Back to Journals » Cancer Management and Research » Volume 10
Identification of key candidate genes and small molecule drugs in cervical cancer by bioinformatics strategy
Authors Tang X, Xu YC, Lu L, Jiao Y, Liu JJ, Wang LL, Zhao HB
Received 29 April 2018
Accepted for publication 5 June 2018
Published 13 September 2018 Volume 2018:10 Pages 3533—3549
DOI https://doi.org/10.2147/CMAR.S171661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Xin Tang,1 Yicong Xu,2,3 Lin Lu,2,3 Yang Jiao,2,3 Jianjun Liu,2,3 Linlin Wang,2,3 Hongbo Zhao2,3
1School of Rehabilitation, Kunming Medical University, Kunming, China; 2Institute of Molecular and Clinical Medicine, Kunming Medical University, Kunming, China; 3Yunnan Key Laboratory of Stem Cell and Regenerative Medicine, Kunming, China
Purpose: Cervical cancer (CC) is one of the most common malignant tumors among women. The present study aimed at integrating two expression profile datasets to identify critical genes and potential drugs in CC.
Materials and methods: Expression profiles, GSE7803 and GSE9750, were integrated using bioinformatics methods, including differentially expressed genes analysis, Kyoto Encyclopedia of Genes and Genomes pathway analysis, and protein–protein interaction (PPI) network construction. Subsequently, survival analysis was performed among the key genes using Gene Expression Profiling Interactive Analysis websites. Connectivity Map (CMap) was used to query potential drugs for CC.
Results: A total of 145 upregulated genes and 135 downregulated genes in CC were identified. The functional changes of these differentially expressed genes related to CC were mainly associated with cell cycle, DNA replication, p53 signaling pathway, and oocyte meiosis. A PPI network was identified by STRING with 220 nodes and 2,111 edges. Thirteen key genes were identified as the intersecting genes of the enrichment pathways and the top 20 nodes in PPI network. Survival analysis revealed that high mRNA expression of MCM2, PCNA, and RFC4 was significantly associated with longer overall survival, and the survival was significantly better in the low-expression RRM2 group. Moreover, CMap predicted nine small molecules as possible adjuvant drugs to treat CC.
Conclusion: Our study found key dysregulated genes involved in CC and potential drugs to combat it, which might provide insights into CC pathogenesis and might shed light on potential CC treatments.
Keywords: cervical cancer, bioinformatics, cell cycle, biomarker, drug
Introduction
Cervical cancer (CC) is the second most common malignant tumor among women, responsible for ~527,600 new cases and >265,700 deaths annually.1 Despite advances in screening detection and new treatment strategies, CC is one of the leading causes of cancer death among females in many developing countries.2,3 Although most patients can be cured if diagnosed at an early stage, poor prognosis is observed with secondary metastatic cancer and tumor relapse.
Although human papillomavirus (HPV) is a prerequisite for CC, only a small number of women infected by this virus develop cancer. Thus, other risk factors should be considered as cofactors contributing to the progression of CC.4 Dysregulated genes play important roles in CC development.5 Several studies have used gene expression profiling to identify key genes between CC samples and normal cervix.6–9 Hundreds of differentially expressed genes (DEGs) were detected. However, DEGs reported in different studies vary enormously with only some of them consistently detected. Therefore, the discovery of novel effective therapeutic targets against CC is urgently required.
A number of chemotherapeutic agents have shown activity against CC, including cisplatin,10 bevacizumab,11 carboplatin,12 paclitaxel,13 ifosfamide,14 and topotecan.15 Various combinations of these agents are recommended as therapies.16 A recent systematic literature review found that carboplatin–paclitaxel is equally effective and less toxic than cisplatin–paclitaxel as the first-line therapy for metastatic CC.17 However, patients overall survival (OS) times remains short, indicating an urgent need to discover some molecular drugs that are more efficient and selective. Based on bioinformatics approaches, several studies found small molecules as potential anticancer agents.18–20
In this study, we selected the following microarray datasets GSE7803 and GSE9750 from the Gene Expression Omnibus (GEO) database to identify DEGs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using the identified DEGs was investigated. A protein–protein interaction (PPI) network was constructed to elucidate the significant relationships among DEGs and to identify key genes. Furthermore, the Kaplan–Meier estimator was used on the Gene Expression Profiling Interactive Analysis (GEPIA) website. Candidate small molecules were identified for their potential use in the treatment of CC.
Materials and methods
Data collection
Two CC microarray datasets were downloaded from the GEO website (http://www.ncbi.nlm.nih.gov/geo/). GSE7803 microarray data contained 21 CC tissues and 10 normal cervical epithelia tissues.6 GSE9750 included 33 tumors samples and 24 healthy cervical samples.7 Both the profile datasets were based on the Affymetrix GPL96 platform (Affymetrix Human Genome U133A Array). Because Connectivity Map (CMap) strictly required all probesets obtained from the Affymetrix Human Genome U133A Array,21 we predicted the drugs for the DEGs measured only in this platform with high accuracy. GSE63514 data included 28 cancer cases and 24 normal cases8 and were chosen to validate RRM2 mRNA expression in our analysis.
Data preprocessing and DEGs screening
The raw data were standardized and transformed into expression values using the affy package of Bioconductor (http://www.bioconductor.org/).22 DEGs between cancer and normal samples were selected by significance analysis using the empirical Bayes methods within limma package.23 False discovery rate (FDR) <0.05 and |log2 (fold change)| >1 were set as the cutoff criteria for the identification of DEGs. Common dysregulated probesets between GSE7803 and GSE9750 were selected for subsequent analyzes.
KEGG pathway analysis
Pathway enrichment analysis was performed using the clusterProfiler package and a pathway with an adjusted P-value <0.05 was considered significantly enriched.24 DEGs that we identified could be involved in multiple pathways, Thus, some overlap was observed among the pathways. We identified the significant pathways that shared the same DEGs and used Cytoscape (version 3.5.1) to construct graphical representations of the interactive relationships among the pathways.25
PPI network construction and analysis
The PPI pairs of the screened DEGs were analyzed using the online database STRING version 10.5 (https://string-db.org/).26 The pairs with combined scores >0.4 were used for the PPI network construction, then the Cytoscape software was used to construct the network and analyze the interaction relationship of the candidate DEGs encoding proteins in CC.
Validation of key genes
Key genes were identified as the intersecting genes of the enrichment pathways and top 20 nodes in PPI network. To confirm the reliability of these genes from our detection, we analyzed their prognostic and expression in CC using GEPIA.27 GEPIA is an interactive web application for gene expression analysis based on 9,736 tumors and 8,587 normal samples from the Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression databases.28,29 We evaluated the expression of key genes in CC tissues and normal tissues. Then the survival curve and boxplot were performed to visualize the relationships.
Identification of candidate small molecules
The CC gene signature was used to query CMap to find potential drugs for use in patients.21 CMap is an in silico method to predict potential drugs that could possibly reverse, or induce, the biological state encoded in particular gene expression signatures. The common differently expressed probesets in GSE7803 and GSE9750 between CC samples and healthy controls were divided into upregulated and downregulated groups. Then, these probesets were used to query the CMap database. Finally, the enrichment score representing similarity was calculated, ranging from –1 to 1. A positive connectivity score indicates that a drug is able to induce the input signature in human cell lines. Conversely, a negative connectivity score indicates that a drug is able to reverse the input signature. Negative connectivity scores were investigated, which indicate potential therapeutic value. After rank ordering all instances, the connectivity score of various instances were filter by the number of instances (N>10) and P-value (<0.05).
Results
DEGs identification
The two mRNA expression profiles, including 54 patients with CC and 34 healthy individuals, were included in our study. Using a FDR <0.05 and |logFC| >1 as cutoff criteria, we extracted 443 and 848 differentially expressed probesets from the expression profile datasets GSE7803 and GSE9750, respectively. In GSE7803, 212 unregulated probes and 231 downregulated probes were identified. A total of 376 unregulated probes and 472 downregulated probes were identified in GSE9750. After being overlapped, the common 149 upregulated and 146 downregulated probesets corresponding to 145 upregulated and 135 downregulated genes were identified from the two profile datasets (Table S1).
CC significant pathways evaluation
A total of 16 pathways with adjusted P-value <0.05 were found enriched including 10 upregulated and 6 downregulated pathways (Table 1). The most significant upregulated pathway was cell cycle; the other significant pathways included DNA replication, oocyte meiosis, p53 signaling pathway, microRNAs in cancer, and cellular senescence. The downregulated pathways included arachidonic acid metabolism, serotonergic synapse, gap junction, estrogen signaling pathway, signaling pathways regulating pluripotency of stem cells, and proteoglycans in cancer (Figure 1A). In order to consider the potentially biological complexities in which a gene may belong to multiple pathways and provide information of numeric changes, we constructed pathway–gene networks to extract the complex association (Figure 1B, C). Cell cycle pathway contained the most significant genes in the network.
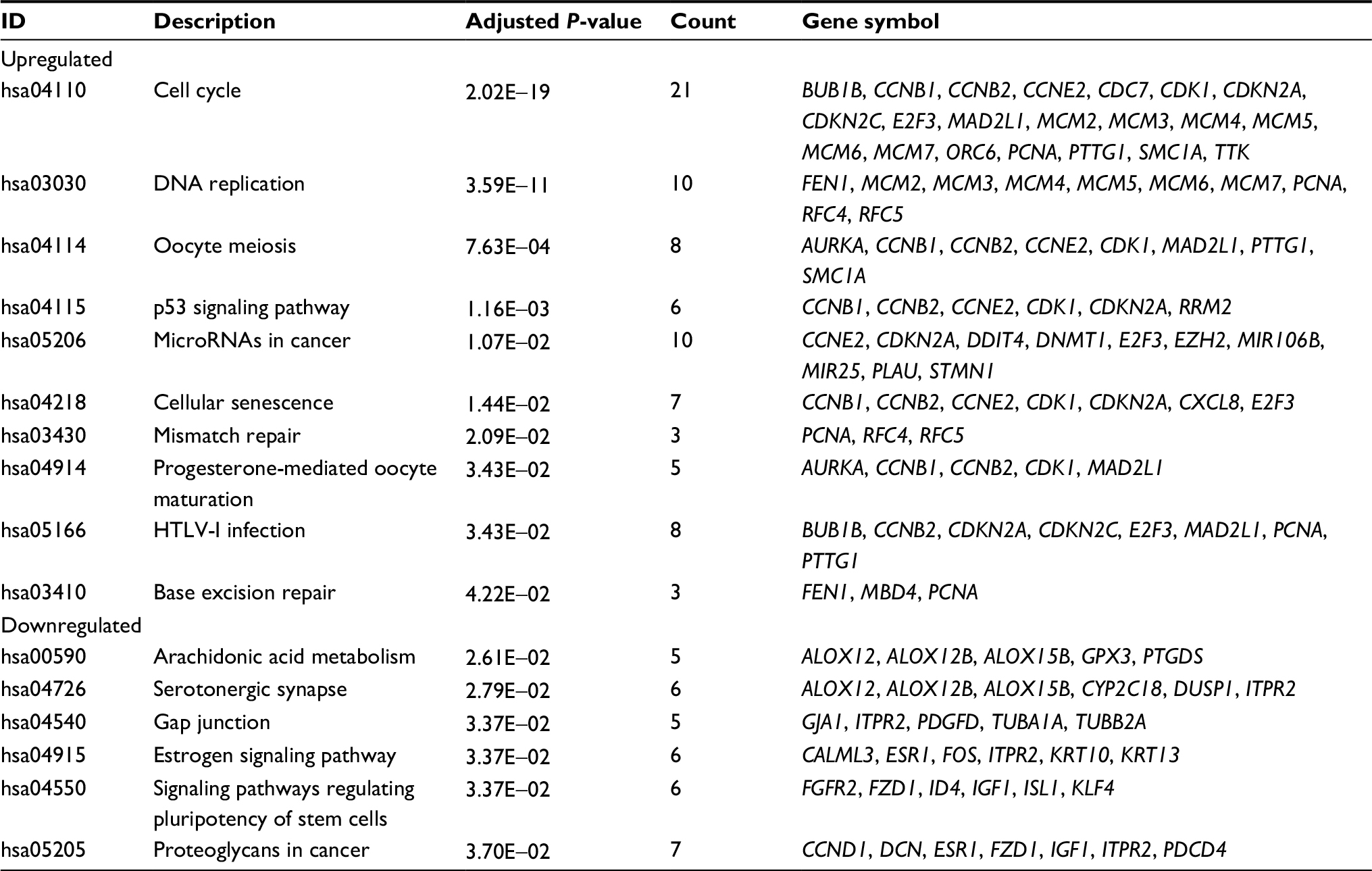 | Table 1 Pathway enrichment analysis of DEGs function in CC Abbreviations: CC, cervical cancer; DEGs, differentially expressed genes; HTLV-I, human T-lymphotropic virus type 1. |
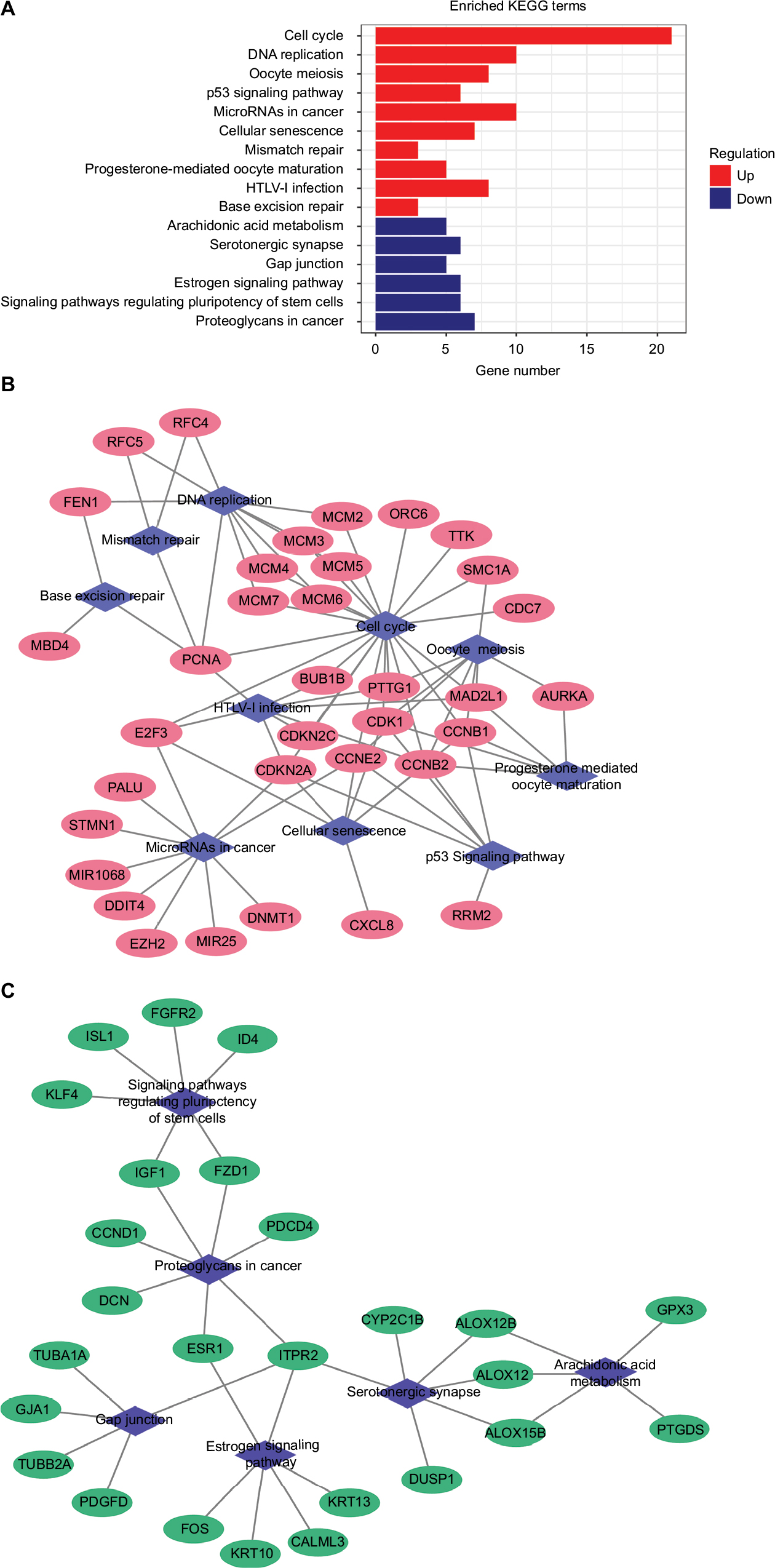 | Figure 1 Significantly enriched pathway terms associated to DEGs in CC. Notes: (A) KEGG pathways in CC DEGs enrichment analysis. (B) Upregulated pathway–gene network including 35 upregulated genes and 10 pathways. (C) Downregulated pathway–gene network including 26 downregulated genes and 6 pathways. Abbreviations: CC, cervical cancer; DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes; HTLV-I, human T-lymphotropic virus type 1. |
PPI network construction
STRING was used for mining proteins expressed by DEGs which can interact with others. At a combined score >0.4, a total of 222 DEGs (118 upregulated and 104 downregulated genes) among the 280 commonly altered DEGs were filtered into the DEGs PPI network, containing 222 nodes and 2,111 edges (Figure 2A). NetworkAnalyzer app in Cytoscape was used to calculate the node degree.25 The genes CDK1, PCNA, TOP2A, CCNB1, RFC4, MAD2L1, NDC80, CCNB2, AURKA, TYMS, MCM2, FEN1, RRM2, NCAPG, TTK, PRC1, MCM4, ZWINT, DTL, and MCM6 were the most significant 20 node degree genes and were selected as the hub nodes, since they might play important roles in CC progression (Figure 2B).
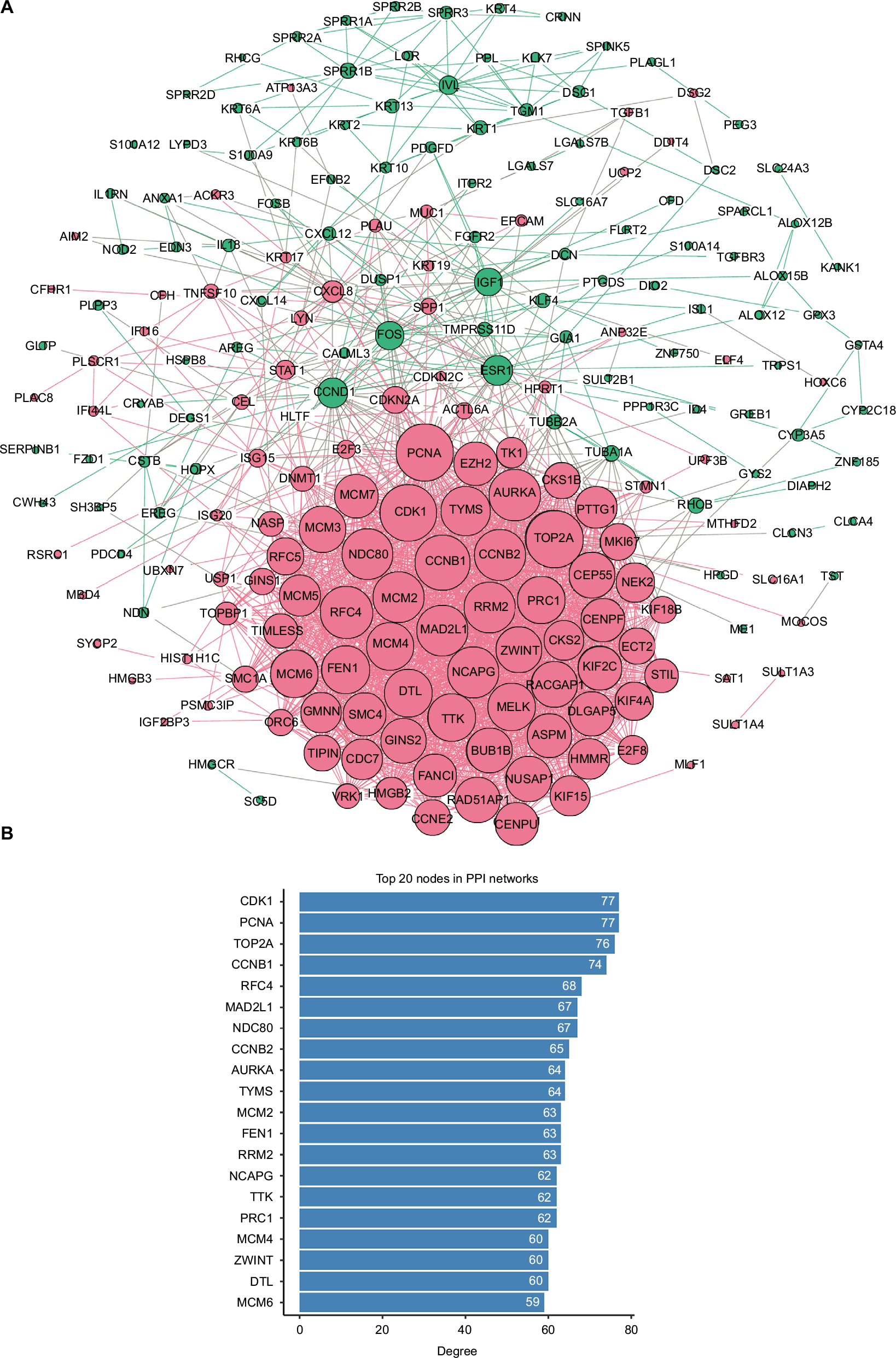 | Figure 2 PPI network analysis. Notes: (A) Using the STRING online database, a total of 222 DEGs (118 upregulated in red standing for upregulation and 104 downregulated genes in green standing for downregulation) were filtered into the DEGs PPI network. Bigger nodes represent genes with more links. (B) Degree of the top 20 nodes in the PPI network. All these nodes are upregulated genes. Abbreviations: DEGS, differentially expressed genes; PPI, protein–protein interaction. |
Key gene signatures identification in CC
Compared with KEGG enrichment genes, 13 of the top 20 nodes in the PPI network, including AURKA, CCNB1, CCNB2, CDK1, FEN1, MAD2L1, MCM2, MCM4, MCM6, PCNA, RFC4, RRM2, and TTK were found as key genes. Further survival analyses on these key genes were employed to evaluate their effects on CC patients’ survival using GEPIA. Expression levels of MCM2, PCNA, RFC4, and RRM2 were significantly related to the OS of patients with cervical squamous cancer (P<0.05). High expression of MCM2, PCNA, and RFC4 could result in a high survival rate, and increased RRM2 expression in CC was significantly associated with shorter patients’ survival (Figure 3A–D). The expression of these four genes was significantly higher in CC tissues compared to that of normal tissues (P<0.01; Figure 3E–H). Together, the high level of these four genes might represent the important prognostic factor to predict the survival of CC. GSE63514 was used to validate RRM2 mRNA expression. The results showed that RRM2 expression was significantly higher in CC compared to that of normal tissues (P<0.01; Figure 4A).The PPI network based on RRM2 found that PCNA and RFC4 have a close relationship with RRM2, and most of the proteins in the network were related to cell cycle (Figure 4B).
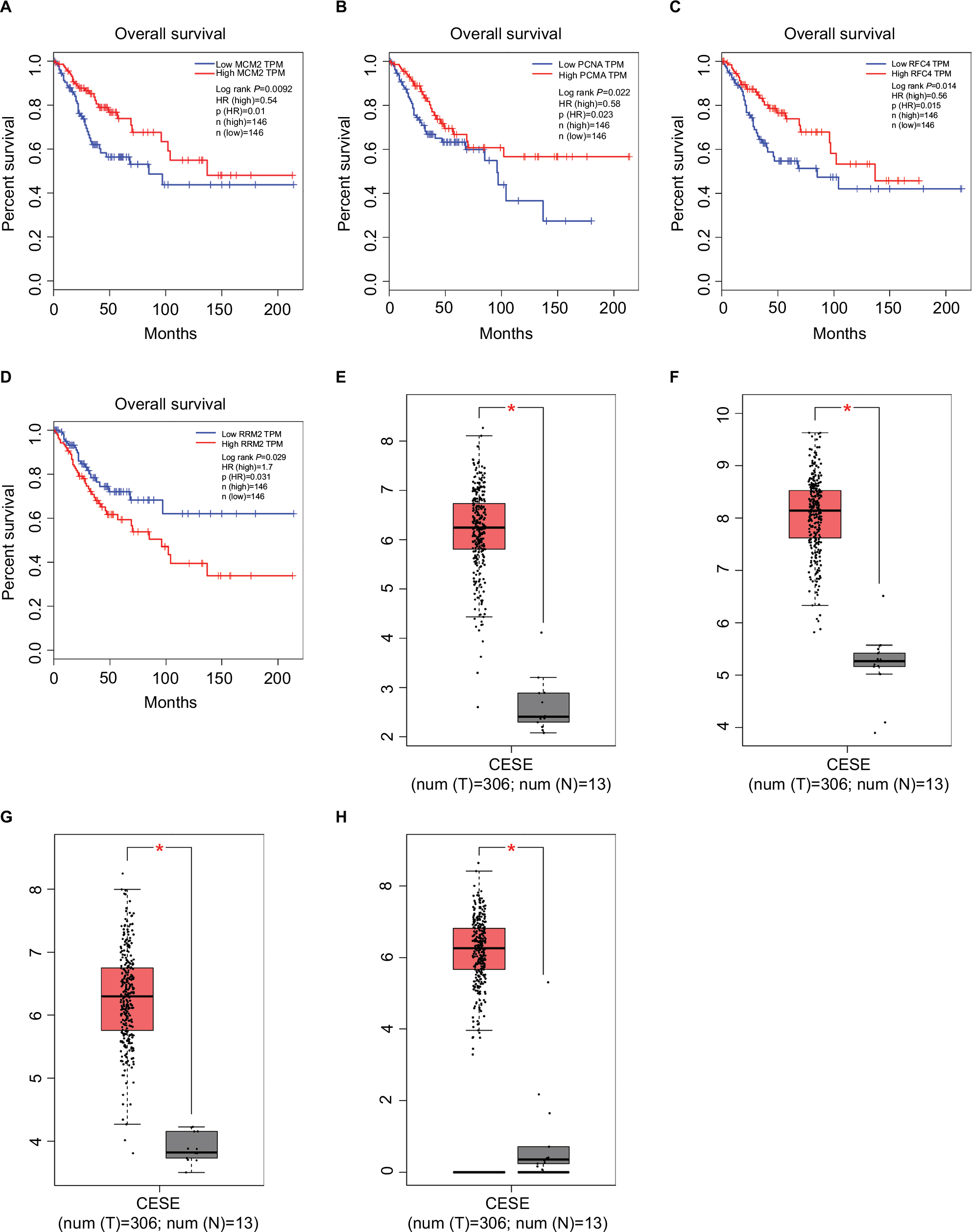 | Figure 3 Survival curves and expression boxplots of key genes using GEPIA website. Notes: (A–D) Expression level of MCM2, PCNA, RFC4, and RRM2 was significantly related to the overall survival of patients with cervical squamous cancer (P<0.05). (E–H) MCM2, PCNA, RFC4, and RRM2 were significantly upregulated in cervical squamous cancer compared with normal tissues (P<0.01). Abbreviations: CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; GEPIA, Gene Expression Profiling Interactive Analysis; TPM, transcripts per million. |
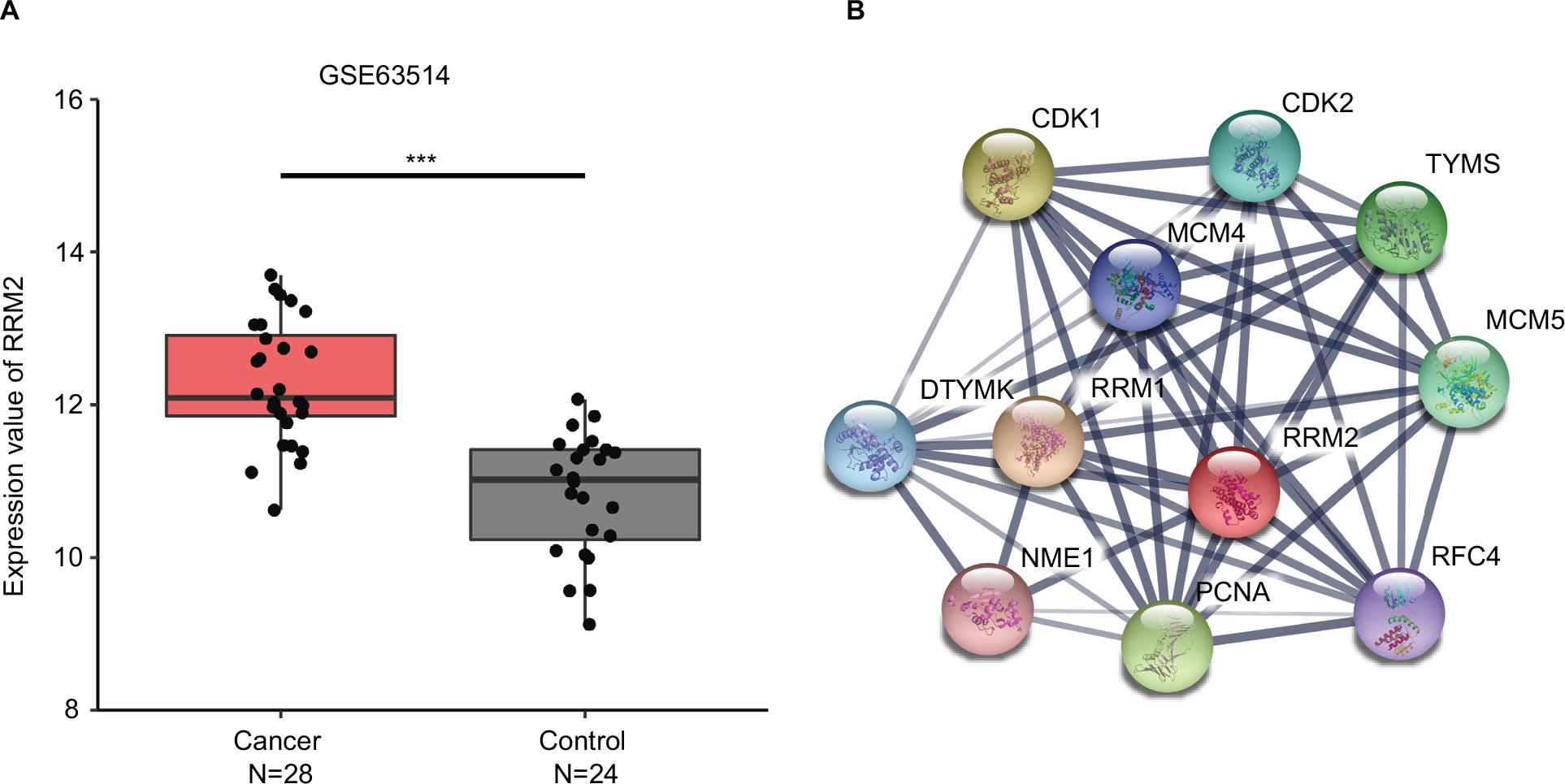 | Figure 4 RRM2 validation using GSE63514 and PPI network. Notes: (A) GSE63514 showed higher expression of RRM2 in CC tissues compared with normal cervical tissues (P<0.01). (B) RRM2 PPI network based on STRING. Abbreviations: CC, cervical cancer; PPI, protein–protein interaction. |
Related small molecule drugs screening
In order to screen out small molecule drugs, consistent differently expressed probesets between CC samples and healthy controls were analyzed with CMap. The related small molecules with highly significant correlations are listed in Table 2. Among these molecules, trichostatin A (TSA), tanespimycin, vorinostat, trifluoperazine, prochlorperazine, and thioridazine showed higher negative correlation and the potential to treat CC.
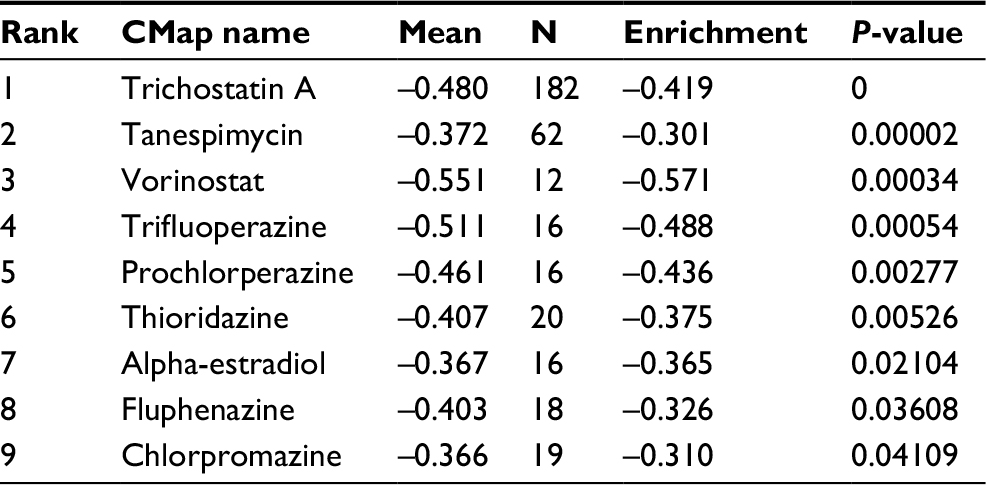 | Table 2 Results of CMap analysis Abbreviation: CMap, Connectivity Map. |
Discussion
Driver genes play vital roles during stages of cancer progression. Although many studies on CC development are available, more efforts are needed to identify driver genes and candidate drugs that may shed light on CC treatments. This study integrated two gene profile datasets based on Affymetrix Human Genome U133A Array, utilized bioinformatics methods to analyze these datasets, and identified 280 commonly changed DEGs (145 upregulated and 135 downregulated). Pathway enrichment analysis indicated that cell cycle, DNA replication, oocyte meiosis, p53 signaling pathway, cellular senescence, and DNA repair-relevant biological pathways were overrepresented among the upregulated genes. The PPI network was constructed including 222 nodes/DEGs and 2,111 edges. Thirteen key genes were identified and chosen for survival analysis. MCM2, PCNA, RFC4, and RRM2 were clearly related to the prognosis of patients. In addition, small molecules that can provide new insights in CC therapeutic studies were identified.
Many researchers have found that four key genes were involved in cell cycle, participating in tumorigenesis and tumor proliferation. MCM2 has been studied in a wide range of human malignancies and is associated with tumor histopathological grade in several malignancies, including colon, oral cavity, ovarian, urothelial, and non-small cell lung carcinoma.30–34 In cervical carcinoma and precancerous lesions, MCM2 is overexpressed and positively correlated with high risk types of HPV.35 Amaro Filho et al also reported an increasing expression of MCM2 in invasive CC compared to control, but they suggested that MCM2 is not a good biomarker when comparing the different clinical stages of CC.36 PCNA acts as a central coordinator of DNA transactions by providing a multivalent interaction surface for factors involved in DNA replication and cell cycle regulation. Owing to its function, PCNA has been widely used as a tumor marker for cancer cell progression and patient prognosis.37–39 A recent systematic literature review found that the expression of PCNA is significantly associated with poor 5-year survival, International Federation of Gynecology and Obstetrics stage, or WHO grade, suggesting its use as a valuable prognostic and diagnostic biomarker in CC and gliomas.40 RFC4 is involved in cancer. Knockdown of RFC4 in HepG2 cells induces apoptosis.41 Similar results were discovered in breast carcinoma.42 In colorectal cancer, overexpression of RFC4 is associated with tumor progression and poor survival outcome.43 Additionally, with gene network reconstruction, RFC4 is regarded as one of the main drivers in cell cycle network in CC.44 Together with our results, MCM2, PCNA, and RFC4 were significantly upregulated in CC compared with normal samples, and in CC patients, the survival rate was positively correlated with the high expression of these genes.
RRM2 is markedly upregulated in many patients’ cancer types and indeed acts as an oncogene.45 RRM2 knockdown reduces cell proliferation and invasive ability in gastric cancer and pancreatic adenocarcinoma.46,47 Wang et al reported that RRM2 expression inhibition significantly increases apoptosis, promotes cell cycle arrest at the G1 phase, and inhibits tumor formation in CC nude mice transplant models.48 Several studies showed that RRM2 is an independent prognostic factor and may predict poor survival in ovarian cancer, bladder cancer, breast cancer, and CC.49–52 In this study, according to the PPI network, RRM2 closely interacts with PCNA and RFC4 involved in CC progression. Therefore, a further exploration of cell cycle and related genes was of enormous significance.
Consistent with our results, recent studies have also reported the identification of DEGs in CC. van Dam et al used three publicly available Affymetrix gene expression datasets (GSE5787, GSE7803, and GSE9750) and identified five cancer hallmarks enriched pathways in CC, showing that cell cycle deregulation is the major component of CC biology. They also identified seven probesets that were highly expressed in both CIN3 samples compared to normal samples and in cancer samples compared to CIN3 samples. From these probesets, six genes (AURKA, DTL, HMGB3, KIF2C, NEK2, and RFC4) were overexpressed in CC cell lines compared to cancer samples, suggesting their potential role as biomarkers in CC early diagnosis.53 One of these genes, such as RFC4, was also identified in our study. Furthermore, our conclusion generated from both expression and survival analysis suggested that RFC4 might have a prognostic value. Another report from Li et al was based on TCGA data.54 They found that MCM2, MCM4, MCM5, PCNA, and RNASEH2A participating in DNA replication pathway might be prognostic biomarkers in CC patients. MCM2 and PCNA were also found in our results.
Several small molecules with potential therapeutic efficacy against CC were identified. The most significant small molecules in our result have been reported to display anticancer activity. TSA, as a histone deacetylase (HDAC) inhibitor, shows a potential therapeutic effect in various types of cancer cells, when combined with radiotherapy or chemotherapy.55,56 In particular, TSA and its hydroxamate analogs can effectively and selectively induce tumor growth arrest at very low concentrations.57 Additionally, TSA can inhibit HeLa cells growth via Bcl-2-mediated and caspase-dependent apoptosis.58 Vorinostat is a hydroxamate-based pan-HDAC inhibitor also known as suberoylanilide hydroxamic acid used for the treatment of cutaneous T-cell lymphoma.59 In HeLa cell, both mRNA and protein levels of HPV18 E6 and E7 were reduced after vorinostat treatment.60 Furthermore, vorinostat promotes SiHa apoptosis through upregulation of p21 and Bax mRNA and protein, leading to cell cycle arrest in G0/G1 phase.61 Thioridazine, a derivative of phenothiazine, displays anticancer abilities in a variety of cancer types and can reverse multidrug resistance.62–64 Kang et al found that thioridazine can inhibit the PI3K/Akt/mTOR/p70S6K signaling pathway and exert cytotoxic effect on CC cells by inducing cell cycle arrest and apoptosis.65 Thus, we might suppose that these identified drugs could play certain roles to combat CC.
Conclusion
Using bioinformatics analysis, 280 DEGs were identified, which were significantly enriched in several pathways, mainly associated with cell cycle, DNA replication, oocyte meiosis, p53 signaling pathway, and cellular senescence. We also identified key genes including MCM2, PCNA, RFC4, and RRM2 that might play important roles in CC and that might represent novel biomarkers in CC diagnosis, prognosis, and therapy. Additionally, a group of small molecules was identified that might be exploited as adjuvant drugs for improved therapeutics for CC. However, further investigations are required to validate the predicted drugs.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (grant no 81360336), the Joint Special Funds for the Department of Science and Technology of Yunnan Province – Kunming Medical University (grant no 2015FB017) and One Hundred Young and Middle - Aged Academic and Technical Backbone Project of Kunming Medical University (grant no. 60117190449).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Forouzanfar MH, Foreman KJ, Delossantos AM, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet. 2011;378(9801):1461–1484. | ||
Maguire R, Kotronoulas G, Simpson M, Paterson C. A systematic review of the supportive care needs of women living with and beyond cervical cancer. Gynecol Oncol. 2015;136(3):478–490. | ||
zur Hausen H. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology. 1991;184(1):9–13. | ||
Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506(7488):371–375. | ||
Zhai Y, Kuick R, Nan B, et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67(21):10163–10172. | ||
Scotto L, Narayan G, Nandula SV, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47(9):755–765. | ||
den Boon JA, Pyeon D, Wang SS, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A. 2015;112(25): E3255–E3264. | ||
Pappa KI, Polyzos A, Jacob-Hirsch J, et al. Profiling of discrete gynecological cancers reveals novel transcriptional modules and common features shared by other cancer types and embryonic stem cells. PLoS One. 2015;10(11):e0142229. | ||
Leisching GR, Loos B, Botha MH, Engelbrecht AM. The role of mTOR during cisplatin treatment in an in vitro and ex vivo model of cervical cancer. Toxicology. 2015;335:72–78. | ||
Tewari KS, Sill MW, Long HJ, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734–743. | ||
Katanyoo K, Tangjitgamol S, Chongthanakorn M, et al. Treatment outcomes of concurrent weekly carboplatin with radiation therapy in locally advanced cervical cancer patients. Gynecol Oncol. 2011;123(3):571–576. | ||
Wang X, Shen Y, Zhao Y, et al. Adjuvant intensity-modulated radiotherapy (IMRT) with concurrent paclitaxel and cisplatin in cervical cancer patients with high risk factors: a phase II trial. Eur J Surg Oncol. 2015;41(8):1082–1088. | ||
Downs LS, Chura JC, Argenta PA, et al. Ifosfamide, paclitaxel, and carboplatin, a novel triplet regimen for advanced, recurrent, or persistent carcinoma of the cervix: a phase II trial. Gynecol Oncol. 2011;120(2):265–269. | ||
Muderspach LI, Blessing JA, Levenback C, Moore JL Jr. A phase II study of topotecan in patients with squamous cell carcinoma of the cervix: a gynecologic oncology group study. Gynecol Oncol. 2001;81(2):213–215. | ||
Eskander RN, Tewari KS. Chemotherapy in the treatment of metastatic, persistent, and recurrent cervical cancer. Curr Opin Obstet Gynecol. 2014;26(4):314–321. | ||
Lorusso D, Petrelli F, Coinu A, Raspagliesi F, Barni S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol Oncol. 2014;133(1):117–123. | ||
Yeh CT, Wu AT, Chang PM, et al. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am J Respir Crit Care Med. 2012;186(11):1180–1188. | ||
Chen MH, Lin KJ, Yang WL, et al. Gene expression-based chemical genomics identifies heat-shock protein 90 inhibitors as potential therapeutic drugs in cholangiocarcinoma. Cancer. 2013;119(2):293–303. | ||
Hassane DC, Guzman ML, Corbett C, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood. 2008;111(12):5654–5662. | ||
Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. | ||
Gautier L, Cope L, Bolstad BM, Irizarry RA. affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. | ||
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. | ||
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. | ||
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. | ||
Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. | ||
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. | ||
Cancer Genome Atlas Research Network; Weinstein JN, Collisson EA, Mills GB, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. | ||
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. | ||
Wang Y, Li Y, Zhang WY, et al. mRNA expression of minichromosome maintenance 2 in colonic adenoma and adenocarcinoma. Eur J Cancer Prev. 2009;18(1):40–45. | ||
Szelachowska J, Dziegiel P, Jelen-Krzeszewska J, et al. Correlation of metallothionein expression with clinical progression of cancer in the oral cavity. Anticancer Res. 2009;29(2):589–595. | ||
Gakiopoulou H, Korkolopoulou P, Levidou G, et al. Minichromosome maintenance proteins 2 and 5 in non-benign epithelial ovarian tumours: relationship with cell cycle regulators and prognostic implications. Br J Cancer. 2007;97(8):1124–1134. | ||
Burger M, Denzinger S, Hartmann A, Wieland WF, Stoehr R, Obermann EC. Mcm2 predicts recurrence hazard in stage Ta/T1 bladder cancer more accurately than CK20, Ki67 and histological grade. Br J Cancer. 2007;96(11):1711–1715. | ||
Zhang X, Teng Y, Yang F, et al. MCM2 is a therapeutic target of lovastatin in human non-small cell lung carcinomas. Oncol Rep. 2015;33(5):2599–2605. | ||
Zheng J. Diagnostic value of MCM2 immunocytochemical staining in cervical lesions and its relationship with HPV infection. Int J Clin Exp Pathol. 2015;8(1):875–880. | ||
Amaro Filho SM, Nuovo GJ, Cunha CB, et al. Correlation of MCM2 detection with stage and virology of cervical cancer. Int J Biol Markers. 2014;29(4):e363–e371. | ||
Tachibana KE, Gonzalez MA, Coleman N. Cell-cycle-dependent regulation of DNA replication and its relevance to cancer pathology. J Pathol. 2005;205(2):123–129. | ||
Zhao H, Lo YH, Ma L, et al. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol Cancer Ther. 2011;10(1):29–36. | ||
Wang LF, Chai CY, Kuo WR, Tai CF, Lee KW, Ho KY. Correlation between proliferating cell nuclear antigen and p53 protein expression and 5-year survival rate in nasopharyngeal carcinoma. Am J Otolaryngol. 2006;27(2):101–105. | ||
Lv Q, Zhang J, Yi Y, et al. Proliferating cell nuclear antigen has an association with prognosis and risks factors of cancer patients: a systematic review. Mol Neurobiol. 2016;53(9):6209–6217. | ||
Arai M, Kondoh N, Imazeki N, et al. The knockdown of endogenous replication factor C4 decreases the growth and enhances the chemosensitivity of hepatocellular carcinoma cells. Liver Int. 2009;29(1):55–62. | ||
Srihari S, Kalimutho M, Lal S, et al. Understanding the functional impact of copy number alterations in breast cancer using a network modeling approach. Mol Biosyst. 2016;12(3):963–972. | ||
Xiang J, Fang L, Luo Y, et al. Levels of human replication factor C4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer. J Transl Med. 2014;12:320. | ||
Mine KL, Shulzhenko N, Yambartsev A, et al. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun. 2013;4:1806. | ||
Xu X, Page JL, Surtees JA, et al. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008;68(8):2652–2660. | ||
Kang W, Tong JH, Chan AW, et al. Targeting ribonucleotide reductase M2 subunit by small interfering RNA exerts anti-oncogenic effects in gastric adenocarcinoma. Oncol Rep. 2014;31(6):2579–2586. | ||
Duxbury MS, Whang EE. RRM2 induces NF-kappaB-dependent MMP-9 activation and enhances cellular invasiveness. Biochem Biophys Res Commun. 2007;354(1):190–196. | ||
Wang N, Li Y, Zhou J. Downregulation of ribonucleotide reductase subunits M2 induces apoptosis and G1 arrest of cervical cancer cells. Oncol Lett. 2018;15(3):3719–3725. | ||
Ferrandina G, Mey V, Nannizzi S, et al. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother Pharmacol. 2010;65(4):679–686. | ||
Morikawa T, Maeda D, Kume H, Homma Y, Fukayama M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology. 2010;57(6):885–892. | ||
Kretschmer C, Sterner-Kock A, Siedentopf F, Schoenegg W, Schlag PM, Kemmner W. Identification of early molecular markers for breast cancer. Mol Cancer. 2011;10(1):15. | ||
Su YF, Wu TF, Ko JL, et al. The expression of ribonucleotide reductase M2 in the carcinogenesis of uterine cervix and its relationship with clinicopathological characteristics and prognosis of cancer patients. PLoS One. 2014;9(3):e91644. | ||
van Dam PA, van Dam PJ, Rolfo C, et al. In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget. 2016;7(3):2780–2795. | ||
Li X, Tian R, Gao H, et al. Identification of significant gene signatures and prognostic biomarkers for patients with cervical cancer by integrated bioinformatic methods. Technol Cancer Res Treat. 2018;17:1–12. | ||
Ranganathan P, Rangnekar VM. Exploiting the TSA connections to overcome apoptosis-resistance. Cancer Biol Ther. 2005;4(4):391–392. | ||
Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene. 2008;27(22):3134–3144. | ||
Vanhaecke T, Papeleu P, Elaut G, Rogiers V. Trichostatin A-like hydroxamate histone deacetylase inhibitors as therapeutic agents: toxicological point of view. Curr Med Chem. 2004;11(12):1629–1643. | ||
You BR, Park WH. Trichostatin A induces apoptotic cell death of HeLa cells in a Bcl-2 and oxidative stress-dependent manner. Int J Oncol. 2013;42(1):359–366. | ||
Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16(7):1111–1120. | ||
He H, Liu X, Wang D, et al. SAHA inhibits the transcription initiation of HPV18 E6/E7 genes in HeLa cervical cancer cells. Gene. 2014;553(2):98–104. | ||
Xing J, Wang H, Xu S, Han P, Xin M, Zhou JL. Sensitization of suberoylanilide hydroxamic acid (SAHA) on chemoradiation for human cervical cancer cells and its mechanism. Eur J Gynaecol Oncol. 2015;36(2):117–122. | ||
Gil-Ad I, Shtaif B, Levkovitz Y, et al. Phenothiazines induce apoptosis in a B16 mouse melanoma cell line and attenuate in vivo melanoma tumor growth. Oncol Rep. 2006;15(1):107–112. | ||
Li J, Yao QY, Xue JS, et al. Dopamine D2 receptor antagonist sulpiride enhances dexamethasone responses in the treatment of drug-resistant and metastatic breast cancer. Acta Pharmacol Sin. 2017;38(9):1282–1296. | ||
Seo SU, Cho HK, Min KJ, et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death Dis. 2017;8(2):e2599. | ||
Kang S, Dong SM, Kim BR, et al. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis. 2012;17(9):989–997. |
Supplementary material
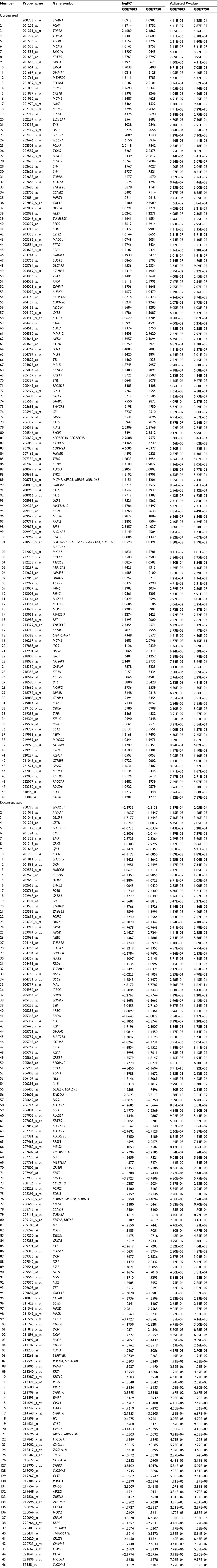 | Table S1 Common dysregulated probes identified in GSE7803 and GSE9750 Abbreviation: FC, fold change. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.