Back to Journals » OncoTargets and Therapy » Volume 13
Identification of Potential Key Genes and Pathways for Inflammatory Breast Cancer Based on GEO and TCGA Databases
Authors Lv Q ![]() , Liu Y, Huang H, Zhu M, Wu J
, Liu Y, Huang H, Zhu M, Wu J ![]() , Meng D
, Meng D
Received 24 March 2020
Accepted for publication 18 May 2020
Published 12 June 2020 Volume 2020:13 Pages 5541—5550
DOI https://doi.org/10.2147/OTT.S255300
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Qing Lv,1,* Yansong Liu,2,* Hu Huang,1 Mingjie Zhu,1 Junqiang Wu,1 Dong Meng1
1Department of Breast Surgery, Affiliated Hospital of Jiangnan University, Wuxi, Jiangsu, People’s Republic of China; 2Department of Breast Surgery, Tumor Hospital of Mudanjiang City, Mudanjiang, Heilongjiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dong Meng; Junqiang Wu
Department of Breast Surgery, Affiliated Hospital of Jiangnan University, Wuxi 214035, Jiangsu, People’s Republic of China
Tel +86-8868 2317
Email [email protected]; [email protected]
Introduction: Inflammatory breast cancer (IBC) is a rare type of breast cancer with poor prognosis, and the pathogenesis of this life-threatening disease is yet to be fully elucidated. This study aims to identify key genes of IBC, which could be potential diagnostic or therapeutic targets.
Methods: Four datasets GSE5847, GSE22597, GSE23720, and GSE45581 were downloaded from the Gene Expression Omnibus (GEO) and differential expression analysis was performed. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted to understand the potential bio-functions of the differentially expressed genes (DEGs). Protein–protein interaction (PPI) network was constructed for functional modules analysis and hub genes identification, and TCGA survival analysis and qRT-PCR of clinical samples were used to further explore and validate the effect of hub genes on IBC.
Results: A total of 114 DEGs were identified from the GEO datasets. GO and KEGG analyses showed that the DEGs were mainly enriched in oncogenesis and cell adhesion. From the PPI network, we screened out five hub genes, including PTPRC, IL6, SELL, CD40, and SPN. Survival analysis and expression validation verified the robustness of the hub genes.
Discussion: The present study provides new insight into the understanding of IBC pathogenesis and the identified hub genes may serve as potential targets for diagnosis and treatment.
Keywords: inflammatory breast cancer, bioinformatic analysis, hub genes, microarray
Introduction
Inflammatory breast cancer (IBC) is a rare type of locally advanced breast cancer, accounting for about 2% to 4% of all breast cancer cases.1 IBC is a life-threatening disease characterized by diffuse erythema, edema, and peau d’orange of the skin.2 Despite its low prevalence, IBC causes 7% of breast cancer-associated deaths.3 According to the data from the Surveillance, Epidemiology, and End Results (SEER) program which was carried out by American National Cancer Institute, the diagnosis rate of IBC is increasing by the year, and it is a much higher rate of increase than the incidence of overall breast cancer diagnosis.4 Thus, it is necessary to further understand the unique biology and pathogenesis underlying this aggressive form of breast cancer.
To date, accumulating evidence has demonstrated that aberrant expression and mutation of genes are involved in the carcinogenesis and progression of IBC, and several markers have been identified. For instance, the tumors of IBC maintain the expression of E-cadherin which is strongly associated with tumor emboli formation.5,6 Another epithelial-mesenchymal transition (EMT) marker zinc-finger E-box-binding homeobox 1 (ZEB1) and the stem cell marker aldehyde dehydrogenase 1 (ALDH1) are also reported to contribute to IBC progression and invasion.7,8 However, the exact mechanism underlying IBC is yet to be fully elucidated.
During the last decades, due to the development of high‐throughput technologies, including microarray and RNA-sequencing, the gene expression profiles have improved the understanding of IBC on its molecular mechanisms.9–12 However, due to the low incidence of IBC, such researches do not have satisfactory sample sizes, and analyses of samples from single centers may lead to biased results. A combined analysis of multiple datasets is urgently needed. Thus, we carried out this study, trying to identify differentially expressed genes (DEGs) in IBC distinct from those in non-IBC by analyzing datasets obtained from Gene Expression Omnibus (GEO) database, and explore the potential bio-functions by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Furthermore, the protein–protein interaction (PPI) network was constructed, and five hub genes were screened out from it. The present study may provide new insight into the understanding of IBC pathogenesis and the identified hub genes may serve as potential targets for diagnosis and treatment.
Materials and Methods
Microarray Dataset Selection and Processing
To identify genes differentially expressed in IBC and non-IBC tissues, microarray datasets (GSE5847,9 GSE22597,10 GSE23720,11 and GSE4558112) were downloaded from Genome Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) database, and probe names in each expression matrix were annotated to Entrez ID according to the corresponding GEO platform files. All of the selected datasets contain transcriptome microarray data of IBC and non-IBC clinical samples, and the diagnosis of IBC was based on both clinical features and pathological results. Details of the four datasets are shown in Supplementary Table 1.
Identification of DEGs
DEGs between IBC and non-IBC samples from the four GEO datasets were screened using R scripts. Background correction was performed using the robust multi-array average (RMA) method. The R package, limma (https://www.bioconductor.org/packages/release/bioc/html/limma.html), was applied to analyze differential expression. Bayes tests were used to evaluate statistical significance. Adjusted P-value (Adj.P) < 0.05 and |log2 fold-change (FC)| > 1 were used as a threshold for DEG identifying. After respective differential expression analysis, the results were intersected to determine DEGs for all datasets.
Functional Enrichment Analyses of DEGs
To investigate the functional roles of the screened DEGs, Database for Annotation, Visualization and Integrated Discovery (DAVID; version 6.8, https://david.ncifcrf.gov/) were employed to perform the Gene Ontology (GO) enrichment analysis under three categories, biological process (BP), cellular component (CC) and molecular function (MF). KEGG pathway analysis was carried out using the KOBAS database (version 3.0, http://kobas.cbi.pku.edu.cn/). False discovery rate (FDR) < 0.05 was considered statistically significant.
Protein–Protein Interaction (PPI) Network Construction and Hub Gene Identification
Basing on the DEGs screened above, we performed a PPI analysis using the Search Tool for the Retrieval of Interacting Genes (STRING; version 11.0, https://string-db.org/). The combined score > 0.4 was used as the cut-off criteria. Subsequently, Cytoscape (version 3.7.1) software was utilized for PPI network analysis and visualization. The plug-in MCODE (version 1.5, http://apps.cytoscape.org/apps/mcode) was employed to identify significant modules of the constructed network. Moreover, to investigate the hub (key) genes, we used cytohubba (version 0.1, apps.cytoscape.org/apps/cytohubba), another plug-in of Cytoscape which could screen the important nodes from a given network by several topology algorithms. In the present study, the maximal clique centrality (MCC) method was selected to identify hub genes, and other parameters of cytohubba were set to default.
The Cancer Genome Atlas (TCGA) Database Survival Analysis
To further explore the effect of hub genes expression on patients’ clinical outcomes, we employed GEPIA2 (URL: https://gepia2.cancer-pku.cn/) online tool to conduct a survival analysis of data from TCGA (URL: https://cancergenome.nih.gov/). Breast cancer samples were derived into hub gene high- and low-expression groups using the median transcript per million (TPM) value as a cutoff, respectively. A Log rank test was used for the hypothesis tests. To further confirm the biological functions of hub genes on tumorigenesis, Cox regression analysis was performed to evaluate the association between hub gene expression and prognosis in multiple malignancies, including breast cancer (BRCA), cervical squamous cell carcinoma (CESC), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), skin cutaneous melanoma (SKCM) and pancreatic adenocarcinoma (PAAD). P-value < 0.05 was considered to be statistically significant.
Clinical Tissues Collection
Eight IBC and twenty non-IBC clinical samples were collected by biopsy or surgery from patients who were primarily diagnosed between 2013 and 2019 at the Affiliated Hospital of Jiangnan University. The clinical and histological features of the specimens are shown in Supplementary Table 2. Immediately after removal, all specimens were transferred into liquid nitrogen and finally stored at −80°C before RNA extraction. Each patient enrolled in the present study has signed informed consent, and this study was approved by the ethical committee of the hospital.
RNA Extraction and Quantitative Reverse Transcriptional PCR (qRT-PCR)
Total RNA of clinical specimens was isolated using the E.Z.N.A Total RNA Kit I (Omega, USA) according to the protocol. Complementary DNA (cDNA) was synthesized subsequently by using the Prime Script RT Reagent Kit (Takara Bio, Japan). Real-time PCR was performed on the CFX96 Touch Real-Time PCR Detection System (Bio-rad, USA) to measure the expression levels of hub genes using the comparative Ct method. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was considered as a normalization control. The sequences of primers are shown in Supplementary Table 3.
Results
Identification of DEGs
In the present study, we performed a multi-step analysis to explore hub genes of inflammatory breast cancer, and further investigated their potential biological functions and effects on the prognosis of breast cancer. “Hub genes” are defined as core genes whose expression products are hubs in the PPI network. Firstly, four IBC datasets (GSE5847, GSE22597, GSE23720, and GSE45581) which contain a total of 131 IBC samples and 238 non-IBC samples, were downloaded from the GEO database. Differential expression analysis was performed for each dataset basing on the cut-off criteria (Figure 1A–D). Furthermore, the overlapping genes among the four datasets were considered as intersecting DEGs, including 53 upregulated and 61 downregulated genes (Figure 1E). The expression heatmap of the top 10 up- and down-regulated DEGs are shown in Figure 1F.
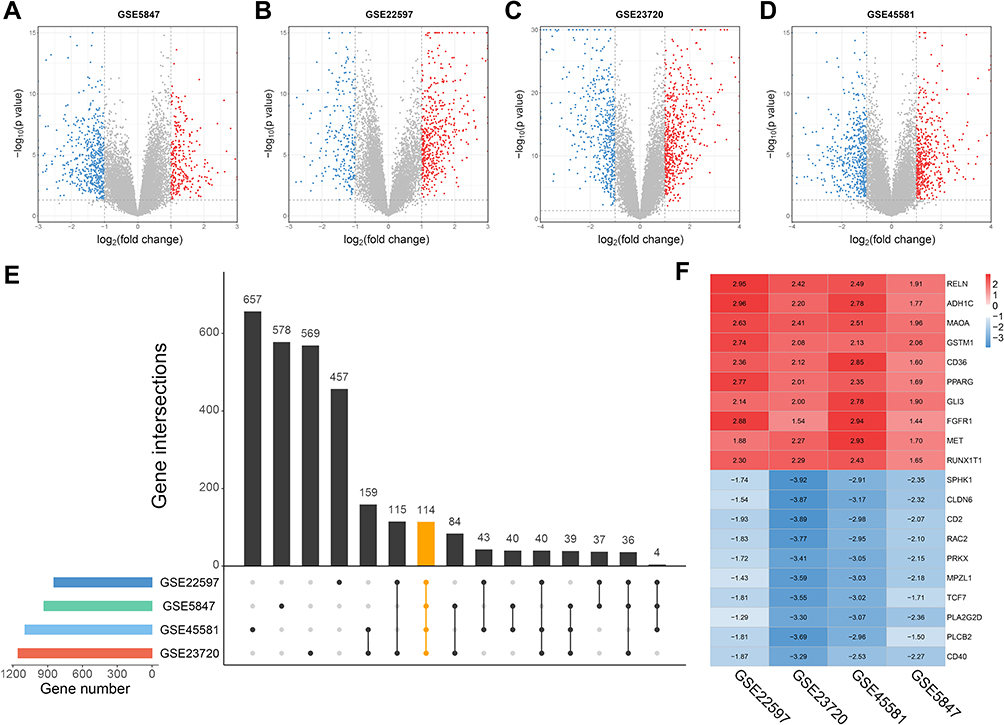 |
Figure 1 Identification of DEGs in four IBC datasets from GEO. Notes: (A–D) Volcano plots of differential expression analysis for GSE5847, GSE22597, GSE23720 and GSE45581. The red dots represent for significantly up-regulated genes, and the blue ones represent for down-regulated genes. The dotted lines indicate thresholds for DEG identification [Adj.P < 0.05 and |log2 fold-change (FC)| > 1]. (E) Upset plot of DEGs overlapped in the four datasets. (F) Expression heatmap of top 10 up- and down-regulated genes.Abbreviations: DEGs, differentially expressed genes; GEO, Genome Expression Omnibus. |
Functional and Pathway Enrichment Analysis
GO and KEGG pathway enrichment analyses were performed to characterize the functional roles of selected DEGs. For category BP of GO analysis, the DEGs were significantly enriched in signal transduction, cell adhesion, inflammatory response, positive regulation of cell proliferation and transcription (Figure 2A). For category CC, the expression product of the DEGs were mainly located in the plasma membrane, exosome, extracellular region (Figure 2B). Moreover, for category MF, the main functions of proteins encoded by the DEGs were DNA binding, protein binding, protein homodimerization activity, etc (Figure 2C).
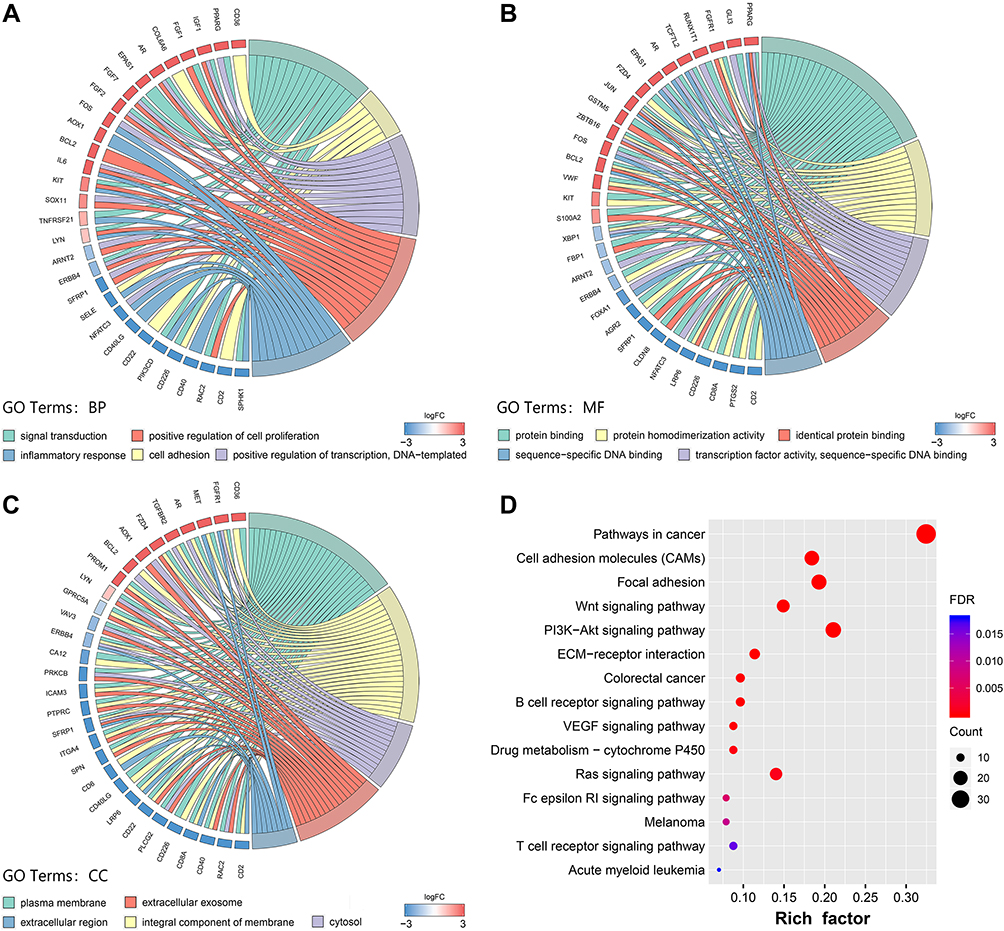 |
Figure 2 GO and KEGG pathway enrichment analyses for DEGs. Notes: (A) Chord plot of GO terms under BP category. (B) Chord plot of GO terms under MF category. (C) Chord plot of GO terms under CC category. (D) Dot plot of KEGG pathway enrichment analyses.Abbreviations: GO, Gene Ontology; BP, biological process; MF, molecular function; CC, cellular component; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
According to KEGG analysis, multiple pathways associated with tumorigenesis were enriched, including focal adhesion, Wnt, PI3K-AKT, VEGF, and Ras signaling pathways, etc. In addition, the DEGs were also enriched in cell adhesion and extracellular matrix (ECM) receptor interaction (Figure 2D).
PPI Network Construction and Module Analysis
To further explore the interactions of DEGs in IBC, a PPI network involving 101 nodes and 471 edges (Figure 3A), was constructed depending on the STRING database. Subsequently, Cytoscape plug-in MCODE was used for clustering analysis, and four functional modules were identified from the whole network (Figure 3B–E). Moreover, we performed pathway enrichment analyses for genes of the four modules to understand their potential bio-functions. According to the results, these modules were mainly associated with oncogenesis and cell adhesion, the enriched pathways included MAPK, ErbB, Wnt, VEGF signaling pathways, ECM-receptor interaction, and focal adhesion, etc (Table 1).
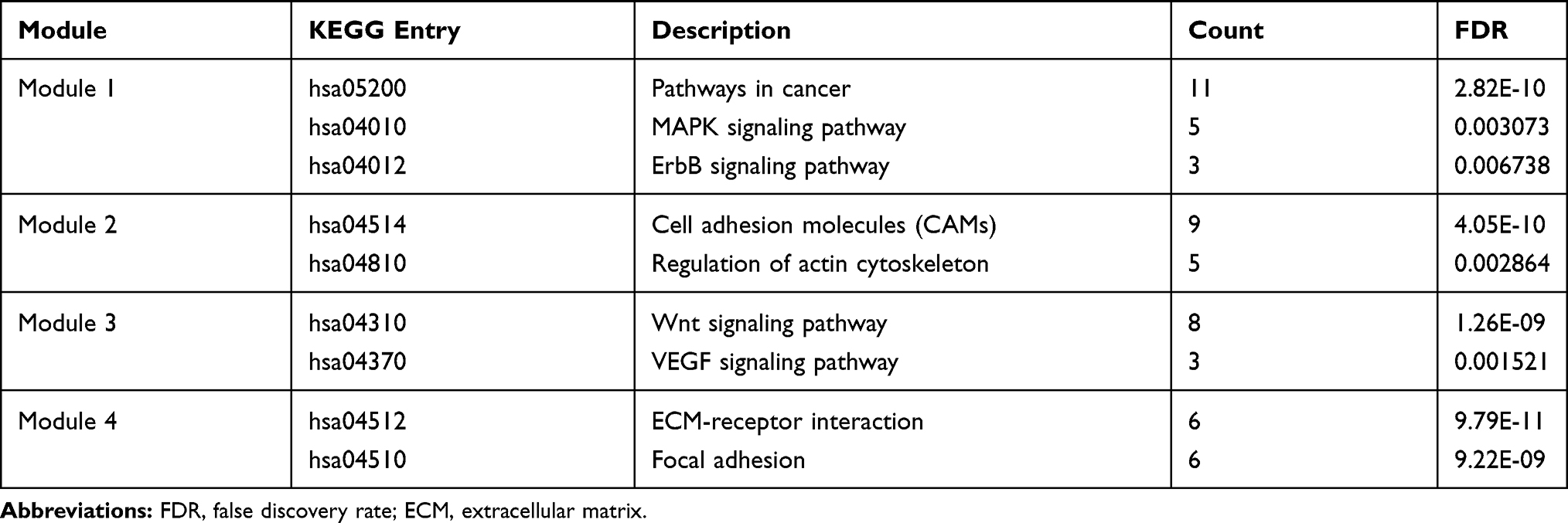 |
Table 1 KEGG Enrichment Analysis of Top 4 Modules Identified from PPI Network |
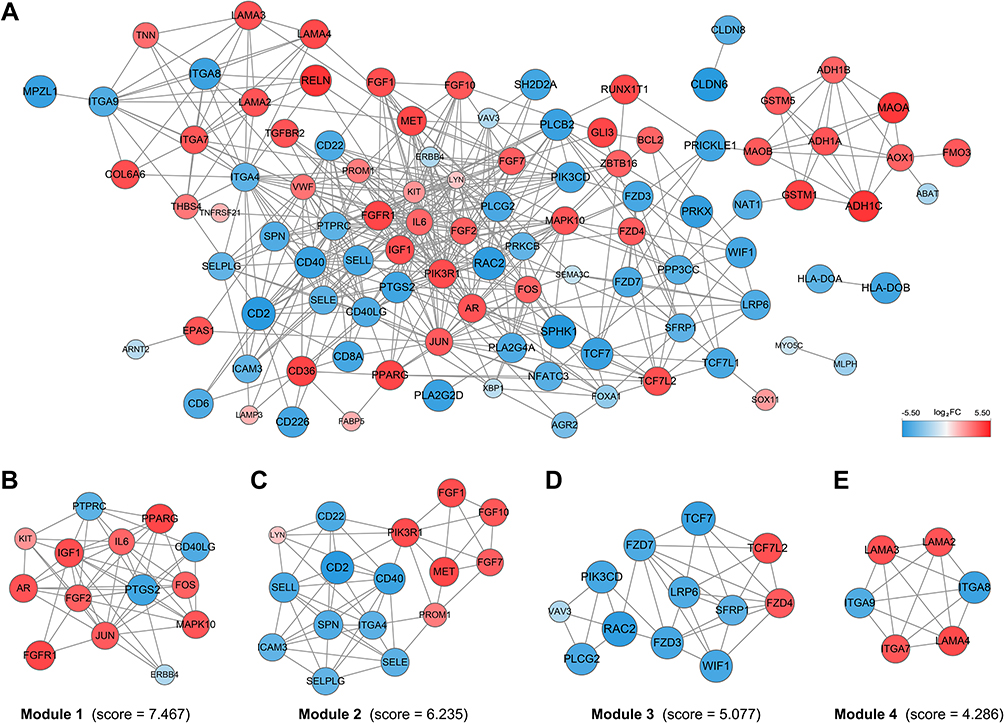 |
Figure 3 PPI network construction and functional module analysis. Notes: (A) The whole PPI network. (B) Network of functional module 1. (C) Network of functional module 2. (D) Network of functional module 3. (E) Network of functional module 4.Abbreviation: PPI, protein–protein network. |
Hub Genes Screening, Survival Analysis, and Expression Validation
Plug-in cytohubba of Cytoscape was used to identify hub genes from the whole PPI network. According to the ranking by the MMC method, five hub genes (PTPRC, IL6, SELL, CD40, and SPN) were filtered out. The sub-network of these genes is demonstrated in Figure 4A.
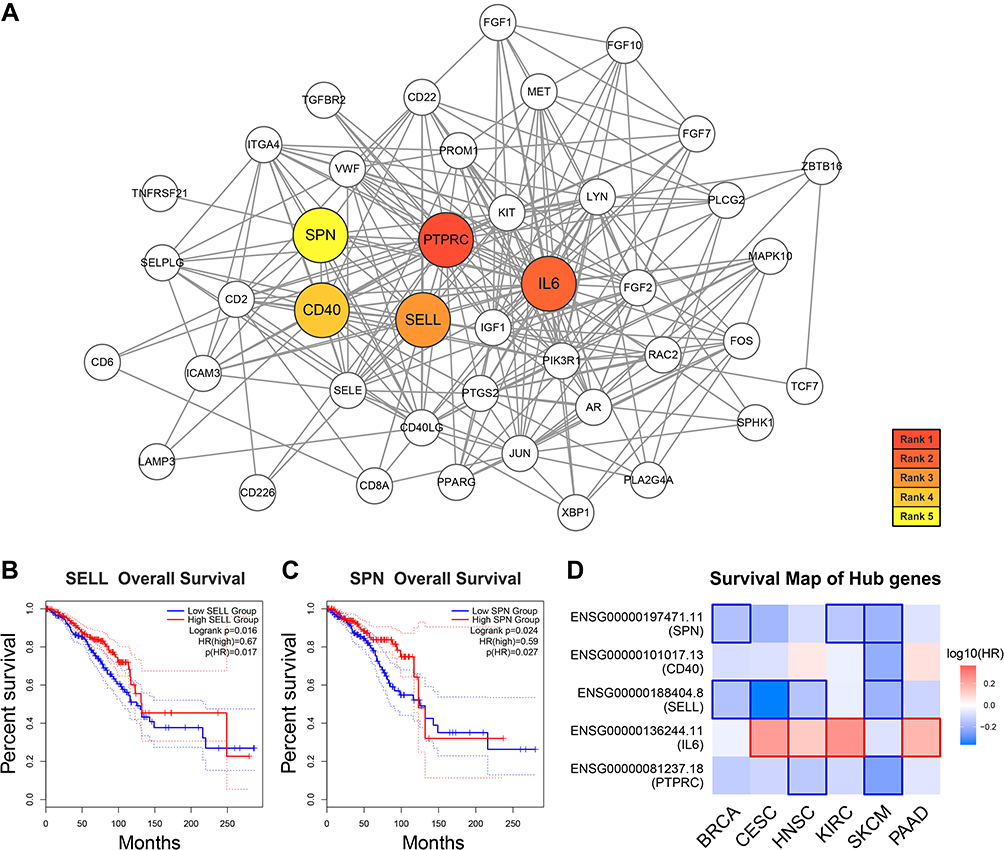 |
Figure 4 Hub genes identification and survival analysis. Notes: (A) PPI sub-network of the five hub genes. (B) Kaplan–Meier survival analysis of SELL. (C) Kaplan–Meier survival analysis of SPN. (D) Survival map of hub genes in multiple malignancies. |
To further explore the effect of hub genes expression on patients’ clinical outcomes, a total of 1069 breast cancer samples were derived into high- and low-expression groups according to the median expression level of each hub gene respectively, and survival analysis was performed by GEPIA2. In the five hub genes, SELL, and SPN were significantly associated with the overall survival of breast cancer (log-rank PSELL = 0.016, PSPN = 0.024; Figure 4B and C). However, PTPRC, IL6 and CD40 were not significantly correlated to breast cancer prognosis (log-rank P > 0.05; Supplementary Figure 1). Moreover, cox regression analysis was performed in multiple malignancies. The survival map showed that PTPRC, SELL, CD40, and SPN tended to reduce the risk of tumor diseases, and IL6 tended to be associated with poorer prognosis (Figure 4D).
In addition, we validated the expression of hub genes in clinical specimens. As shown in Figure 5, the relative expression level of PTPRC, SELL, CD40 and SPN in IBC samples were significantly lower than in non-IBC samples (P < 0.05), and IL6 expression in IBC samples were significantly higher (P < 0.05). This result was consistent with the bioinformatics results.
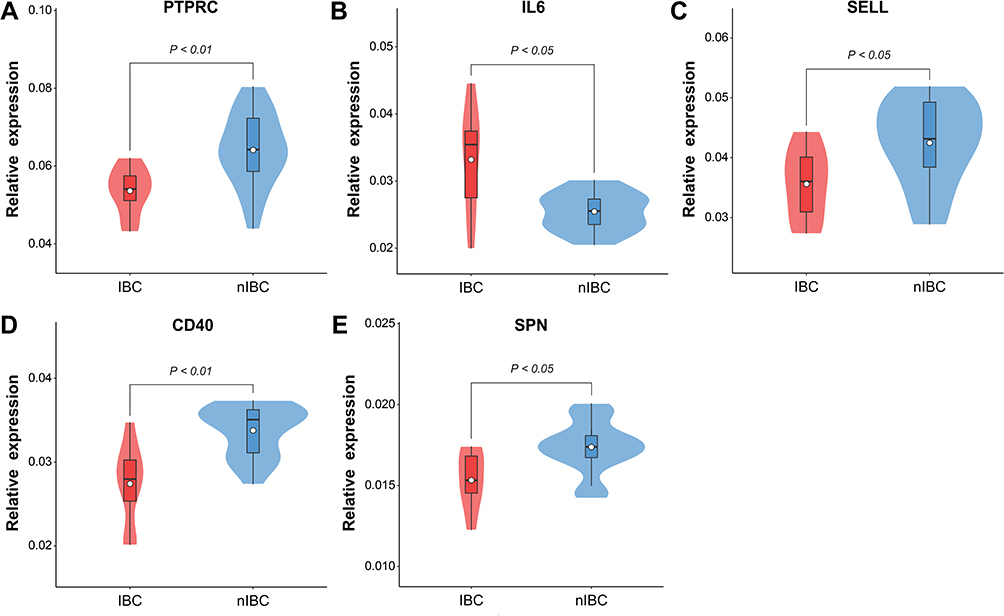 |
Figure 5 Validation of hub genes expression in IBC and non-IBC clinical samples by qRT-PCR. Notes: The relative expression level of the five hub genes PTPRC (A), IL6 (B), SELL (C), CD40 (D) and SPN (E) are shown in violin plot.Abbreviation: qRT-PCR, quantitative reverse transcriptional polymerase chain reaction. |
Discussion
In the present study, a total of 114 DEGs, including 53 upregulated and 61 downregulated genes, were identified from four GEO datasets of IBC. The results of GO and KEGG enrichment analyses indicated that the DEGs were strongly associated with multiple cancer-related functions and pathways, such as cell adhesion, cell proliferation, Wnt, PI3K-AKT, VEGF, and Ras signaling pathways, etc. The PPI network of selected DEGs was constructed basing on the STRING database, and module analysis was performed to further explore functional sub-networks. Finally, five hub genes were screened out from the whole PPI network, including PTPRC, IL6, SELL, CD40, and SPN. Survival analysis and expression validation of the five hub genes verified the robustness of the above results.
In recent years, an increasing number of studies working on DEGs in human cancers have been published. Due to the development of high-throughput technologies, including microarray and RNA-sequencing, current researches have been paying more attention to tumor genomes and transcriptomes instead of isolated genes. In a study basing on the original IBC dataset GSE5847 from GEO,13 368 DEGs were identified. According to GO and KEGG analyses, the DEGs were mainly enriched in cell adhesion, ECM-receptor interaction, angiogenesis, and focal adhesion, which were also involved in our results. However, for hub genes, there is no intersection between this study and ours. Compared with similar published researches on IBC, our study has a much larger sample size, consisting of 131 IBC and 238 non-IBC samples from up to four unique datasets. This ensures the robustness and high confidence in our results.
Based on the PPI network, the five hub genes were identified to be significantly associated with IBC pathogenesis and progression. Among these genes, IL-6 has been reported to be a candidate oncogene of IBC.14 IL-6 encodes a cytokine that functions in inflammation and the maturation of B cells, and is implicated in a wide range of inflammation-associated diseases, including almost all types of tumors.15,16 Molecular evidence has suggested that inflammatory signaling pathways are upregulated in IBC, especially NF-κB and IL-6.17 Moreover, aberrant IL-6 expression is proved to increase tumor stage, lymph node involvement, recurrence risk, and distant metastasis of breast cancer.18–20 In this study, IL-6 was identified to be a core gene of IBC and was strongly associated with poor prognosis of breast cancer. These findings are consistent with the previous ones.
Another inflammation associated gene, CD40, was also screened as a hub gene in our study. It is a member of the TNF-receptor superfamily, is essential for mediating a broad variety of immune and inflammatory responses.21,22 It is widely recognized that CD40 plays key roles in tumor immunity by inducting productive anti-tumor T cell immune responses, and its agonists have been applied to cancer immunotherapy in various malignancies.23–25 Interestingly, a study of gene expression in IBC reported that, compared with non-IBC samples, a series of NF-κB-regulated genes (including CD40) were observed to be upregulated in IBCs.26 Our study showed that CD40 is significantly downregulated in IBC, and is associated with favorable prognosis. In combination with the majority of previous studies, we tend to believe the tumor suppression effect of CD40 on IBC.
For hub gene PTPRC, SELL, and SPN, there have been few reports about their effect on IBC. PTPRC encodes a protein which is a member of protein tyrosine phosphatase (PTP) family, and has been recognized as an essential regulator of T- and B-cell antigen receptor signaling.27,28 SELL, which is also known as CD62L, is also related to inflammation and immunity.29 The protein it encodes is a cell surface adhesion molecule and takes part in selectin-induced immune modulation in cancer progression.30,31 SPN is also named CD43, it encodes a glycoprotein that functions in antigen-specific activation of T cells, and is reported to be a candidate biomarker of tumor diseases.27,32 In the present study, these genes are down-regulated in IBC and encode key proteins according to the PPI network. However, more molecular evidence is still required to verify the association between these genes and IBC.
To the best of our knowledge, our study is the first one to perform a combined bioinformatic analysis of multiple transcriptome microarray datasets on IBC. Compared with the small sample sized, single centered researches, the results of our study are less interfered by confounding factors and are more credible. On the other hand, our study has several limitations. Firstly, since TCGA database does not provide enough information for us to identify IBC samples from non-IBC ones, the survival analysis of the identified hub genes was performed using samples of all breast cancer subtypes. Therefore, although this result contributes to the understanding of the biological relevance of the hub genes, further study is still required to evaluate their prognostic values on IBC. Secondly, the association between the hub genes and clinicopathological features of IBC was not analyzed in this study due to the incomplete clinical information of GEO samples. We will perform these analyses when we collect enough specimens of IBC.
Take together, the identified five hub genes, PTPRC, IL6, SELL, CD40, and SPN, have been suggested to be potential key genes in IBC development and progression. These findings may provide a novel understanding of the pathogenesis of IBC, and these genes may be further considered as therapeutic targets. Moreover, high SELL and SPN expression were associated with a favorable prognosis of breast cancer, suggesting that the identified hub genes may have prognostic value. Further study is still needed to investigate the association between hub genes and survival in IBC samples and elucidate the underlying mechanisms of their effect on IBC.
Conclusion
In the present study, 114 DEGs between IBC and non-IBC were identified by bioinformatic analysis based on transcriptome microarray data from GEO database, and five hub genes (PTPRC, IL6, SELL, CD40, and SPN) were screened out. These genes are related to inflammation and immunity processes and may play key roles in IBC development and progression. Further study is needed to validate the results of our research.
Disclosure
The authors report no conflicts of interest in this work.
References
1. van Uden DJ, van Laarhoven HW, Westenberg AH, et al. Inflammatory breast cancer: an overview. Crit Rev Oncol Hematol. 2015;93(2):116–126. doi:10.1016/j.critrevonc.2014.09.003
2. Menta A, Fouad TM, Lucci A, et al. Inflammatory breast cancer: what to know about this unique, aggressive breast cancer. Surg Clin North Am. 2018;98(4):787–800. doi:10.1016/j.suc.2018.03.009
3. Hance KW, Anderson WF, Devesa SS, et al. Trends in inflammatory breast carcinoma incidence and survival: the surveillance, epidemiology, and end results program at the National Cancer Institute. J Natl Cancer Inst. 2005;97(13):966–975. doi:10.1093/jnci/dji172
4. Lim B, Woodward WA, Wang X, et al. Inflammatory breast cancer biology: the tumour microenvironment is key. Nat Rev Cancer. 2018;18(8):485–499. doi:10.1038/s41568-018-0010-y
5. Nguyen DM, Sam K, Tsimelzon A, et al. Molecular heterogeneity of inflammatory breast cancer: a hyperproliferative phenotype. Clin Cancer Res. 2006;12(17):5047–5054. doi:10.1158/1078-0432.CCR-05-2248
6. Kleer CG, van Golen KL, Braun T, et al. Persistent E-cadherin expression in inflammatory breast cancer. Mod Pathol. 2001;14(5):458–464. doi:10.1038/modpathol.3880334
7. Xiao Y, Ye Y, Zou X, et al. The lymphovascular embolus of inflammatory breast cancer exhibits a Notch 3 addiction. Oncogene. 2011;30(3):287–300. doi:10.1038/onc.2010.405
8. Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16(1):45–55. doi:10.1158/1078-0432.CCR-09-1630
9. Boersma BJ, Reimers M, Yi M, et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer. 2008;122(6):1324–1332. doi:10.1002/ijc.23237
10. Iwamoto T, Bianchini G, Qi Y, et al. Different gene expressions are associated with the different molecular subtypes of inflammatory breast cancer. Breast Cancer Res Treat. 2011;125(3):785–795. doi:10.1007/s10549-010-1280-6
11. Bekhouche I, Finetti P, Adelaïde J, et al. High-resolution comparative genomic hybridization of inflammatory breast cancer and identification of candidate genes. PLoS One. 2011;6(2):e16950. doi:10.1371/journal.pone.0016950
12. Woodward WA, Krishnamurthy S, Yamauchi H, et al. Genomic and expression analysis of microdissected inflammatory breast cancer. Breast Cancer Res Treat. 2013;138(3):761–772. doi:10.1007/s10549-013-2501-6
13. Chai F, Liang Y, Zhang F, et al. Systematically identify key genes in inflammatory and non-inflammatory breast cancer. Gene. 2016;575(2 Pt 3):600–614. doi:10.1016/j.gene.2015.09.025
14. Morrow RJ, Etemadi N, Yeo B, et al. Challenging a misnomer? The role of inflammatory pathways in inflammatory breast cancer. Mediators Inflamm. 2017;2017:4754827. doi:10.1155/2017/4754827
15. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harb Perspect Biol. 2018;10(2):a028415. doi:10.1101/cshperspect.a028415
16. Kumari N, Dwarakanath BS, Das A, et al. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016;37(9):11553–11572. doi:10.1007/s13277-016-5098-7
17. Drygin D, Ho CB, Omori M, et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem Biophys Res Commun. 2011;415(1):163–167. doi:10.1016/j.bbrc.2011.10.046
18. Badache A, Hynes NE. Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of T47D breast cancer cells. Cancer Res. 2001;61(1):383–391.
19. Chen L, Shulman LM, Revel MIL-6. receptors and sensitivity to growth inhibition by IL-6 in clones of human breast carcinoma cells. J Biol Regul Homeost Agents. 1991;5(4):125–136.
20. Tamm I, Cardinale I, Krueger J, et al. Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. J Exp Med. 1989;170(5):1649–1669. doi:10.1084/jem.170.5.1649
21. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell. 2018;33(4):563–569. doi:10.1016/j.ccell.2018.03.008
22. Elgueta R, Benson MJ, de Vries VC, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229(1):152–172. doi:10.1111/j.1600-065X.2009.00782.x
23. Beatty GL, Li Y, Long KB. Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther. 2017;17(2):175–186. doi:10.1080/14737140.2017.1270208
24. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–1043. doi:10.1158/1078-0432.CCR-12-2064
25. Mangsbo SM, Broos S, Fletcher E, et al. The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin Cancer Res. 2015;21(5):1115–1126. doi:10.1158/1078-0432.CCR-14-0913
26. Lerebours F, Vacher S, Andrieu C, et al. NF-kappa B genes have a major role in inflammatory breast cancer. BMC Cancer. 2008;8:41. doi:10.1186/1471-2407-8-41
27. Junghans V, Santos AM, Lui Y, et al. Dimensions and Interactions of large T-cell surface proteins. Front Immunol. 2018;9:2215. doi:10.3389/fimmu.2018.02215
28. Nosho K, Baba Y, Tanaka N, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222(4):350–366. doi:10.1002/path.2774
29. Ivetic A, Hoskins Green HL, Hart SJ. L-selectin: a major regulator of leukocyte adhesion, migration and signaling. Front Immunol. 2019;10:1068. doi:10.3389/fimmu.2019.01068
30. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). 2016;8(3):36. doi:10.3390/cancers8030036
31. Borsig L. Selectins in cancer immunity. Glycobiology. 2018;28(9):648–655. doi:10.1093/glycob/cwx105
32. Tuccillo FM, de Laurentiis A, Palmieri C, et al. Aberrant glycosylation as biomarker for cancer: focus on CD43. Biomed Res Int. 2014;2014:742831. doi:10.1155/2014/742831
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.