Back to Journals » Infection and Drug Resistance » Volume 11
Identification of key genes and pathways using bioinformatics analysis in septic shock children
Authors Yang J, Zhang S, Zhang J, Dong J, Wu J, Zhang L, Guo P ![]() , Tang S, Zhao Z, Wang H, Zhao Y, Zhang W, Wu F
, Tang S, Zhao Z, Wang H, Zhao Y, Zhang W, Wu F
Received 16 November 2017
Accepted for publication 30 May 2018
Published 14 August 2018 Volume 2018:11 Pages 1163—1174
DOI https://doi.org/10.2147/IDR.S157269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Joachim Wink
Junting Yang,1,* Shunwen Zhang,1,2,* Jie Zhang,3 Jiangtao Dong,3 Jiangdong Wu,1 Le Zhang,1 Peng Guo,3 Suyu Tang,3 Zhengyong Zhao,1 Hongzhou Wang,1 Yanheng Zhao,3 Wanjiang Zhang,1 Fang Wu1
1Department of Pathophysiology, Shihezi University School of Medicine Shihezi, Xinjiang, China; 2The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China; 3The First Affiliated Hospital of Shihezi University School of Medicine, Shihezi, Xinjiang, China
*These authors contributed equally to this work
Background and hypothesis: Sepsis is still one of the reasons for serious infectious diseases in pediatric intensive care unit patients despite the use of anti-infective therapy and organ support therapy. As it is well-known, the effect of single gene or pathway does not play a role in sepsis. We want to explore the interaction of two more genes or pathways in sepsis patients for future works. We hypothesize that the discovery from the available gene expression data of pediatric sepsis patients could know the process or improve the situation.
Methods and results: The gene expression profile dataset GSE26440 of 98 septic shock samples and 32 normal samples using whole blood-derived RNA samples were generated. A total of 1,108 upregulated and 142 downregulated differentially expressed genes (DEGs) were identified in septic shock children using R software packages. The Gene Ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway were analyzed using DAVID software; Gene Set Enrichment Analysis method was also used for enrichment analysis of the DEGs. The protein–protein interaction (PPI) network and the top 10 hub genes construction of the DEGs were constructed via plug-in Molecular Complex Detection and cytoHubba of Cytoscape software. From the PPI network, the top 10 hub genes, which are all upregulated DEGs in the septic shock children, were identified as GAPDH, TNF, EGF, MAPK3, IL-10, TLR4, MAPK14, IL-1β, PIK3CB, and TLR2. Some of them were involved in one or more significant inflammatory pathways, such as the enrichment of tumor necrosis factor (TNF) pathway in the activation of mitogen-activated protein kinase activity, toll-like receptor signaling pathway, nuclear factor-κB signaling pathway, PI3K-Akt signaling pathway, and TNF signaling pathway. These findings support future studies on pediatric septic shock.
Keywords: pediatric septic shock, microarray, differentially expressed gene, bioinformatics analysis
Introduction
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection.1,2 Sepsis and septic shock are major healthcare problems affecting millions of people around the world each year and killing as many as one in four and even more patients.3,4 Septic shock is a heterogeneous syndrome within which several biological subclasses probably exist. The discovery and identification of septic shock subclasses could provide the foundation for the design of more specifically targeted therapies.5,6 Furthermore, it has been demonstrated that multiple genes and signaling pathways participate in the occurrence and development of pediatric septic shock. So, it is extremely important to study the molecular mechanism or the pathological process of pediatric septic shock in the development of more effective diagnosis, therapy, and prognosis. Nowadays, microarray technology combined with bioinformatics to analyze disease has made it feasible to analyze the changes in DNA or RNA expression during the development and prognosis of pediatric septic shock. We can now also investigate the interactions among differentially expressed genes (DEGs) and analyze the functional annotation and the enrichment pathways in the interaction network.
In the present study, the GSE26440 data were downloaded from Gene Expression Omnibus (GEO).5 The expression data were generated from 98 children with septic shock and 32 normal children using whole blood-derived RNA samples who were admitted to the pediatric intensive care unit within the first 24 hours.
Gene expression profiles of septic shock patients were compared with normal children to identify DEGs. First, the DEGs were identified by limma packages of R software from the GEO database and their involvement in biomedical process (BP), cell component (CC), molecular function (MF) through Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) in DAVID, which is an online tool, and Gene Set Enrichment Analysis (GSEA) software (3.0 version) were also identified. Ultimately, we aimed to explore the candidate biomarkers for diagnosis, clinical care, and prognosis in septic shock children.
Materials and methods
Microarray data and identification of DEGs
The microarray expression dataset GSE26440 in the septic shock and normal children was obtained from Affymetrix GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array), which was submitted by Wong et al in January 04, 2011, and updated in September 15, 2017. Unlike Wong et al, we mainly focused on the effects of the body’s immune response, apoptosis cells, and changes in intestinal function in septic shock children. The packages affy and affyPLM of R software were used to read the data with the k-nearest neighbor method to fill in missing value and the RMA algorithm was used to normalize and correct the data. Then, the data were screened for DEGs between the septic shock and normal children with criteria of fold change (FC) >2 and adjusted P-value <0.05 for statistically significant difference by using the limma package of Bioconductor.
DEGs analyze via GO, KEGG, and GSEA
The GO project develops and uses a set of structured, controlled vocabularies for community use in annotating genes, gene products, and gene sequences.7–9 The expression matrix of all genes and phenotypes of the samples were submitted to the GSEA software to analyze the GO enrichment and to determine whether the DEGs show statistically significant difference between the septic shock and normal children. The KEGG is a knowledge base used for systematic analysis of gene functions, linking genomic information with higher order functional information of the DEGs.10,11 To analyze the DEGs at the functional level, the DAVID 9th database for annotation, visualization, and integrated discovery, which is an online tool for the analysis of the relevant biological annotation of gene lists,12 was used to provide functional interpretation of the GO enrichment and KEGG pathway analysis for DEGs. The P-value <0.05 was considered statistically significant.
Protein–protein interaction network and modules analysis
The STRING database is an online tool designed to evaluate the protein–protein interaction (PPI) information.13–17 The STRING (version 10.5) database currently covers 9,643,763 proteins from 2,031 organisms. To assess the interactive relationships, the identified DEGs were mapped via STRING, and the experimentally validated interactions were ensured as statistically significant with a combined score of >0.4. Then, the Cytoscape software (version 3.6.0) was used to construct the PPI networks between the DEGs. The plug-in named Molecular Complex Detection (MCODE) and cytoHubba were individually used to determine the modules and the top 10 hub genes of the PPI network in Cytoscape. The criteria were set as MCODE scores >6 and the number of nodes >6.
Results
Identification of DEGs
We obtained a total of 1,250 DEGs in the GSE26440 dataset, including 1,108 upregulated DEGs and 142 downregulated DEGs, via R software using criteria of the FC>2 and adjusted P-value <0.05 from 98 children with septic shock and 32 normal children. For example, we found that glyceraldehyde-3-phosphate dehydrogenase (GAPDH), tumor necrosis factor (TNF), and epidermal growth factor (EGF) were highly expressed in children with septic shock. Table 1 contains the top 25 downregulated DEGs and the top 25 upregulated DEGs with FC>2 and adjusted P<0.05.
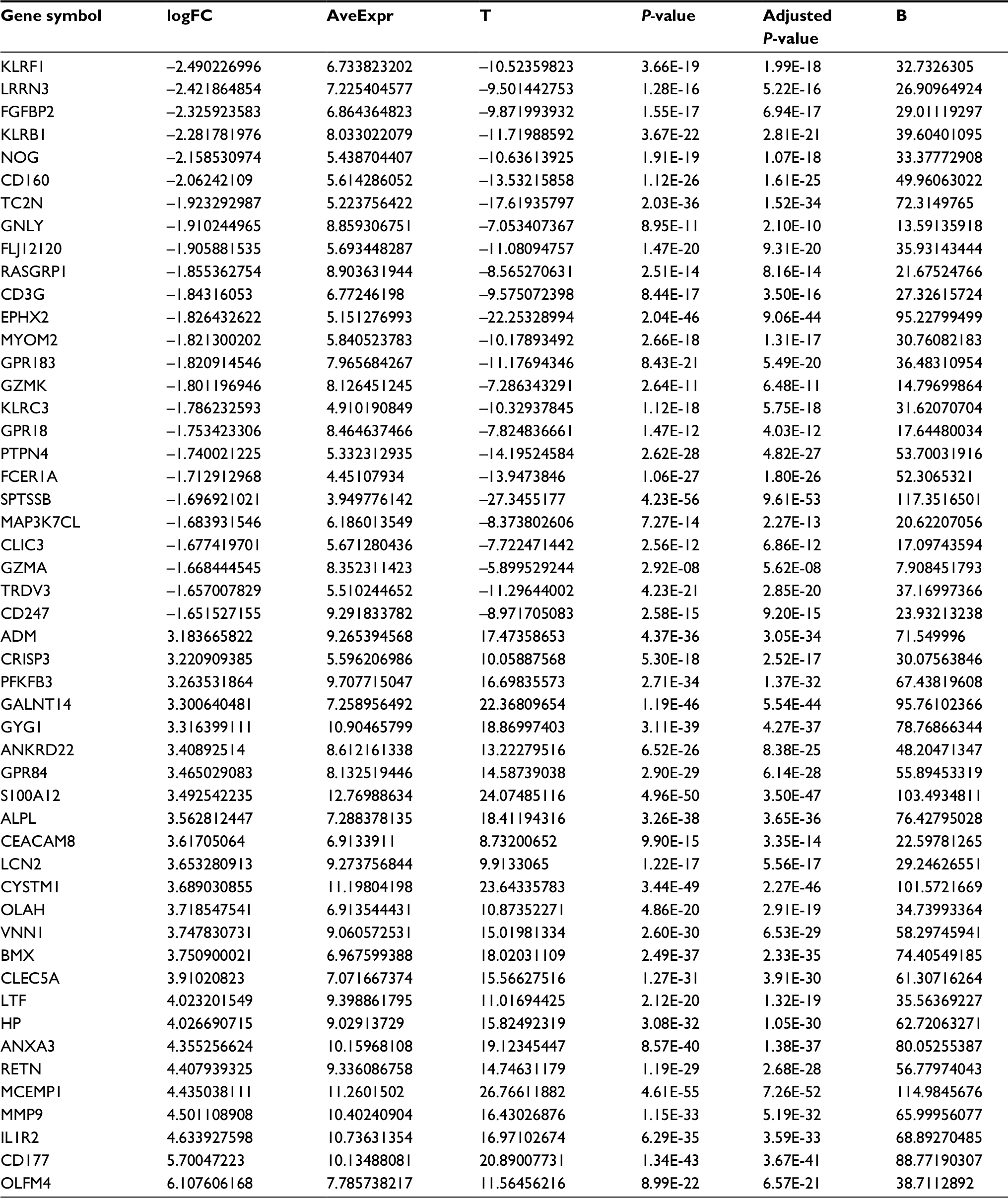 | Table 1 The fold change of the top 25 downregulated DEGs and the top 25 upregulated DEGs Abbreviation: DEG, differentially expressed gene. |
The heat map and volcano map of all DEGs are displayed in Figure 1A and B. The top 50 upregulated and the top 50 downregulated DEGs from GSEA software are displayed in Figure 2.
 | Figure 1 Heat map and the expression changes of all genes. Notes: (A) Heat map of all genes, yellow is upregulated genes and blue is downregulated genes, (B) volcano map displays the expression changes of all genes. Red is the differentially expressed genes. Abbreviation: FC, fold change. |
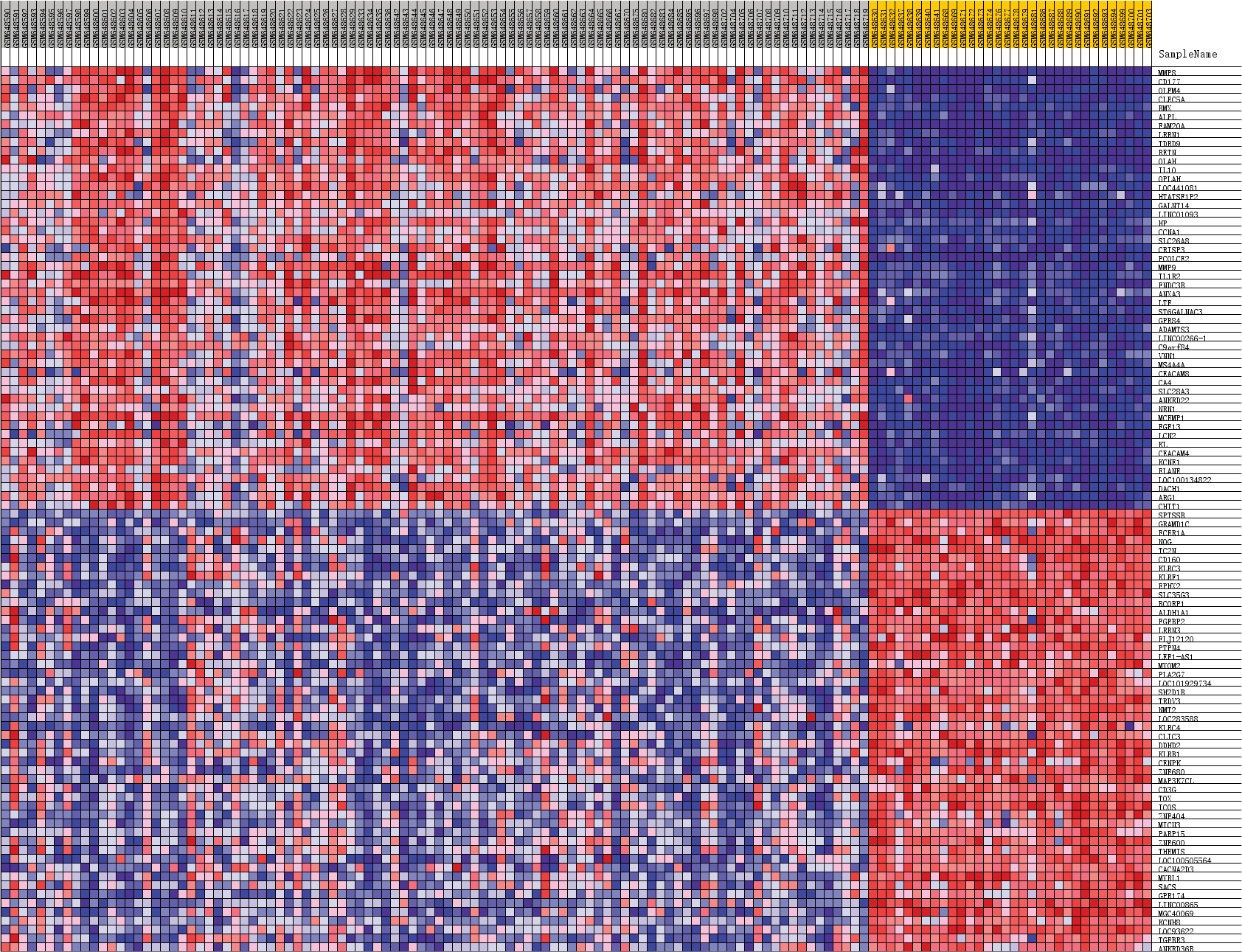 | Figure 2 Heat map of the top 50 upregulated DEGs and the top 50 downregulated DEGs from Gene Set Enrichment Analysis. Abbreviation: DEGs, differentially expressed genes. |
Enrichment function and KEGG pathway analysis via DAVID and GSEA software
All DEGs were uploaded into the online software DAVID to evaluate the GO categories and KEGG pathways. The results revealed that the DEGs are significantly enriched in BP, such as inflammatory response, immune response, cellular defense response, innate immune response, lipopolysaccharide-mediated signaling pathway (Figure 3A), and in CC, such as the extracellular exosome, extracellular space, plasma membrane, membrane, and integral component of plasma membrane (Figure 3B), as well as in MF, such as protein binding, receptor activity, carbohydrate binding, arachidonic acid binding, and protein homodimerization activity (Figure 3C). The KEGG pathway results revealed that the DEGs are significantly enriched in TNF signaling pathway, NOD-like receptor signaling pathway, nuclear factor (NF)-κB signaling pathway, and Jak-STAT signaling pathway (Figure 3D).
 | Figure 3 The enrichment function and pathway analysis. (A) Biomedical process (BP), (B) cell component (CC), (C) molecular function (MF), (D) KEGG pathway. |
However, the results of DEGs enriched by the functional analysis of GO with the GSEA software revealed that DEGs involved in phosphatidylinositol 3 kinase binding, regulation of vascular permeability, vitamin transport, and organic acid transmembrane transporter activity were significantly enriched in gene sets (Figure 4). In those results, we will focus on regulation of vascular permeability with core genes such as ADM, AZU1, PDE2A, AMOT, PED3A, PEPRJ, TEK, HRH1, VEGFA, UCN, TACR1, SRC, SLIT2, and TACR2 (Figures 5 and 6). As we know, when the permeability of blood vessels increases, bacteria and toxins can pass through the enteric cavity into the intestinal lymph nodes, blood, liver, spleen, and other organs, causing a greater inflammatory response and can even lead to failure of multiple organ dysfunction.18 The lower permeability of the intestinal tissue can effectively reduce or limit the translocation of pathogenic bacteria/endotoxins and reduce or delay the onset of sepsis.19,20 We do not know yet if the change in the expression levels of these genes could reduce the transportation of bacteria and endotoxin from intestinal cavity to other tissues and blood, contributing to good conditions for pediatric septic shock through the influence on intestinal permeability. Perhaps, some experiments are needed to prove this.
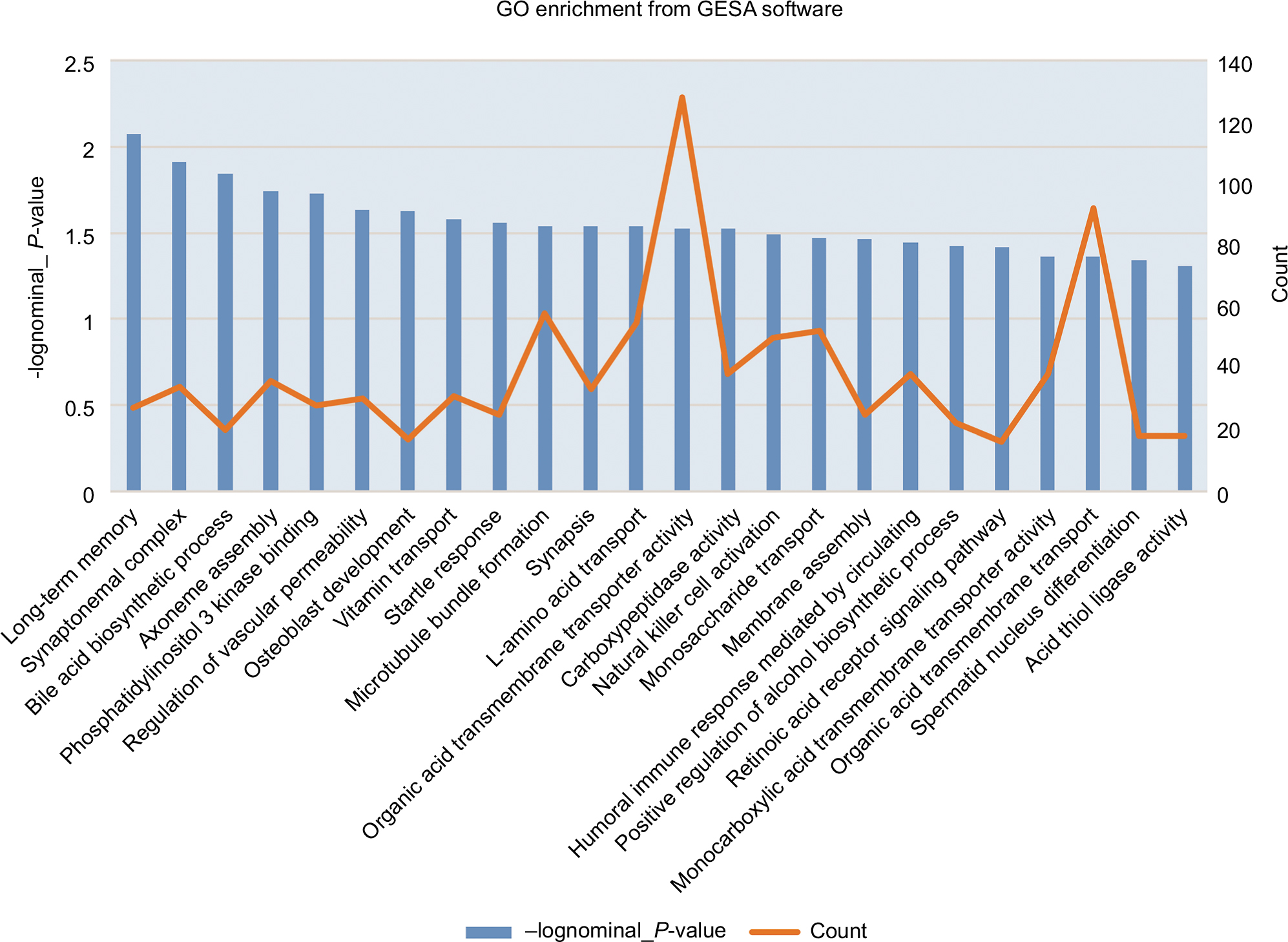 | Figure 4 GO enrichment analysis from GSEA. Abbreviations: GO, Gene Ontology; GSEA, Gene Set Enrichment Analysis. |
 | Figure 5 Enrichment plot of regulation of vascular permeability from Gene Set Enrichment Analysis. Abbreviation: GO, Gene Ontology. |
 | Figure 6 Heat map of gene set of regulation of vascular permeability from Gene Set Enrichment Analysis. |
Screen modules and the top 10 hub genes of the PPI network via Cytoscape software
The interactive information from the analysis with the STRING database (Figure 7) was exported to the Cytoscape software. After that the modules and top 10 hub genes with higher degrees were individually screened via Cytoscape software. In all the modules, a total of 312 nodes and 1,420 edges were analyzed with the plug-in MCODE. The top four modules were selected, and the enriched pathways of the DEGs involved in the modules were analyzed. The results showed that the DEGs included in the top four modules were mainly associated with chemokine signaling pathway. For example, in module1, they included the pathways such as cytokine–cytokine receptor interaction, Ras signaling pathway, PI3K-Akt signaling pathway, cMAP signaling pathway, and neuroactive (Figure 8). In addition, the top 10 hub DEGs were analyzed by the plug-in cytoHubba, and a subnetwork of the top 10 hub genes was obtained. The network revealed that there were interactions among these genes, and the interactions may affect the pathophysiologic process, diagnosis, therapy, and prognosis (Figure 9A). More experiments will be needed to verify this. These included GAPDH, TNF, EGF, mitogen-activated protein kinase 3 (MAPK3), interleukin 10 (IL-10), toll-like receptor 4 (TLR4), mitogen-activated protein kinase 14 (MAPK14), interleukin 1 beta (IL-1β), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (PIK3CB), and toll-like receptor (TLR2). Among these DEGs, GAPDH showed the highest score of 159 (Figure 9B). All the hub genes were analyzed for the enrichment function (Figure 9C) and pathways (Figure 9D).
 | Figure 7 Network of differentially expressed genes from STRING. |
 | Figure 8 Top four modules from protein–protein interaction networks. (A) module 1, (B) module 2, (C) module 3, (D) module 4, (E) the enriched pathway of module 1, (F) the enriched pathway of module 2, (G) the enriched pathway of module 3, and (H) the enriched pathway of module 4. |
 | Figure 9 Subnetwork of hub gene from protein–protein interaction networks; red color is the upregulated genes. Notes: (A) The top ten hub genes, (B) the gene score of the top ten hub genes, (C) the GO terms of the top ten hub genes, (D) and the pathway terms of the top ten hub genes. |
Discussion
Septic shock is an infection-based disease entity. It is not a singular, homogenous disease in the traditional sense.5 To investigate the molecular mechanism of pediatric septic shock is critically important for the diagnosis, treatment, and prognosis.
The expression profiling by microarray can provide information about the expression differences of thousands of genes in human genome. In this study, we used it to predict and find the key or potential genes for pediatric septic shock. We screened the microarray data GSE26440 and obtained 1,108 upregulated DEGs and 142 downregulated DEGs from 98 samples of septic shock children and 32 normal children via bioinformatics analysis.
In this study, our main concern was about the inflammatory process and immune response of the total DEGs, which included upregulated DEGs and downregulated DEGs. The GO enrichment analysis of DEGs revealed that some of them were significantly involved in inflammatory response, immune response, cellular defense response, innate immune response, lipopolysaccharide-mediated signaling pathway, all the above can significantly influence the biomedical process of sepsis.21–23 Such a research reported that the delivery of miR-126 of extracellular exosome prevents microvascular dysfunction and refines the outcomes of sepsis.24
In our PPI network construction with the DEGs, top four modules and top 10 hub genes were identified. Further study on these candidate DEGs could provide new information and ideas to research the process or development, even for the diagnostic and therapeutic studies in pediatric septic shock. The top 10 hub DEGs with higher degree are as follows: GAPDH, TNF, EGF, MAPK3, IL-10, TLR4, MAPK14, IL-1β, PIK3CB, and TLR2. The results of the GO enrichment analysis showed that some DEGs were involved in lipopolysaccharide-mediated signaling pathway, activation of MAPK activity, positive regulation of gene expression, positive regulation of NF-κB import into nucleus, extracellular space, plasma membrane, extracellular exosome, cell surface, and so on. The enriched pathway analysis showed that some DEGs were involved in toll-like receptor signaling pathway, Hypoxia-inducible factor-1 (HIF-1) signaling pathway, T-cell receptor signaling pathway, TNF signaling pathway, FoxO signaling pathway, NF-κB signaling pathway, MAPK signaling pathway, and PI3K-Akt signaling pathway.
The immune cells, such as macrophages, dendritic cells, T cells, and regulatory T cells (Tregs), are involved in the regulation of proinflammatory responses and promote inflammation by secreting or producing proinflammatory mediators. The inflammatory response can also be balanced by secreting anti-inflammatory cytokines. In advanced stages, patients with sepsis exhibit an immunosuppressive response, which is mainly manifested by a decrease in the number of immune cells and a decline in immune function.25
In these hub genes, GAPDH, acting on a transfer receptor on the cell surface of macrophage, has recently been demonstrated in several nonmetabolic processes, such as initiation of apoptosis, and transcription.26 It can lead to cell death because of the NO-induced GAPDH aggregation, mitochondrial dysfunction, and impaired fibrinolysis.27,28 The results of GO terms and enriched pathways mainly showed in plasma membrane, extracellular exosome, and HIF-like receptor signaling pathway. These studies suggested that TNF-α and IL-6 can increase the permeability of intestinal tissue; however, IL-10 has a protective effect on the intestinal wall tissue.29,30 TNF-α is mainly produced by activated macrophages. Proinflammatory cytokine involved in systemic inflammation make up the acute phase reaction and play a primary role in the regulation of immune cell.31 EGF with EGFR leads to cellular proliferation, differentiation, and survival.32 It plays an important physiological role in the maintenance of gastric tissue, healing of gastroesophageal ulcers, and inhibiting bacterial agents.33,34 MAPK3 (ERK1) and MAPK14 (P38-α) are involved in directing cellular response to a diverse array of stimuli, such as proinflammatory cytokines and heat shock, and regulated cell function such as proliferation, gene expression, cell survival, and apoptosis.35 Toll-like receptors (TLRs) are a class of proteins that play an important role in the innate immune system. The antigens can be recognized by TLR2 and lipopolysaccharides by TLR4.TLR4 plays a fundamental role in pathogen recognition and activation of innate immunity and inhibits the TLR4/NF-κB signaling pathway, thereby alleviating sepsis-induced acute lung injury in rats.36–38 So, the lipid A analog eritoran is being developed as a drug against severe sepsis, which acts as a TLR4 antagonist.39 IL-1β reported as one of the proinflammatory cytokines is expressed by IL-1 and works on initiating the inflammatory response to TNF-α.40,41 In the body of sepsis patients, macrophages are overactivated and a large number of proinflammatory cytokines are released when the proinflammatory and anti-inflammatory responses are out of balance, such as IL-10 is reported as an anti-inflammatory cytokines and mainly secreted by M2 subtype macrophages.42 Also PIK3CB, the p110delta isoform of the kinase PI3K, can protect from endotoxic shock.43 Currently, the role of EGF and PIK3CB in sepsis has not been studied clearly yet.
Conclusion
The results of this research identified DEGs in septic shock children through a widespread bioinformatics analysis. Our study indicates that these DEGs may play important roles in the progress of septic shock patient. The results revealed a set of useful candidate genes for the investigation of biomarkers or molecular mechanisms of septic shock. In the future, we need to carry out further experiments to research on the change in expression levels and the function and influence of these DEGs involved in one or more pathways of the DEGs in children with septic shock.
Acknowledgments
We thank Wong HR who uploaded the GSE26440 data.
This research received no specific grant from any funding agency in the public or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–377. | ||
Shankar-Hari M, Phillips GS, Levy ML, et al; Sepsis Definitions Task Force. Developing a new definition and assessing new clinical criteria for septic shock: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):775–787. | ||
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. | ||
Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–1554. | ||
Wong HR, Cvijanovich N, Lin R, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. | ||
Wynn JL, Cvijanovich NZ, Allen GL, et al. The influence of developmental age on the early transcriptomic response of children with septic shock. Mol Med. 2011;17(11–12):1146–1156. | ||
Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. | ||
Harris MA, Clark J, Ireland A, et al; Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Database issue):D258–D261. | ||
Lewis SE. Gene Ontology: looking backwards and forwards. Genome Biol. 2005;6(1):103. | ||
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. | ||
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999; 27(1):29–34. | ||
Dennis G, Sherman BT, Hosack DA, et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. | ||
Franceschini A, Lin J, von Mering C, Jensen LJ. SVD-phy: improved prediction of protein functional associations through singular value decomposition of phylogenetic profiles. Bioinformatics. 2016;32(7):1085–1087. | ||
Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. | ||
Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. | ||
Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. | ||
von Mering C, Jensen LJ, Snel B, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Database issue):D433–D437. | ||
Tian W, Biao WU. Research progress of the protective effect of proto-oncogene on intestinal mucosal barrier. Medical Recapitulate. 2012. | ||
Daigo K, Yamaguchi N, Kawamura T, et al. The proteomic profile of circulating pentraxin 3 (PTX3) complex in sepsis demonstrates the interaction with azurocidin 1 and other components of neutrophil extracellular traps. Mol Cell Proteomics. 2012;11(6(6):):M111.015073. | ||
Ouyang SM, Liu XF, Yin K. The effect and prognostic analysis of continuous blood purification on pro-ADM with sepsis. Medical Innovation of China. 2014. | ||
Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34(3):129–136. | ||
Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14(4):417–427. | ||
Lockyer P, Mao H, Fan Q, et al. LRP1-Dependent BMPER signaling regulates lipopolysaccharide-induced vascular inflammation. Arterioscler Thromb Vasc Biol. 2017;37(8):1524–1535. | ||
Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, Fan H. Exosomes from endothelial progenitor cells improve the outcome of a murine model of sepsis. Mol Ther. 2018;26(5):1375–1384. | ||
Inoue S, Sato T, Suzuki-Utsunomiya K, et al. Sepsis-induced hypercytokinemia and lymphocyte apoptosis in aging-accelerated Klotho knockout mice. Shock. 2013;39(3):311–316. | ||
Tarze A, Deniaud A, Le Bras M, et al. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene. 2007;26(18):2606–2620. | ||
Nakajima H, Itakura M, Kubo T, et al. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) aggregation causes mitochondrial dysfunction during oxidative stress-induced cell death. J Biol Chem. 2017;292(11):4727–4742. | ||
Bazan-Socha S, Mastalerz L, Cybulska A, et al. Prothrombotic state in asthma is related to increased levels of inflammatory cytokines, IL-6 and TNFα, in peripheral blood. Inflammation. 2017;40(4):1225–1235. | ||
Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G422–430. | ||
Sun X, Yang H, Nose K, et al. Decline in intestinal mucosal IL-10 expression and decreased intestinal barrier function in a mouse model of total parenteral nutrition. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G139–G147. | ||
Sanz MG, Loynachan A, Horohov DW. Rhodococcus equi hyperimmune plasma decreases pneumonia severity after a randomised experimental challenge of neonatal foals. Vet Rec. 2016;178(11):261. | ||
Ai G, Shao X, Meng M, et al. Epidermal growth factor promotes proliferation and maintains multipotency of continuous cultured adipose stem cells via activating STAT signal pathway in vitro. Medicine. 2017;96(30):e7607. | ||
Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–26. | ||
Venturi S, Venturi M. Iodine in evolution of salivary glands and in oral health. Nutr Health. 2009;20(2):119–134. | ||
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183. | ||
Hajjar AM, Ernst RK, Yi J, Yam CS, Miller SI. Expression level of human TLR4 rather than sequence is the key determinant of LPS responsiveness. PLoS One. 2017;12(10):e0186308. | ||
Nicholas DA, Zhang K, Hung C, et al. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1β. PLoS One. 2017;12(5):e0176793. | ||
Wang Y, Yang W, Zhao X. Experimental study of the protective effect of simvastatin on lung injury in rats with sepsis. Inflammation. 2017;41(1):1–10. | ||
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973–983. | ||
Chen J, Xuan J, Gu YT, et al. Celastrol reduces IL-1β induced matrix catabolism, oxidative stress and inflammation in human nucleus pulposus cells and attenuates rat intervertebral disc degeneration in vivo. Biomed Pharmacother. 2017;91:208–219. | ||
Zabrodskii PF, Gromov MS, Maslyakov VV. Role of β2-Adrenoreceptors in adrenergic anti-inflammatory mechanism in sepsis. Bull Exp Biol Med. 2017;163(6):710–713. | ||
Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10(5):520–529. | ||
Aksoy E, Taboubi S, Torres D, et al. The p110δ isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat Immunol. 2012;13(11):1045–1054. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.