disease, as their deceased father had been similarly affected. A novel mutation, E280K, was described in the three siblings. Additional family members with various neuropsychiatric manifestations such as learning disabilities, epilepsy, and schizophrenia were tested and found to be negative for PSEN1 E280K. Interestingly, however, genetic testing for PRNP revealed a novel prion mutation at G127S in one relative, that was suggested as a probable pathogenic variant based on the in silico predictions, but the confidence of these predictions might be low.
Materials and methods
Patients
Initially, we tested three siblings from Malaysia whose deceased father had been diagnosed with dementia. The first patient (III;10) was a 55-year-old female with the onset of symptoms at 48 years of age, beginning initially with mood and behavioral changes associated with progressive dementia. She also presented visual and auditory hallucinations (visual hallucination is not a part of AD clinical symptoms; we should consider this symptom as visual illusion or phantom border phenomenon) and myoclonic jerks. Cognitive decline worsened during the last 2 years with signs and symptoms of parkinsonism becoming obvious during the past year. The family history revealed that her two biological siblings (III;6 and III;8), deceased father (II;3), and two deceased stepbrothers (III;3 and III;5) from her father’s first marriage were also affected with some variation in their manifestations of the disease. She scored 11/30 in her mini-mental state examination and had fleeting eye contact with signs of parkinsonism. Biochemical profiles such as full blood count, liver function test, renal and coagulation profile, thyroid function test, Venereal Disease Research Laboratory, and vitamin B12 level were normal. The peripheral blood film was normal with no evidence of acanthocytes. An electroencephalogram showed mild-to-moderate encephalopathic changes with epileptiform spikes corresponding to myoclonus. There was generalized brain volume loss of the supratentorial region including the hippocampus on her cranial magnetic resonance imaging (MRI) imaging with no preferential pattern. Additionally, six symptomatic and four asymptomatic family members were also tested. The family pedigree and a summary of the clinical phenotypes are depicted in Figure 1 and Table 1, respectively. This research was approved by Internal Review Board at Seoul National University Budang Hospital, Sungnamsi, South Korea. Patient consents were received at Kuala Lumpur Hospital, Kuala Lumpur, Malaysia.
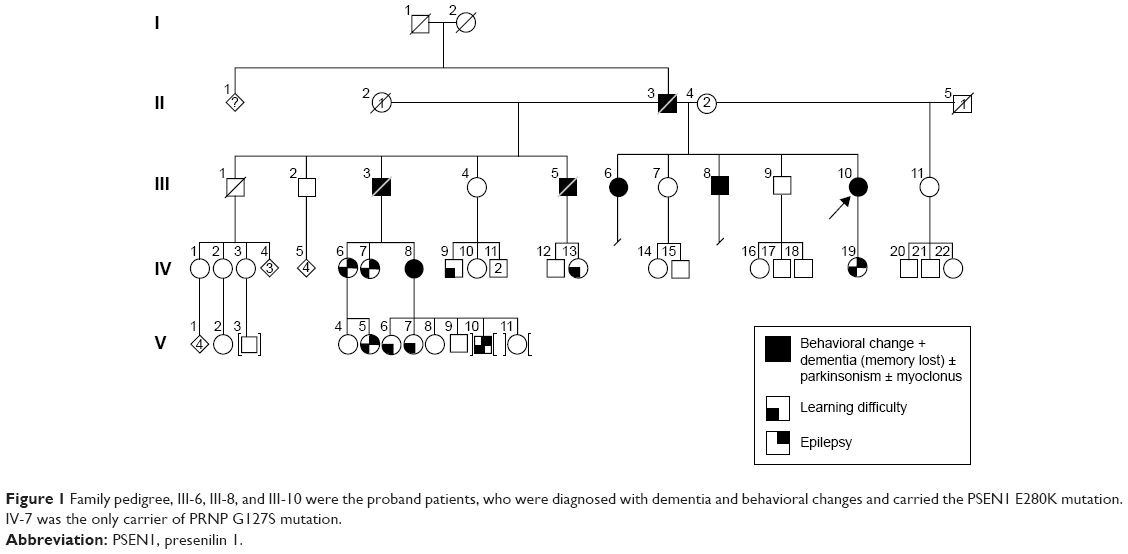 | Figure 1 Family pedigree, III-6, III-8, and III-10 were the proband patients, who were diagnosed with dementia and behavioral changes and carried the PSEN1 E280K mutation. IV-7 was the only carrier of PRNP G127S mutation. |
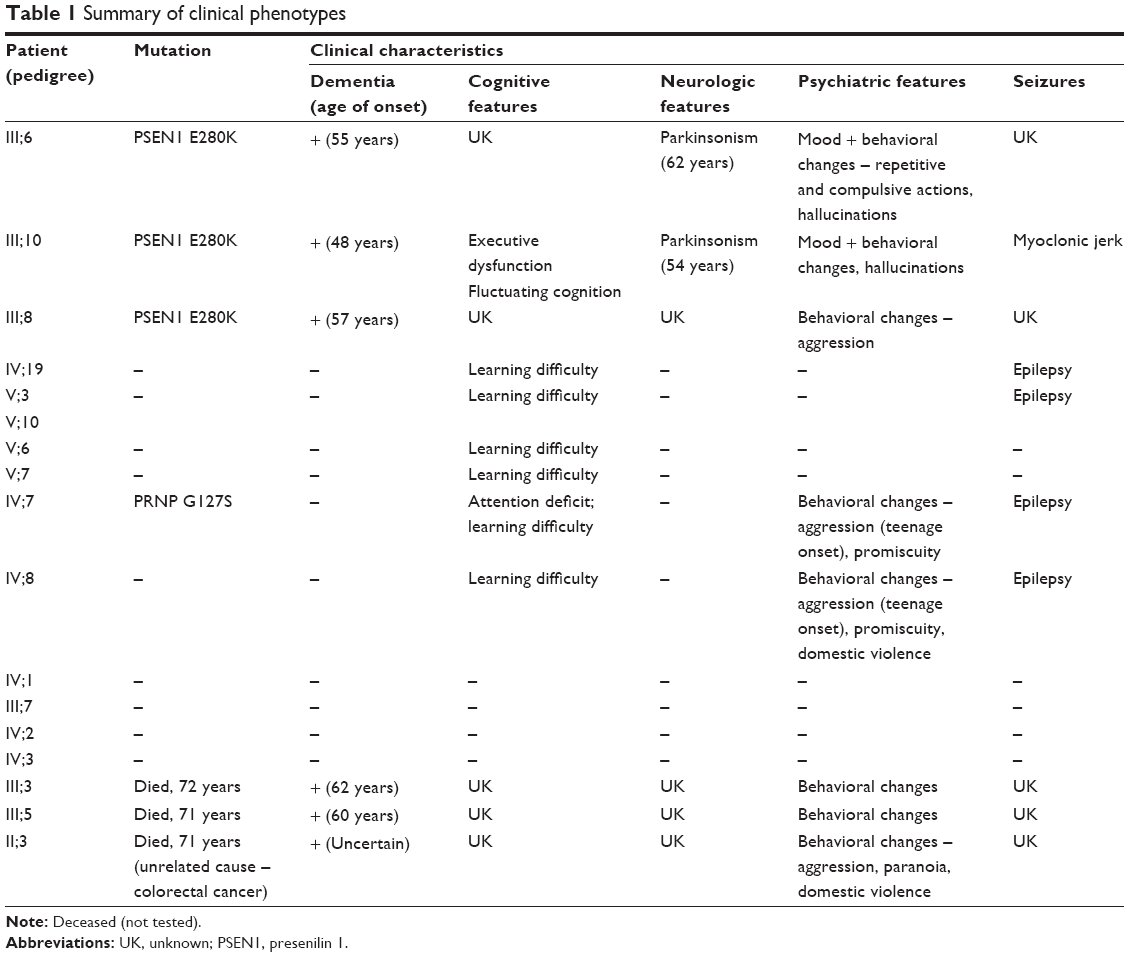 | Table 1 Summary of clinical phenotypes |
Amplification of APP, PSEN1, PSEN2, and the coding region of PRNP
Blood samples were drawn into tubes containing ethylenediaminetetraacetic acid (EDTA) as the anticoagulant and centrifuged at 800× g for 30 minutes. Immediately following centrifugation, the white blood cells were separated, and the buffy coat was kept at −80°C prior to the analysis. DNA was isolated from the buffy coat using the GeneAll blood kit (Seoul, Republic of Korea) following the manufacturer’s protocol. Analysis based on polymerase chain reaction (PCR) was performed with the selected exons of APP, PSEN1, PSEN2, and the coding region of PRNP. For APP, exons 16 and 17 were selected.7,8 For PSEN1, we screened exons 4, 5, 6, 7, 8, and 11, where the majority of the mutations had been detected previously. For PSEN2, we selected exons 4, 5, 6, 7, and 12, where pathogenic mutations had been reported.9–11 For PRNP, the coding exon (exon 2) was tested.12 Patients were also screened for mutations in the microtubule-associated protein tau (MAPT) and progranulin genes (PGRN).13,14 PCR was performed with the selected primers as previously reported.
Single-strand conformation polymorphism
A simple and fast PCR-based mutation detection method was performed with PCR amplicons from the patients. PCR products were denatured by adding formamide solution and heating (Figure 2A).15 After incubation for 10 minutes at 98°C, samples were cooled down (on ice or placed at −20°C in a freezer) for an additional 10 minutes. Native polyacrylamide gel electrophoresis was performed for 17 hours at 100 V with the electrophoresis device on ice. The gels were stained with SYBR Gold (Invitrogen, Seoul, Republic of Korea) and visualized under light.
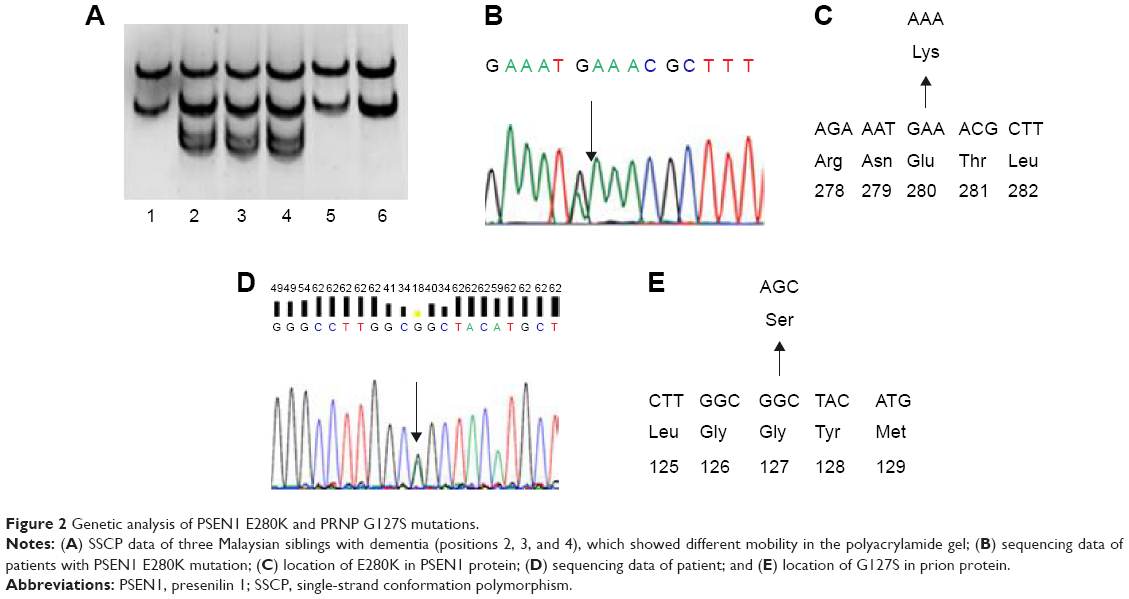 | Figure 2 Genetic analysis of PSEN1 E280K and PRNP G127S mutations. |
Sequencing
Since the error rate of SSCP can be high and confirmations of the mutations would be needed for identification by direct sequencing of all PCR products (Figure 2B and C). Prior to sequencing, the GeneAllExpin PCR kit (Seoul, Republic of Korea) was used to purify the PCR products. Sequencing was performed by BioNeer Inc. (Dajeon, Republic of Korea). Sequencing reactions were set up using the BigDye Terminator Cycle Sequencing kit and resolved on the ABI 3730XL DNA Analyzer (Bioneer Inc.). Sequences were aligned by NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and screened with DNA Baser software (http://www.dnabaser.com). Mutations and sequence variants were identified and compared against the NCBI Gene (http://www.ncbi.nlm.nih.gov/gene) and UniProt (http://www.uniprot.org) databases.
Bioinformatic analysis
The damaging nature of missense mutations could be checked using a simple online software tool called PolyPhen2 (http://genetics.bwh.harvard.edu/pph2). PolyPhen2 uses a clustering algorithm and performs a multiple alignment to select the sequences. The algorithm uses eight sequence-based and three structure-based features for the prediction, which are based on the comparison of wild type and mutant proteins. Two types of datasets can be used, HumDiv and HumVar data. HumDiv data should be used to find out the role of rare alleles in complex disease phenotypes and in natural selection. Alleles that might be less damaging should also be treated as damaging. HumVar data should be used in the diagnosis of Mendelian disorders, which requires the differentiation of highly damaging mutations from less damaging variants.16 Sorting intolerant from tolerant (SIFT) algorithm (http://sift.jcvi.org/) predicts the potential effects of amino acid substitution on protein function. SIFT uses different protein databases, such as SWISS-PROT, SWISS-PROT/TrEMBL, or protein databases of NCBI, and calculates the possibility on the pathogenic nature of mutations by comparing the mutant and normal alleles. It scores the amino acid substitutions, and under and over the score of 0.05, mutations could be defined as deleterious or tolerated, respectively.17 We also performed prediction with the “screening for nonacceptable polymorphisms” (SNAP) algorithm, which uses sequence-based computationally acquired information for data analysis. This algorithm is network-derived tool, which predicts the structural and functional effects of variants. Two categories are available, such as nonneutral and neutral variant, depending on the potential effect on the protein functions. SNAP provides a reliability index (ranging between 0 and 9), which reflects the level of confidence in the prediction. This algorithm identified ~80% of the nonneutral variants with the accuracy of 77%.18 The three-dimensional (3-D) structures of normal and mutant PSEN1 were constructed by the Raptor X web server (http://raptorx.uchicago.edu/), a protein structure prediction server using amino acid sequences. The full sequence of PSEN1 (amino acids 1–467) was analyzed, while the N-terminal fragment subunit of PSEN2 (amino acids 1–297) was used for better structure prediction. Discovery Studio 3.5 Visualizer from Accelrys was used to display superimposed images (Figure 3).19
 | Figure 3 In silico 3-D modeling for PS1 E280K, comparing to the normal PSEN1. |
Results
Figure 1 reveals the family tree, and Table 1 shows the clinical phenotypes of examined patients and relatives. The proband patients were III-6, III-8, and III-10, who were diagnosed with dementia and personality changes. Parkinsonism and seizures could also be present in them. Their father (II-3) and two additional stepbrothers (III;3, III;5) were also affected with dementia phenotypes.
SSCP revealed differential mobility of the PSEN1 exon 8 PCR products from each of the three siblings, suggesting the presence of a mutation (Figure 2A). Upon sequencing, a heterozygous G > A exchange was identified in PSEN1 exon 8, resulting in a GAA (E) > AAA (K) exchange at codon 280 (Figure 2B and C). All three siblings with dementia carried this mutation (Table 1). This mutation did not appear in the AD and FTD mutation database (http://www.molgen.ua.ac.be/ADMutations/) nor in the Alzheimer’s Research Forum (http://www.alzforum.org/mutations), which supported this mutation as a novel variant in PSEN1. PSEN1 (chromosome 14, 73, 603, 142–173, 690, 399) E280K was also checked in the database of Korea Centers for Disease Control and Prevention ([KCDC]; http://www.cdc.go.kr) and the Exome Aggregation Consortium ([ExAC]; http://exac.broadinstitute.org/about) dataset. In the KCDC, whole genome sequencing was performed in 622 control individuals. In ExAC, dataset sequence of 60,706 unrelated individuals were screened, which could be used in the various disease-specific and population genetic studies. PSEN1 E280K was absent in both of these databases, which also supports the novelty of the mutation.
E280 is located in the membrane-associated loop region (HL-VI) in the PS1 protein. In silico analysis with PolyPhen2 and 3-D structure prediction suggested that E280K might be involved in AD or other dementia disorders. PolyPhen2 scores for this mutation were 0.999 using the HumDiv data and 0.998 using the HumVar data. SIFT also predicted E280K as a damaging variant with the score of 0. SNAP results suggested that PSEN1 E280K is a nonneutral (probably pathogenic) variant, with the reliability index of three and with 78% of expected accuracy. Figure 3 shows the 3-D model of PSEN1 with mutation, comparing to the normal PSEN1. Differences between the normal and mutant protein were highlighted in the black circle. Glutamic acid was labeled with blue since lysine was labeled with green. Significant changes could be seen in the structure of mutant PSEN1 due to the charge differences between glutamic acid and lysine. In the mutant PSEN1, extra helices could be seen due to the loop region due to the lysine. An E280K mutation would result in disturbances of the helical structure by counter electrostatic interaction (Figure 3).
Additional family members agreed for the genetic test (Figure 1 and Table 1), of them, six (IV;19, V;3, V;6, V;7, IV;7, and IV;8) were diagnosed with learning difficulties. Additional clinical phenotypes, such as epilepsy, behavioral changes, and aggression, were also prominent. In addition, four asymptomatic relatives (IV;1, III;7, IV;2, and IV;3) also agreed for the genetic test. None of these family members who were tested carried the E280K mutation. The coding region of PRNP was also analyzed for each available family member (IV;7), and a novel PRNP G127S (GGC > AGC) mutation was found in one of the relative of the three patients (Figure 2D and E). This PRNP mutation was not reported in any of the PRNP mutation or Entrez databases. PRNP G127S mutation (PRNP locations: ch20. 4, 667, 156-4, 682, 234) was also checked against the KCDC and ExCA databases. It was not found in the genomes of 622 healthy individuals of KCDC database; however, it appeared in the ExAC database, with the frequency of 0.000008273. G127S was suggested as “singleton variant”. PolyPhen2 analysis suggested that this mutation is likely a damaging variant with 1.000 and 0.999 HumDiv and HumVar scores, respectively. SIFT prediction also suggested G127S as damaging variant with the score of 0, however, with low confidence. SNAP algorithm predicted G127S as a neutral variant with the reliability index of four and with 85% of expected accuracy. All screened patients and asymptomatic relatives were negative for any pathogenic mutations in PGRN MAPT genes.
Discussion
A PSEN1 E280K mutation was discovered in three Malaysian siblings (two sisters and one brother) whose father passed away at age 67 from colorectal cancer diagnosed at age 65. He had experienced apparent behavioral changes such as paranoia, aggression, and domestic violence associated with a decline in memory, but the age of onset was uncertain. In silico and 3-D structural analyses suggested that E280K would be a probable damaging mutation. As further strong evidence for this novel mutation on codon 280 having a pathogenic nature, two additional mutations for codon 280 have been reported, E280A and E280G (Table 2).
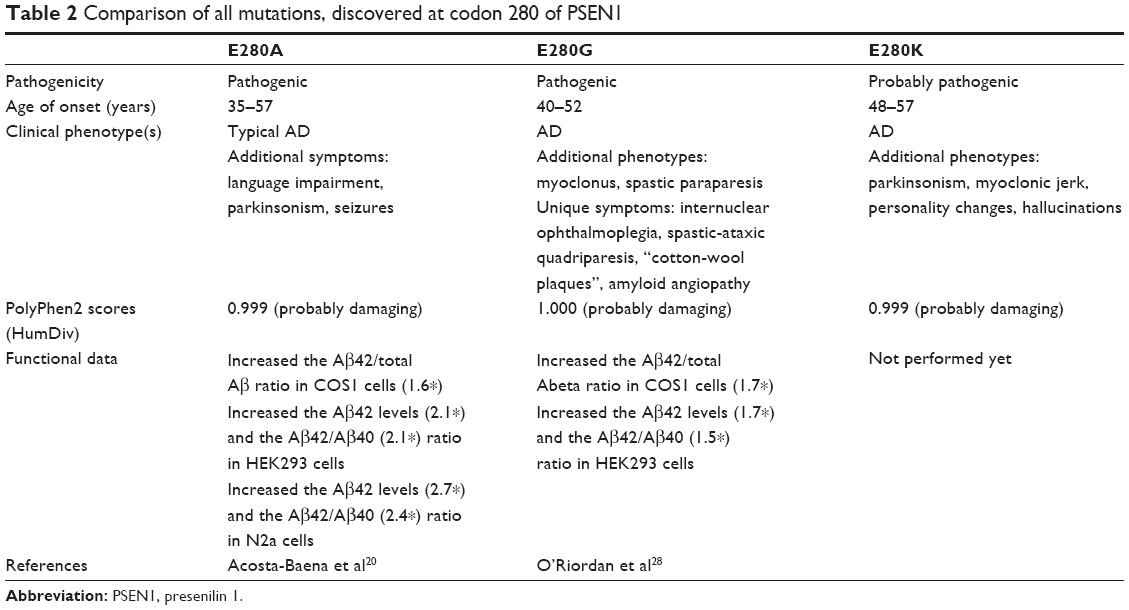 | Table 2 Comparison of all mutations, discovered at codon 280 of PSEN1 |
The PSEN1 E280A variant was described in several patients and families, where E280A was one of the major causative mutations for EOAD (AD and FTD mutation database). This mutation is also called the “Paisa mutation”, since it was dominant in people from the Paisa region in Columbia.20 AD patients harboring the E280A mutation presented with memory deficits as early as their 30s, but the average age of AD onset was between 45 and 50 years. This mutation might be independent from the APOE genotypes. The symptoms in the patients with E280A showed the typical AD phenotype, such as progressive memory impairment and personality changes. However, additional symptoms could also be present, such as gait difficulties, headache, language impairment, seizures, or parkinsonism.21,22 This mutation was also detected in a Japanese family, where the mean age of onset was 57 years.23 In vitro studies revealed that E280A could increase the Aβ1–42/totalAβ (1.6*) ratio in APP co-transfected COS1 cells.24 Elevated Aβ1–42 (2.7*) levels were reported after the transfection of additional cell lines, such as Neuro2A mouse neuroblastoma cells glioblastoma cells.25
The PSEN1 E280G mutation has been reported in AD patients of European descent, from countries such as France, Ireland, and the UK. In these patients, the disease onset occurred in their 40s or early 50s.26,27 E280G could be involved in EOAD. Myoclonus and spastic paraparesis with white matter abnormalities on cranial MRI were present. Unique symptoms, such as internuclear ophthalmoplegia, spastic-ataxic quadriparesis, and “cotton-wool plaques” with amyloid angiopathy on brain biopsy or white matter abnormalities, were also present.28 MRI analyses were used to diagnose patients for ischemic leukoencephalopathy due to amyloid angiopathy affecting meningocortical vessels.25 PSEN1 with E280G was transfected into HEK-293 cell lines, showing both elevated A42 levels and an elevated A…42/A…40 ratio (1.5*).24 Cotranfection with APP into COS1 cells revealed 1.7* elevation in the A…42/total A ratio.25 These results suggested that E280 could be an important residue in PSEN1, since two mutations were described previously and validated as being involved in EOAD.
Interestingly, a novel PRNP mutation (G127S) was also discovered in the step-niece of the three siblings with PSEN1 E280K. This 38-year-old woman (IV;7) presented primarily with neuropsychiatric symptoms, namely, attention deficits, learning difficulties, epilepsy, behavioral changes such as aggression, which had begun during her teenage years, and promiscuity. Her deceased father (III;3) was the stepbrother of the three siblings, and they shared the same father. Her father, together with another deceased paternal uncle (III;5), also had behavioral changes and dementia, with the onset of symptoms occurring between the ages of 60 years and 65 years, and subsequently passed away in their early seventies. Her only two symptomatic female siblings presented with variable neuropsychiatric symptoms, and one of them (IV;8) was tested but was not found to carry the mutation. This is likely a novel mutation, since it did appear neither in the KCDC, neither in nor in the prion mutation database (http://www.federationofscientists.org/pmpanels/tse/priprogene.asp) or in the Entrez database. However, a low frequency was detected in the ExCA database. A missense variant, G127V (rs267606980), was reported for the residue 127 in inhabitants of Eastern Highlands province in Papua New Guinea. Heterozygous G127V was suggested as a protective variant against kuru.29 In addition, for codon 127, a silent mutation was described (GGC > GGT; rs11538754). SIFT and PolyPhen2 scores suggested that PRNP G127S might be a probable damaging variant. However, SNAP prediction suggested that it might be a neutral mutation. Genetic prion disorders usually occur in adulthood, with the age of onset typically over 40–50 years. The PRNP G127S mutation might not directly cause a prion disorder. Further investigations would be needed to determine whether this novel mutation could be associated with any pathogenic phenotype, which could reveal a potential mechanism of pathogenesis. PRNP G127S might be a de novo mutation, since no additional family members have been shown to carry the mutation.
In conclusion, we suggest that PSEN1 E280K might be a novel, probably pathogenic mutation. Guerreiro et al30 designed an algorithm for the novel mutations in the causative genes for EOAD. Different parameters were analyzed, such as segregation, and the family history of patients and the importance of mutant residue.30 PSEN1 E280K was missing in normal controls due to the KCDC and ExCA databases. This mutation seemed to segregate with the disorder, since it was found only in the three affected siblings with EOAD. The additional two pathogenic mutations for the glutamic acid at position 280 (E280G and E280A) could enhance the evidence on the importance of this residue and the pathogenicity of E280K. Online predictions were suggested that E280K mutation could be a probably deleterious variant. However, we could not confirm the pathogenic nature of PRNP G127S. The patient did not show any prion disease associated phenotype and G127S also appeared in ExAC database. Additional studies are needed on for this variant since its pathogenic nature cannot rely on the in silico predictions. Combination of clinical and association studies, prediction and 3-D modeling softwares could enhance the estimation on the pathogenic nature of novel variants. However, in genetic studies, only in silico predictions cannot be used for the clinical prognosis.
Acknowledgment
This work was supported by the grants from the Korea Health Technology R&D Project (HI14C3331) through the Korea Health Industry Development Institute (KHIDI), Korea Ministry of Health & Welfare, and the Korean National Research Foundation (grant number NRF-2010-0024004).
Disclosure
The authors report no conflicts of interest in this work.
References
Bagyinszky E, Youn YC, An SS, Kim S. The genetics of Alzheimer’s disease. Clin Interv Aging. 2014;9:535–551. | ||
Zou Z, Liu C, Che C, Huang H. Clinical genetics of Alzheimer’s disease. Biomed Res Int. 2014;2014:291862. | ||
Ertekin-Taner N. Genetics of Alzheimer’s disease: a centennial review. Neurol Clin. 2007;25(3):611–667. | ||
Bernardi L, Anfossi M, Gallo M, et al. PSEN1 and PRNP gene mutations: co-occurrence makes onset very early in a family with FTD phenotype. J Alzheimers Dis. 2011;24(3):415–419. | ||
Finckh U, Müller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66(1):110–117. | ||
Stevens JC, Beck J, Lukic A, et al. Familial Alzheimer’s disease and inherited prion disease in the UK are poorly ascertained. J Neurol Neurosurg Psychiatry. 2011;82(9):1054–1057. | ||
Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid β-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet. 1992;51:273–282. | ||
Schellenberg GD, Pericak-Vance MA, Wijsman EM, et al. Linkage analysis of familial Alzheimer disease, using chromosome 21 markers. Am J Hum Genet. 1991;48(1):563–583. | ||
Cruts M, van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7(1):43–51. | ||
Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160:76–81. | ||
Honda M, Kaname T, Igata-Yi R, et al. Novel intronic polymorphisms in the presenilin-2 gene and a case-control association study of Alzheimer’s disease. Psychiatry Clin Neurosci. 1999;53(5):579–585. | ||
Jeong BH, Ju WK, Huh K, et al. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998;13(3):234–240. | ||
Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64(2):414–421. | ||
Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. | ||
Hayashi K. PCR-SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991;1(1):34–38. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. | ||
Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24(20):2397–2398. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol. 2012;10(3):213–220. | ||
Sepulveda-Falla D, Glatzel M, Lopera F. Phenotypic profile of early-onset familial Alzheimer’s disease caused by presenilin-1 E280A mutation. J Alzheimers Dis. 2012;32(1):1–12. | ||
Lopera F, Ardilla A, Martínez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277(10):793–799. | ||
Tanahashi H, Kawakatsu S, Kaneko M, Yamanaka H, Takahashi K, Tabira T. Sequence analysis of presenilin-1 gene mutation in Japanese Alzheimer’s disease patients. Neurosci Lett. 1996;218(2):139–141. | ||
Murayama O, Tomita T, Nihonmatsu N, et al. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci Lett. 1999;265(1):61–63. | ||
Kaneko H, Kakita A, Kasuga K, et al. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci. 2007;27(48):13092–13097. | ||
Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Alzheimer’s Disease Collaborative Group. Nat Genet. 1995;11(2):219–222. | ||
Hutton M, Busfield F, Wragg M, et al. Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. Neuroreport. 1996;297(3):801–805. | ||
O’Riordan S, McMonagle P, Janssen JC, et al. Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities. Neurology. 2002;59(7):1108–1110. | ||
Mead S, Whitfield J, Poulter M, et al. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med. 2009;361(21):2056–2065. | ||
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.