Back to Journals » Cancer Management and Research » Volume 13
Identification of Hub Genes as Potential Prognostic Biomarkers in Cervical Cancer Using Comprehensive Bioinformatics Analysis and Validation Studies
Authors Xue H, Sun Z, Wu W, Du D, Liao S
Received 22 September 2020
Accepted for publication 24 November 2020
Published 8 January 2021 Volume 2021:13 Pages 117—131
DOI https://doi.org/10.2147/CMAR.S282989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Han Xue,1 Zhaojun Sun,2 Weiqing Wu,1 Dong Du,1 Shuping Liao1
1Department of Health Management, Shenzhen People’s Hospital, Shenzhen City, Guangdong Province, People’s Republic of China; 2Department of Dermatology, Shenzhen People’s Hospital, Shenzhen City, GuangdongProvince, People’s Republic of China
Correspondence: Zhaojun Sun
Department of Dermatology, Shenzhen People’s Hospital, Shenzhen City, Guangdong Province 518001, People’s Republic of China
Tel +86-13422876297
Email [email protected]
Background: Cervical cancer belongs to one of the most common female cancers; yet, the exact underlying mechanisms are still elusive. Recently, microarray and sequencing technologies have been widely used for screening biomarkers and molecular mechanism discovery in cancer studies. In this study, we aimed to analyse the microarray datasets using comprehensive bioinformatics tools and identified novel biomarkers associated with the prognosis of patients with cervical cancer.
Methods: The differentially expressed genes (DEGs) from Gene Expression Omnibus (GEO) datasets including GSE138080, GSE113942 and GSE63514 were analysed using GEO2R tool. The functional enrichment analysis was performed using g:Profiler tool. The protein–protein interaction (PPI) network construction and hub genes identification were performed using the STRING database and Cytoscape software, respectively. The hub genes were subjected to expression and survival analysis in the cervical cancer. The EdU incorporation and Cell Counting Kit-8 assays were performed to evaluate the effects of hub gene knockdown on the proliferation of cervical cancer cells.
Results: A total of 89 overlapping DEGs (63 up-regulated and 26 down-regulated genes) were identified in the microarray datasets. The functional enrichment analysis indicated that the overlapping DEGs were mainly associated with “DNA replication” and “cell cycle”. Furthermore, the PPI network analysis revealed that the network contains 87 nodes and 309 edges. Sub-module analysis using the Molecular Complex Detection tool identified 21 hub genes from the PPI network. The expression levels of the 21 hub genes were all up-regulated in the cervical cancer tissues when compared to normal cervical tissues as analysed by GEPIA tool. The survival analysis showed that the low expression of cell division cycle 45 (CDC45), GINS complex subunit 2 (GINS2), minichromosome maintenance complex component 2 (MCM2) and proliferating cell nuclear antigen (PCNA) was significantly correlated with the shorter overall survival of patients with cervical cancer. Moreover, the protein expression levels of GINS2, MCM2 and PCNA, but not CDC45, were significantly up-regulated in the cervical cancer tissues when compared to normal cervical tissues. Finally, knockdown of MCM2 significantly suppressed the proliferation of HeLa and SiHa cells.
Conclusion: In conclusion, we screened a total of 89 overlapping DEGs from the GEO datasets, and further analysis identified four hub genes (CDC45, GINS2, MCM2 and PCNA) that were likely associated with the prognosis of patients with cervical cancer. MCM2 knockdown repressed the cervical cancer cell proliferation. The current findings may provide novel insights into understanding the pathophysiology of cervical cancer and develop therapeutic targets for patients with cervical cancer.
Keywords: cervical cancer: bioinformatics, DEGs, prognosis, hub genes, overall survival
Introduction
Cervical cancer belongs to one of the most common female cancers, and persistent infection of high-risk human papillomavirus is one of the major causal factors for the development of cervical cancer.1 In 2019, there were more than 500,000 new cases and 300,000 deaths globally, with about 90% of cases occurring in low-income areas.2 Though vaccination and routine physical examination have been effective in preventing the development of cervical cancer, the mortality of cervical cancer is still high particularly in low-income areas.2 The high mortality of cervical cancer is mainly due to the lack of early diagnosis, as the patients with advanced cervical cancer miss the opportunity for surgeries and are often resistant to radio/chemotherapy.3–5 Great efforts have been invested to uncover the pathophysiology of cervical cancer, however, the exact underlying mechanisms are still elusive, which requires further exploration.
High throughput technology has been a powerful tool to study the human genomics. Recently, microarray and sequencing technologies have been widely used for screening biomarkers and molecular mechanism discovery in cancer studies.6,7 The analysis of multi-omics databases like the Gene Expression Omnibus (GEO) as well as The Cancer Genome Atlas (TCGA) makes it possible to access microarray data and compare cancer profiles to normal profiles.8,9 In the cervical cancer studies, the bioinformatics analysis has been employed to identify the biomarkers and signalling pathways associated with the pathophysiology of cervical cancer. Dai et al, performed the integrated bioinformatics analysis from GEO datasets and identified that the lower expression of Krüppel-like factor 4 and estrogen receptor 1 was closely correlated with the poor prognosis of patients with cervical cancer.10 Liang et al, showed that three microRNA's (miR-145, miR-200c and miR-281-1) signatures predicted survival in cervical cancer using bioinformatics analysis.11 Lin et al, performed genome-wide analysis of aberrant gene expression and methylation profiles and indicated that endothelin-3 and endothelin receptor B might play important roles in the progression of cervical cancer, and LIM homeobox 2, acyl-coenzyme A oxidase 3, cytochrome P450 family 39 subfamily A member 1 and dihydropyrimidinase might be susceptible genes and potential risk markers in cervical cancer.12 To date, the public microarray datasets regarding cervical cancer are still not fully explored, and further analysis of these microarray datasets may be helpful for us to identify novel biomarkers associated with cervical cancer progression.
In this study, we integrated three microarray datasets including GSE138080, GSE113942 and GSE63514 from the GEO database and screened the differentially expressed genes (DEGs) from these datasets. The functional enrichment analysis was performed to annotate these DEGs. Protein-protein interaction (PPI) network analysis of the DEGs was performed to identify the hub genes. Moreover, the expression and survival analysis of the hub genes were further analysed using the online databases, which may provide novel insights into understanding the prognostic role of the identified hub genes in cervical cancer. Finally, the biological role of the hub gene (MCM2) in the cervical cancer cells was validated by in vitro functional assays.
Materials and Methods
Microarray Data Collection
By searching the GEO database using “cervical cancer” as the keywords, three microarray datasets including GSE138080, GSE113942 and GSE63514 were downloaded for analysis in this study. The platform for GSE138080 is GPL4133, Agilent-014850 Whole Human Genome Microarray 4x44K G4112F (Feature Number version), which contains 10 normal cervical samples and 10 cervical cancer specimens; the platform for GSE9750 is GPL96, [HG-U133A] Affymetrix Human Genome U133A Array, which contains 22 normal cervical samples and 33 cervical cancer tissues; the platform for GSE63514 is the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, which contains 24 normal cervical epithelial tissues and 28 cervical cancer tissues.
Identification of DEGs from GEO Datasets
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE40435) was used to compare gene expression profiles between normal cervical tissues and cervical cancer tissues. The Benjamini-Hochberg method was used to determine the false discovery rate and the adjusted P-value was used to reduce the likelihood of false positive errors. The selection criteria for the DEGs was based on an adjusted P-value of < 0.05 and |log fold change (FC)|≥1.5. Finally, the overlapping DEGs among the three datasets was identified by a Venn diagram tool.13
Functional Enrichment Analysis
Gene ontology (GO) analyses focuses on three domains: biological processes, cellular components, and molecular functions, and such analyses are used to understand the biological functions, pathways, or localization of DEGs. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis database serves as a valuable resource for assessing how particular DEGs may be involved in or influenced by specific signalling pathways and disease states. The g: Profiler tool was utilized for functional enrichment analyses, with a P < 0.05 cut-off for significance.14
PPI Network of DEGs
The PPI network of the overlapping DEGs was analysed using the STRING database (http://www.bork.embl-heidelberg.de/STRING/),15 which is a computerized and powerful global resource for studying the interactions between the predicted and experimental interactions of proteins. A combined score of ≥ 0.7 was used as the cut-off value. A Molecular Complex Detection (MCODE) plug-in in Cytoscape (version 3.6.1) was used to select the PPI network modules, with a cutoff = 2, node score cutoff = 0.2, k-core = 2 and maximum depth = 100 as the selection criteria. The hub genes were then extracted from the PPI network modules.
Expression Analysis of Hub Genes in Cervical Cancer
The expression analysis of hub genes in cervical cancer was performed using the Gene Expression Profiling Interactive Analysis (GEPIA) tool,16 which is a new web-based tool that uses standardized, integrated processing to analyse tumor and normal gene expression from The Cancer Genome Atlas (TCGA) and genotype tissue expression (GTEx) and contains 319 cervical tissue samples (306 tumor samples and 13 normal tissue samples) from the TCGA database, and the differential expression of the hub genes was defined when P < 0.01. The protein expression of hub genes in the cervical tissue was obtained from the online database (Human Protein Atlas database, https://www.proteinatlas.org/).
Survival Analysis of Hub Genes in Cervical Cancer
The overall survival analysis of the hub genes in cervical cancer was analysed by the GEPIA tool, Human Protein Atlas database (https://www.proteinatlas.org/) and KM plotter (https://kmplot.com/analysis/index.php?p=background),17 respectively. The GEPIA database contains survival data of 292 patients with cervical cancer. The Human Protein Atlas database includes survival data of 291 patients with cervical cancer. The KM plotter database contains survival data of 304 patients with cervical cancer. Log rank P < 0.05 was considered to be statistically significant.
Cell Culture
Two human cervical cancer cell lines (HeLa and SiHa) were obtained from ATCC (ATCC, USA). HeLa and SiHa cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, USA) in a humidified atmosphere with 5% CO2 at 37 °C.
Quantitative Real-Time PCR (qRT-PCR)
Cervical cancer cells were collected at 24 h after siRNA transfection for RNA extraction. Total RNA was reverse transcribed to cDNA using a First Strand cDNA kit (TaKaRa, Japan). qRT-PCR was performed by using SYBR Green PCR Master Mix (Applied Biosystems, USA). GAPDH was used as the endogenous control for mRNA expression. Relative gene expression levels were determined by the Ct method. The primers were as follow: MCM2, forward: 5ʹ-TGCCACTGTCATCCTAGCCA-3ʹ and reverse: 5ʹ-GATGGAAGGAGCAATGCTGG-3ʹ; GADPH, forward: 5ʹ-5ʹ-GCAAATTCCATGGCACCGT-3ʹ and reverse: 5ʹ-TCGCCCCACTTGATTTTGG-3ʹ.
Small Interfering RNA (siRNA) and Cell Transfection
HeLa and SiHa cells were transfected with siRNAs against MCM2 (siMCM2) or scrambled siRNA (siNC) by using Lipofectamine 3000 (Thermo Fisher Scientific), when HeLa and SiHa cells were up to ~80% confluence. The MCM2 siRNA sequence was 5ʹ-GGAGCUCAUUGGAGAUGGCAUGGAA-3ʹ.
EdU Incorporation Assay
The EdU incorporation assay was performed using an EdU DNA Cell Proliferation Kit (RiboBio, Guangzhou, China) to detect cell proliferation by following the manufacturer’s protocol. The number of EdU-positive cells was counted in 6 randomly selected fields.
Cell Counting Kit-8 (CCK-8) Assay
Cell proliferation was evaluated by CCK-8 assay (Dojindo, Tokyo, Japan). Briefly, the transfected HeLa and SiHa cells were seeded into 96-well plates. At indicated time points, 10 μL of CCK-8 solution was added to each well followed by incubation at 37 °C for another 2 h. The cell proliferative index was measured by detecting the absorbance at 450 nm using an automatic spectrophotometer.
Statistical Analysis
All analyses were performed using GraphPad Prism version 6.0 (GraphPad Software, USA). Data are expressed as the mean ± standard deviation, and differences between groups were analysed by one-way ANOVA followed by Bonferroni’s multiple comparison tests. Statistical significance was set at P < 0.05.
Results
Screening of DEGs from GEO Datasets Including GSE138080, GSE113942 and GSE63514
The workflow of our bioinformatics analysis and experimental validation is shown in Figure 1. We screened the DEGs from three datasets including GSE138080, GSE113942 and GSE63514, and presented the DEGs in each dataset using the volcano plots (Figure 2A–C). Pink and blue dots represented the up-regulated and down-regulated genes, respectively. In the GSE138080, a total of 813 DEGs with 308 up-regulated and 505 down-regulated genes were identified; in the GSE113942, a total of 3370 DEGs with 2079 up-regulated and 1291 down-regulated genes were identified; in the GSE63514, a total of 3023 DEGs with 1897 up-regulated and 1126 down-regulated genes were identified. The overlapping DEGs were identified in the up-regulated and down-regulated genes. As illustrated by the Venn diagrams, a total of 89 overlapping DEGs with 63 up-regulated and 26 down-regulated genes were identified among these datasets (Figure 2D; Supplemental Table 1).
 |
Figure 1 Workflow of the bioinformatics analysis and experimental validation. |
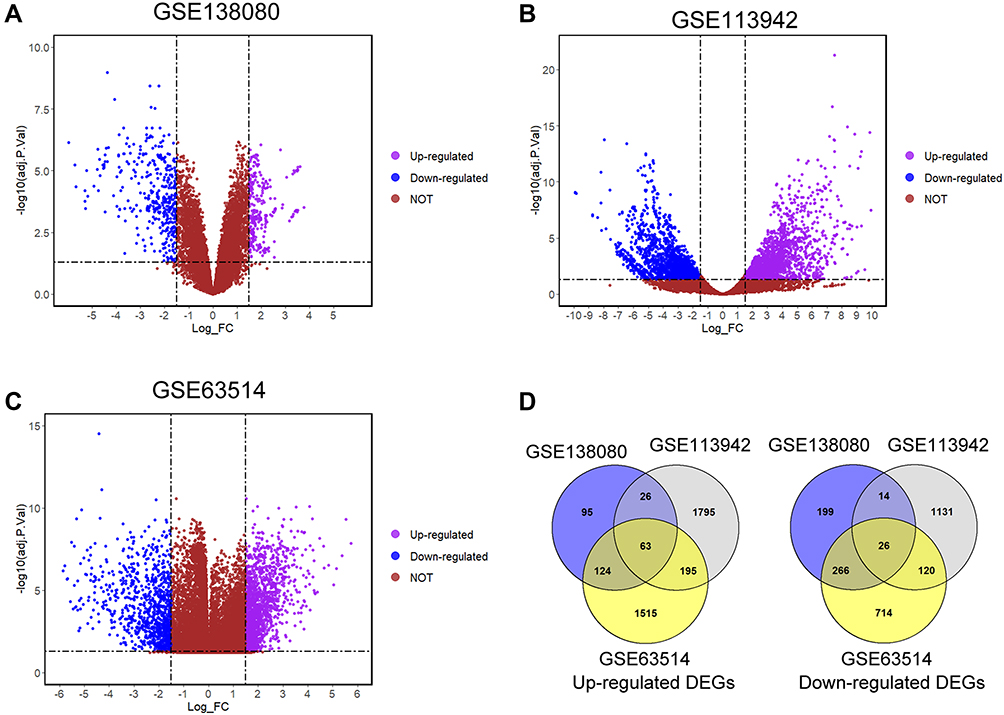 |
Figure 2 Screening of the overlapping DEGs among GSE138080, GSE113942 and GSE63514 datasets. The volcano plots of DEGs in (A) GSE138080, (B) GSE113942 and (C) GSE63514; blue dots and pink dots represent the significantly down-regulated and up-regulated DEGs respectively. (D) The up-regulated and down-regulated overlapping DEGs were illustrated in the Venn diagram. |
Functional Enrichment Analysis of Overlapping DEGs
The 89 overlapping DEGs were firstly subjected to the GO enrichment analysis. The top 10 enriched GO terms from biological process, cellular component and molecular function are shown in Figure 3A–C. In the biological process, the DEGs were significantly enriched in the terms including “DNA-dependent DNA replication”, “mitotic cell cycle process”, “cell cycle”, “mitotic cell cycle” and so on (Figure 3A). In the cellular component, the DEGs were enriched in the terms including “chromosome”, “chromosomal region”, “replication fork protection complex”, “replication fork” and so on (Figure 3B). In the molecular function, the DEGs were mainly classified into terms, including “3ʹ-5ʹ DNA helicase activity”, “catalytic activity, acting on DNA”, “DNA-dependent ATPase activity”, “DNA helicase activity” and so on (Figure 3C). For the KEGG analysis, the DEGs were significantly enriched in the pathways including “cell cycle, mitotic”, “cell cycle”, “DNA strand elongation”, “Unwinding of DNA” and so on (Figure 3D).
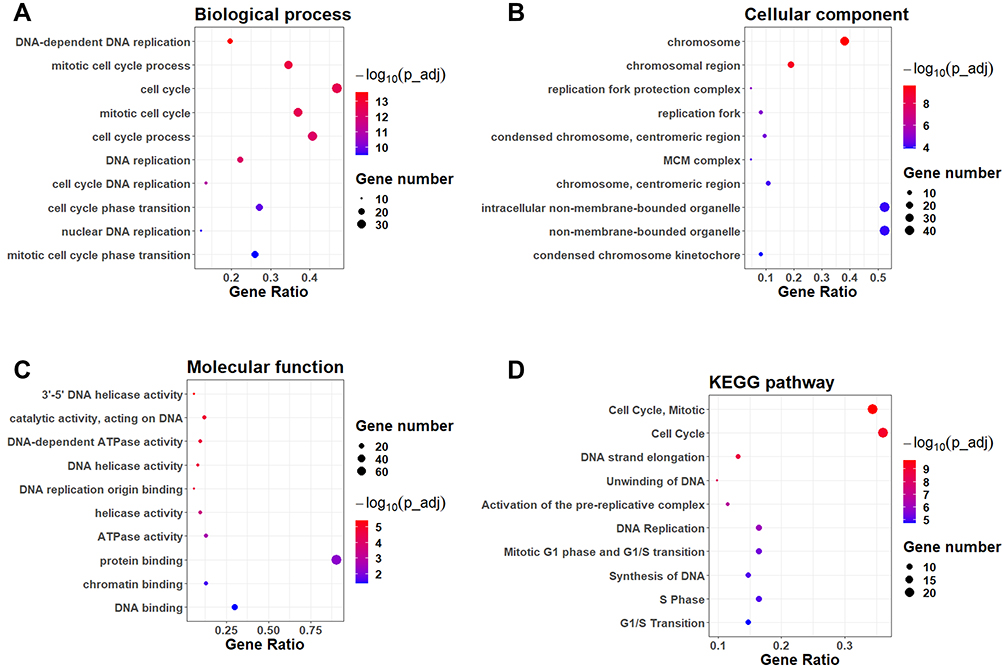 |
Figure 3 Functional enrichment analysis of the overlapping DEGs. The GO enrichment analysis of DEGs in the categories of (A) biological process, (B) cellular component and (C) molecular functions. The top 10 enriched GO terms in each category were shown. (D) The KEGG pathway enrichment analysis of the overlapping DEGs. The top 10 enriched KEGG pathways were shown. |
PPI Network Construction and Identification of Hub Genes
Firstly, the PPI network of the overlapping DEGs was constructed using the STRING database. As shown in Figure 4, the constructed PPI network contains 87 nodes and 309 edges with an average node degree of 7.1. Furthermore, the PPI network was constructed using the Cytoscape software (Figure 5A), and the sub-modules of the PPI network was further analysed using the MCODE tool. Based on the MCODE analysis, one sub-module with a score of 16.5 was identified. The sub-module network contains 21 genes with 21 nodes and 165 edges, and the constructed PPI network was shown in Figure 5B. The genes identified in the sub-module network were defined as hub genes for further analysis.
 |
Figure 4 PPI network construction of the overlapping DEGs using STRING database. |
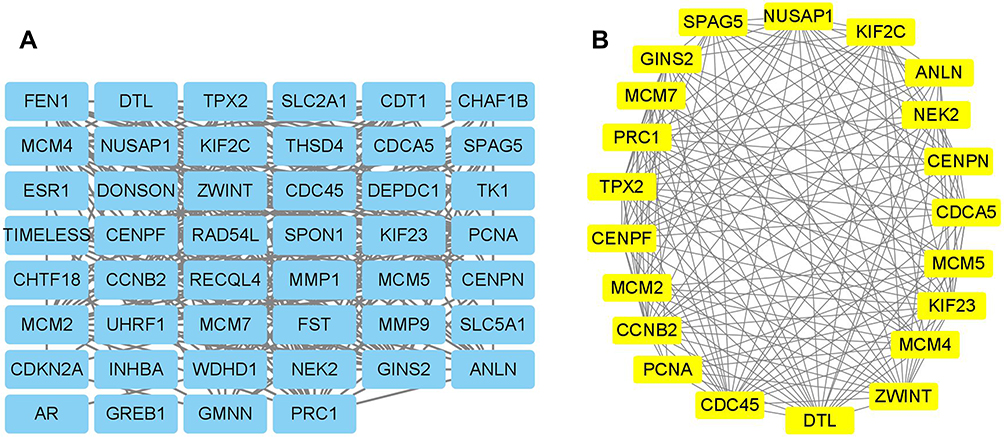 |
Figure 5 PPI network construction and hub genes identification using Cytoscape. (A) The PPI network of the overlapping DEGs were constructed in Cytoscape software. (B) The sub-module of the PPI network was illustrated using the MCODE tool. |
Expression Analysis of Hub Genes
The expression of the hub genes in the normal cervical tissues and cervical cancer tissues was analysed by the GEPIA. In the GEPIA datasets, 13 normal cervical tissues and 306 cervical cancer tissues were included in the analysis. As shown in Figure 5, all the 21 hub genes were significantly up-regulated in the cervical cancer tissues when compared to the normal cervical tissues (Figure 6).
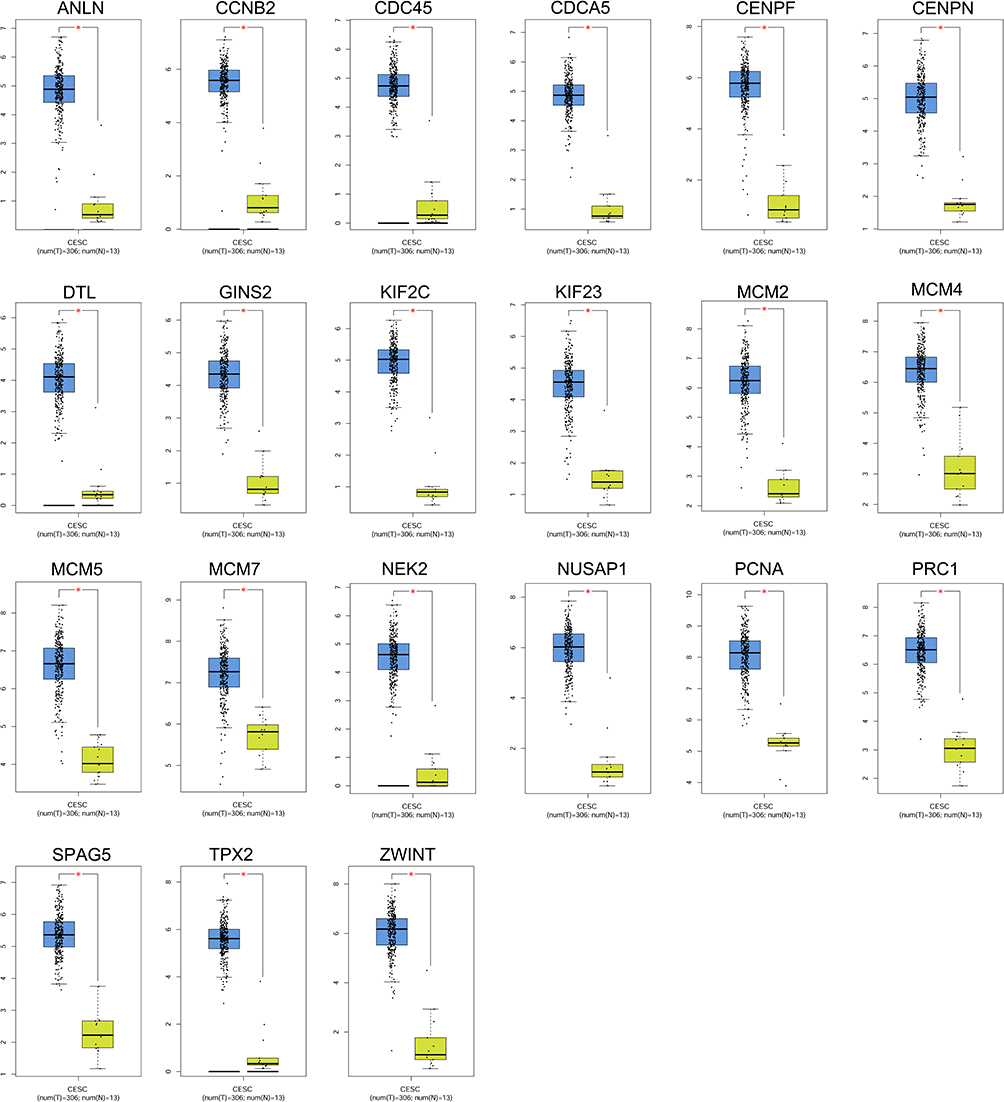 |
Figure 6 Expression analysis of hub genes in the cervical tissues. The expression levels of ANLN, CCNB2, CDC45, CDCA5, CENPF, CENPN, DTL, GINS2, KIF2C, KIF23, MCM2, MCM4, MCM5, MCM7, NEK2, NUSAP1, PCNA, PRC1, SPAG5, TPX2 and ZWINT in the normal cervical tissues and cervical cancer tissues were analysed using the GEPIA tool. |
Overall Survival Analysis of Hub Genes in Patients with Cervical Cancer
The correlation between hub genes and the overall survival of patients with cervical cancer was analysed by the GEPIA. In the analysed datasets, a total of 292 patients with cervical cancer were included in the analysis. Based on the median values of hub gene expression, the patients were classified into low expression and high expression groups. As shown in Figure 7, the low expressions of cell division cycle 45 (CDC45), GINS complex subunit 2 (GINS2), minichromosome maintenance complex component 2 (MCM2) and proliferating cell nuclear antigen (PCNA) were significantly correlated with the shorter survival of patients with cervical cancer (Figure 7A–D); whereas the other hub genes were not associated with the overall survival of the patients with cervical cancer (Supplemental Figure S1). Further validation studies using Human Protein Atlas database and KM plotter showed that low expression of CDC45, GINS2, MCM2 and PCNA predicted worse prognosis of patients with cervical cancer (Figures 8 and 9). Furthermore, the protein expression of CDC45, GINS2, MCM2 and PCNA in the cervical cancer tissues and normal cervical tissues was evaluated by immunohistochemistry using the Human Protein Atlas database; as shown in Figure 9, the protein expression levels of GINS2, MCM2 and PCNA, but not CDC45 were significantly higher in the cervical cancer tissues than that in the normal cervical tissues (Figure 10).
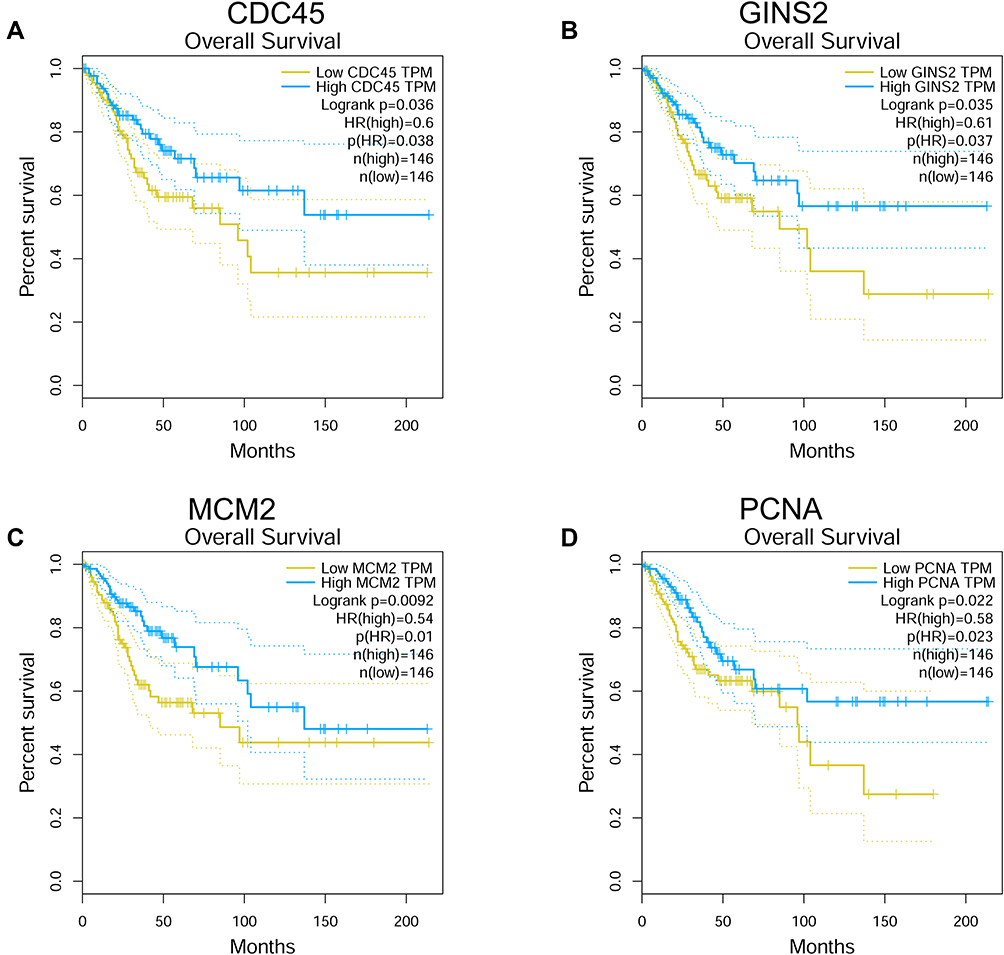 |
Figure 7 Overall survival analysis of hub genes using the GEPIA tool. The association between the expression levels of CDC45 (A), GINS2 (B), MCM2 (C) and PCNA (D) and overall survival of the patients with cervical cancer was analysed by using the GEPIA tool. |
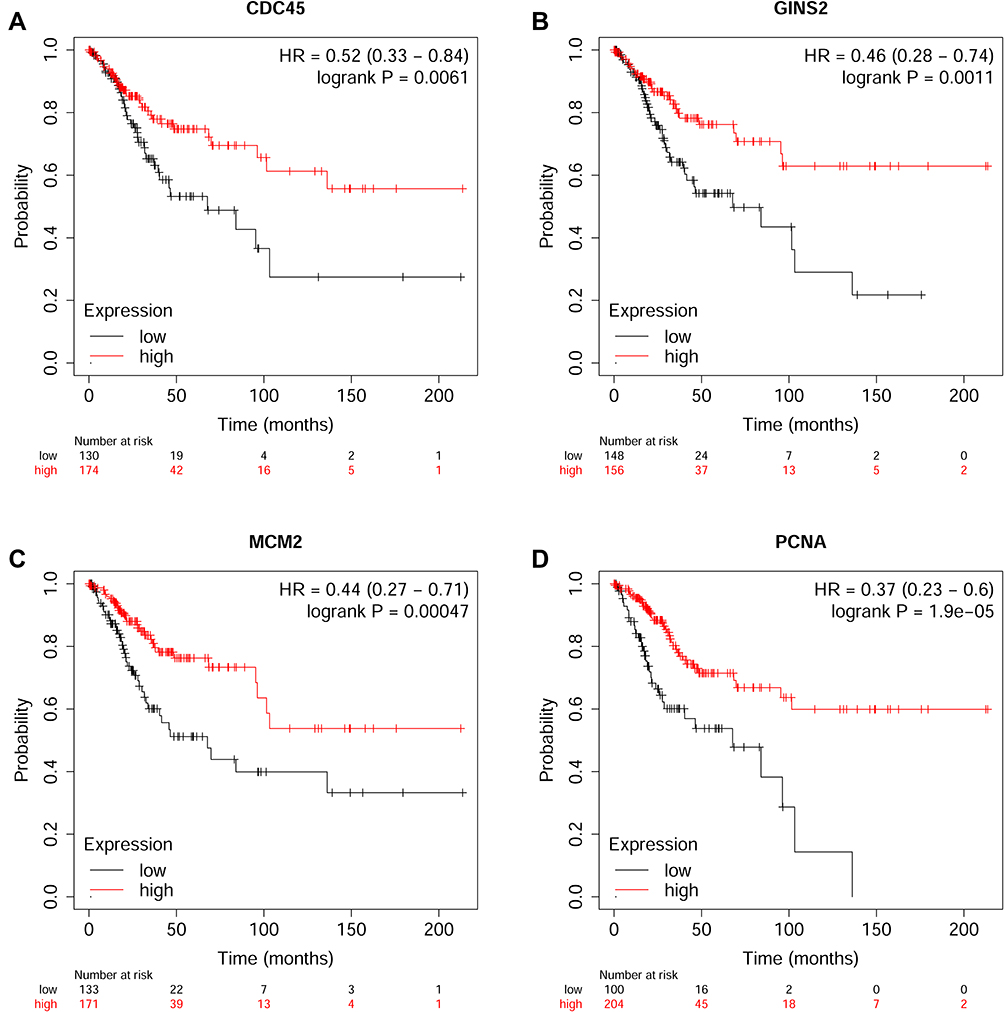 |
Figure 8 Overall survival analysis of hub genes using the KM plotter (https://kmplot.com/analysis/index.php?p=background). The association between the expression levels of CDC45 (A), GINS2 (B), MCM2 (C) and PCNA (D) and overall survival of the patients with cervical cancer was analysed by using the KM plotter. |
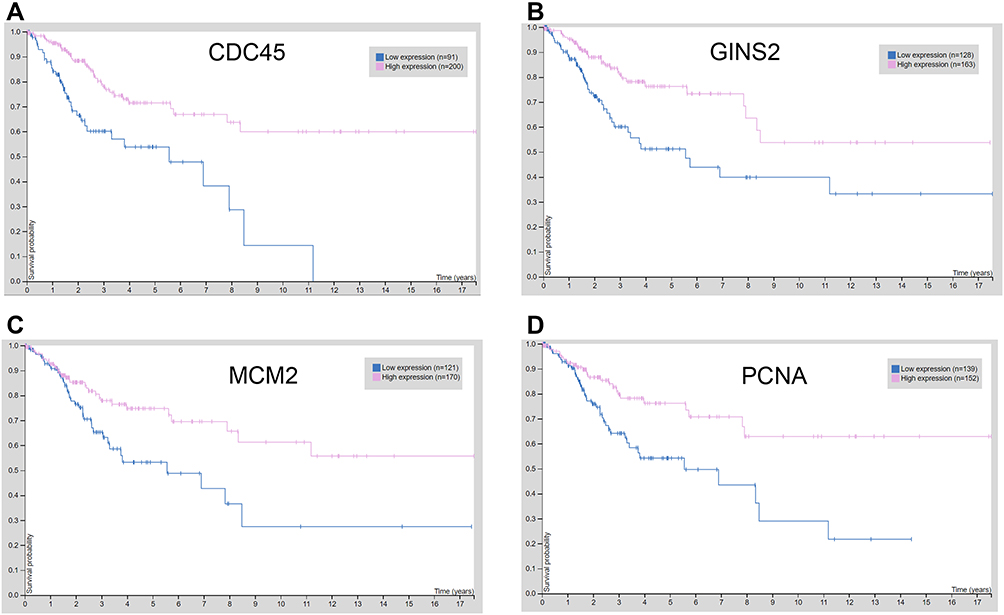 |
Figure 9 Overall survival analysis of hub genes using the Human Protein Atlas database (https://www.proteinatlas.org/). The association between the expression levels of CDC45 (A), GINS2 (B), MCM2 (C) and PCNA (D) and overall survival of the patients with cervical cancer was analysed by using the Human Protein Atlas database. |
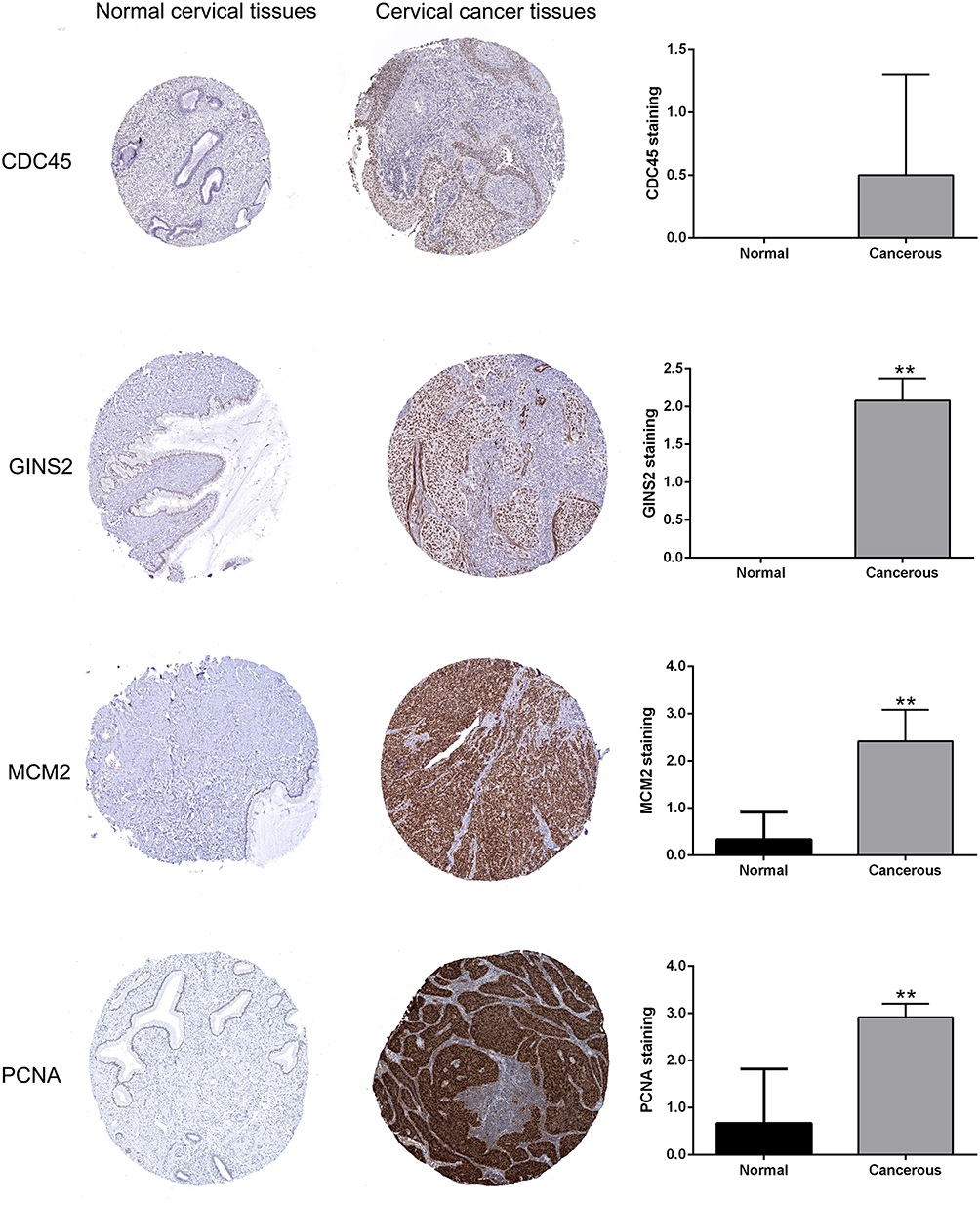 |
Figure 10 Protein expression levels of hub genes (CDC45, GINS2, MCM2 and PCNA) in the normal cervical tissues and cervical cancer tissues were analysed using the Human Protein Atlas database (https://www.proteinatlas.org/).Notes: **P < 0.01. |
Effects of MCM2 Knockdown on the Cell Proliferation of HeLa and SiHa Cells
Among these hub genes, MCM2 belongs to the MCM family, and emerging evidence implied that the MCM family members were key mediators in the cancer biology.18,19 Among the MCM members, the role of MCM2 in cervical cancer development and progression remains unclear, thus, we performed validation studies on MCM2. The effects of MCM2 knockdown on the cervical cancer cell proliferation were determined by CCK-8 and EdU incorporation assays, respectively. As shown in Figure 11A, the transfection of MCM2 siRNA (siMCM2) significantly down-regulated the MCM2 mRNA expression in both HeLa and SiHa cells. CCK-8 assay showed that MCM2 knockdown suppressed the HeLa and SiHa cell proliferation (Figure 11B and C). The EdU incorporation assay consistently showed that MCM2 siRNA transfection significantly reduced the EdU-positive HeLa and SiHa cells as compared to the siNC group (Figure 11D).
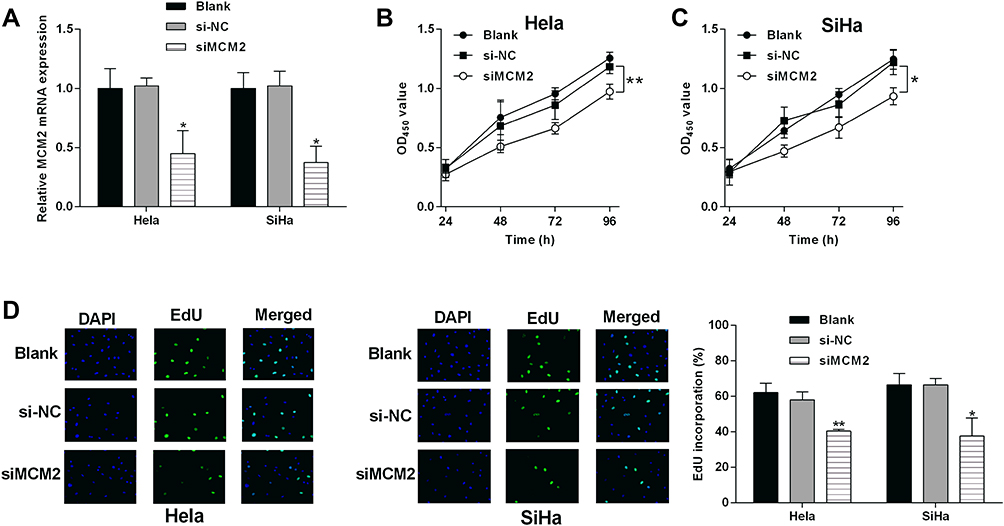 |
Figure 11 Effects of MCM2 knockdown on the cervical cancer cell proliferation. (A) The mRNA expression of MCM2 in HeLa and SiHa without any treatment, with siNC or siMCM2 transfections were determined by qRT-PCR. (B) HeLa and (C) SiHa cells after being treated with siNC or siMCM2. (D) The cell proliferation of HeLa and SiHa cells in different groups were determined by EdU incorporation assay. CCK-8 assay evaluated the proliferative potential of N = 3, *P < 0.05 and **P < 0.01. |
Discussion
Cervical cancer is one of the most common female malignancies, and great improvements have been made in the prevention and treatment of cervical cancer.1 However, the molecular mechanisms underlying cervical cancer progression are not clear. With the advancement of the high throughput technologies, the novel biomarkers for cancer progression have been identified via analysing the microarray datasets. In the present study, we analysed 3 GEO datasets including GSE138080, GSE113942 and GSE63514; and a total of 89 overlapping DEGs (63 up-regulated and 26 down-regulated genes) were identified among the datasets. The functional enrichment analysis indicated that the overlapping DEGs were mainly associated with “DNA replication” and “cell cycle”. Furthermore, the PPI network analysis revealed a network with 87 nodes and 309 edges. Sub-module analysis using MCODE identified 21 hub genes from the PPI network. The expressions of the 21 hub genes were all up-regulated in the cervical cancer tissues when compared to normal cervical tissues as analysed by the GEPIA tool. The survival analysis showed that the low expression of CDC45, GINS2, MCM2 and PCNA was significantly correlated with the shorter survival of patients with cervical cancer. Moreover, the protein expression levels of GINS2, MCM2 and PCNA, but not CDC45 were also significantly up-regulated in the cervical cancer tissues when compared to normal cervical tissues. Based on the above results, our analysis revealed four hub genes including CDC45, GINS2, MCM2 and PCNA may be associated with the prognosis of patients with cervical cancer.
In our study, we found that CDC45 was up-regulated in the cervical cancer tissues and predicted a better prognosis of patients with cervical cancer. In the KEGG pathway enrichment analysis, CDC45 was classified into cell cycle pathway. CDC45 has a critical role in the initiation and elongation steps of DNA replication to ensure that chromosomal DNA is replicated only once per cell cycle.20 In the esophageal squamous cell carcinoma, CDC45 can be suppressed ring finger protein 1 and Yin Yang 1-binding protein, which exerted the tumor-suppressive effects on this malignancy.21 CDC45 could promote papillary thyroid cancer progression via regulating cell cycle.22 Studies from Huang et al, indicated that CDC45 functioned as an oncogenes in non-small cell lung cancer.23 KNK437 could destabilize CDC45, which lead to the suppression of colorectal cancer growth and metastasis.24 In the bioinformatics analysis, CDC45 was found to be up-regulated in colorectal cancer,25 hepatocellular carcinoma,26 and breast cancer tissues.27 In colorectal cancer, low expression of CDC45 predicts poor prognosis of patients with colorectal cancer, which can be explained as CDC45 being down-regulated in colorectal cancer tissues with advanced stage.25 The bioinformatics studies showed that CDC45 was up-regulated in the cervical cancer tissues and a high expression of CDC45 was correlated with longer overall survival of patients with cervical cancer.28,29 Our findings were consistent with the previous studies, suggesting the robust bioinformatics analysis in this study. However, the expression of CDC45 may be investigated in the cervical cancer tissues with different clinical stages, which can further reveal the prognostic role of CDC45 in the cervical cancer.
GINS2 belongs to the tetrameric complex family and functions as the replicative helicase, unwinding duplex DNA ahead of the moving replication fork. The role of GINS2 has been deciphered in various types of cancers. A high GINS2 transcript level predicts poor prognosis and correlates with high histological grade and endocrine therapy resistance through mammary cancer stem cells in breast cancer patients,30 and GINS2 regulates matrix metallopeptidase 9 expression and cancer stem cell property in human triple negative breast cancer.31 GINS2 promotes cell proliferation and inhibits cell apoptosis in thyroid cancer by regulating Cbp/p300-interacting transactivator 2 and lysyl oxidase like 2;32 knockdown of GINS2 inhibits cell proliferation and tumorigenesis in human gliomas;33 GINS2 promotes EMT in pancreatic cancer via specifically stimulating extracellular signal-regulated kinase 1/2/mitogen-activated protein kinase signaling.34 In the cervical cancer, Chen et al, found that GINS2 was up-regulated in the SiHa cells and cervical cancer tissues.35 However, the prognostic role of GINS2 in cervical cancer remains unknown. Our bioinformatics analysis revealed that low expression of GINS2 was associated with poor prognosis of patients with cervical cancer; however, the detailed mechanisms of GINS2 in cervical cancer progression still requires further examination.
PCNA plays a key role in the DNA replication of mammalian cells and their small DNA tumour viruses. In the cervical cancer studies, Up-regulation of PCNA is closely associated with high-risk human papillomavirus and progression of cervical intraepithelial neoplasia (CIN), but does not predict disease outcome in cervical cancer.36 Kim et al, demonstrated that PCNA immunostaining enhanced diagnostic accuracy for high-grade cervical squamous intraepithelial lesion.37 Madhumati et al, found that PCNA labelling index was increased with increasing severity of CIN lesions.38 In agreement with the above studies, our results consistently identified the up-regulation of PCNA in the cervical cancer tissues. Although the analysis of the public database from this study indicated that low expression of PCNA predicted poor prognosis of patients with cervical cancer, the prognostic role of PCNA is still unclear; and future clinical studies are warranted to validate the prognostic role of PCNA in cervical cancer.
MCM2 belongs to the MCM protein complex which consists of six highly conserved proteins (MCM2-7) and plays an important role in the initiation of DNA replication and DNA unwinding due to its replicative helicase activity. The prognostic role of MCM2 has been demonstrated in various types of cancers including breast cancer,39 colon cancer,40 liver cancer26 and so on. In the cervical cancer studies, Kuku et al, performed the immunostaining for MCM2 in cervical cancer tissues and suggested that MCM2 does not appear to predict disease grade, stage, or outcome of patients with cervical cancer.41 Filho et al, showed that HPV-infected cells strongly express MCM2; nevertheless, their data suggests that MCM2 is not a good biomarker when comparing the different clinical stages of cervical cancer.42 Studies also showed that MCM2 was over expressed in cervical cancer irrespective of clinicopathological parameters,43 which was consistent with our findings regarding the MCM2 protein expression in cervical cancer using the Human Protein Atlas database. A recent study suggested that a strong expression of MCM2 protein in a high grade squamous intraepithelial lesion may aid as a concatenated screening tool in detecting pre-cancerous cervical lesions.44 In our study, the bioinformatics result showed that up-regulated MCM2 in the cervical cancer tissues may predict longer overall survival of patients with cervical cancer. The in vitro studies have shown that MCM2 mRNA was enriched in HeLa cells to approximately four times the level in WI-38 cells.45 In other types of cancers such as ovarian and lung cancer, MCM2 was found to promote the proliferative potential of these cancer cells.46,47 In our in vitro functional studies, we consistently showed that MCM2 knockdown suppressed the proliferation of HeLa and SiHa cells, suggesting that MCM2 may play an oncogenic role in the cervical cancer progression. However, the prognostic role of MCM2 in cervical cancer is still controversial among studies, as a high expression of MCM2 showed better prognosis for patients with cervical cancer as shown in our bioinformatics analysis. Indeed, studies have found a strong correlation between the MCM2-positive cells and the presence of HPV DNA detected by in situ hybridization. The database for the cervical patients may not be stratified according to the clinical characteristics of the patients, and future studies should stratify the patients based on clinical characteristics such as HPV-infection, tumor stage, etc to further confirm the prognostic role of MCM2.
The evidence regarding the prognostic role of MCM2 was mainly based on the online bioinformatics tools in our study, and the exact prognostic role of MCM2 still requires detailed examination using clinical data from a large cohort of patients with cervical cancer from multiple centres. The present study only analysed three GEO datasets, and future studies may screen more DEGs to further explore novel targets or confirm the role of MCM2 in cervical cancer progression. Unfortunately, our experimental studies were limited to the in vitro assays, and further in vivo assays should be considered to examine the role of MCM2 in tumor growth. Finally, the regulatory mechanism of MCM2 in cervical cancer is worthy of further investigation.
In conclusion, we screened a total of 89 overlapping genes from the GEO datasets, and further analysis identified four hub genes (CDC45, GINS2, MCM2 and PCNA) that were likely associated with the prognosis of patients with cervical cancer. MCM2 knockdown repressed the cervical cancer cell proliferation. The current findings may provide novel insights into understanding the pathophysiology of cervical cancer and develop therapeutic targets for patients with cervical cancer. Future validation studies are required to confirm the prognostic role and mechanistic actions of the hub genes in cervical cancer.
Disclosure
The authors report no conflict of interest in this work.
References
1. Cohen PA, Jhingran A, Oaknin A, Denny L. Cervical cancer. Lancet. 2019;393(10167):169–182. doi:10.1016/S0140-6736(18)32470-X
2. Abbas KM, van Zandvoort K, Brisson M, Jit M. Effects of updated demography, disability weights, and cervical cancer burden on estimates of human papillomavirus vaccination impact at the global, regional, and national levels: a PRIME modelling study. Lancet Global Health. 2020;8(4):e536–e544. doi:10.1016/S2214-109X(20)30022-X
3. Canfell K, Kim JJ, Brisson M, et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):591–603. doi:10.1016/S0140-6736(20)30157-4
4. The L. Eliminating cervical cancer. Lancet. 2020;395(10221):312. doi:10.1016/S0140-6736(20)30247-6
5. Ward ZJ, Grover S, Scott AM, et al. The role and contribution of treatment and imaging modalities in global cervical cancer management: survival estimates from a simulation-based analysis. Lancet Oncol. 2020;21(8):1089–1098. doi:10.1016/S1470-2045(20)30316-8
6. Huang X, Liu S, Wu L, Jiang M, Hou Y. High throughput single cell RNA sequencing, bioinformatics analysis and applications. Adv Exp Med Biol. 2018;1068:33–43.
7. Merrick BA, London RE, Bushel PR, Grissom SF, Paules RS. Platforms for biomarker analysis using high-throughput approaches in genomics, transcriptomics, proteomics, metabolomics, and bioinformatics. IARC Sci Publ. 2011;163:121–142.
8. Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncol. 2015;19(1a):A68–77.
9. Wang Z, Lachmann A, Ma’ayan A. Mining data and metadata from the gene expression omnibus. Biophys Rev. 2019;11(1):103–110. doi:10.1007/s12551-018-0490-8
10. Dai F, Chen G, Wang Y, et al. Identification of candidate biomarkers correlated with the diagnosis and prognosis of cervical cancer via integrated bioinformatics analysis. Onco Targets Ther. 2019;12:4517–4532. doi:10.2147/OTT.S199615
11. Liang B, Li Y, Wang T. A three miRNAs signature predicts survival in cervical cancer using bioinformatics analysis. Sci Rep. 2017;7(1):5624. doi:10.1038/s41598-017-06032-2
12. Lin H, Ma Y, Wei Y, Shang H. Genome-wide analysis of aberrant gene expression and methylation profiles reveals susceptibility genes and underlying mechanism of cervical cancer. Eur J Obstet Gynecol Reprod Biol. 2016;207:147–152. doi:10.1016/j.ejogrb.2016.10.017
13. Choi K, Ratner N. iGEAK: an interactive gene expression analysis kit for seamless workflow using the R/shiny platform. BMC Genomics. 2019;20(1):177. doi:10.1186/s12864-019-5548-x
14. Raudvere U, Kolberg L, Kuzmin I, et al. g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–w198. doi:10.1093/nar/gkz369
15. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
16. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–w102. doi:10.1093/nar/gkx247
17. Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8(1):9227. doi:10.1038/s41598-018-27521-y
18. Fei L, Xu H. Role of MCM2-7 protein phosphorylation in human cancer cells. Cell Biosci. 2018;8:43. doi:10.1186/s13578-018-0242-2
19. Ishimi Y. Regulation of MCM2-7 function. Genes Genet Syst. 2018;93(4):125–133. doi:10.1266/ggs.18-00026
20. Pollok S, Bauerschmidt C, Sänger J, Nasheuer HP, Grosse F. Human CDC45 is a proliferation-associated antigen. FEBS J. 2007;274(14):3669–3684. doi:10.1111/j.1742-4658.2007.05900.x
21. Ke Y, Guo W, Huang S, et al. RYBP inhibits esophageal squamous cell carcinoma proliferation through downregulating CDC6 and CDC45 in G1-S phase transition process. Life Sci. 2020;250:117578. doi:10.1016/j.lfs.2020.117578
22. Sun J, Shi R, Zhao S, et al. Cell division cycle 45 promotes papillary thyroid cancer progression via regulating cell cycle. Tumour Biology. 2017;39(5):1010428317705342. doi:10.1177/1010428317705342
23. Huang J, Li Y, Lu Z, et al. Analysis of functional hub genes identifies CDC45 as an oncogene in non-small cell lung cancer - a short report. Cellular Oncol. 2019;42(4):571–578. doi:10.1007/s13402-019-00438-y
24. Yang S, Ren X, Liang Y, et al. KNK437 restricts the growth and metastasis of colorectal cancer via targeting DNAJA1/CDC45 axis. Oncogene. 2020;39(2):249–261. doi:10.1038/s41388-019-0978-0
25. Hu Y, Wang L, Li Z, et al. Potential prognostic and diagnostic values of CDC6, CDC45, ORC6 and SNHG7 in colorectal cancer. Onco Targets Ther. 2019;12:11609–11621. doi:10.2147/OTT.S231941
26. Sang L, Wang XM, Xu DY, Zhao WJ. Bioinformatics analysis of aberrantly methylated-differentially expressed genes and pathways in hepatocellular carcinoma. World J Gastroenterol. 2018;24(24):2605–2616. doi:10.3748/wjg.v24.i24.2605
27. Wu J, Lv Q, Huang H, Zhu M, Meng D. Screening and identification of key biomarkers in inflammatory breast cancer through integrated bioinformatic analyses. Genet Test Mol Biomarkers. 2020;24(8):484–491. doi:10.1089/gtmb.2020.0047
28. Liu J, Nie S, Gao M, et al. Identification of EPHX2 and RMI2 as two novel key genes in cervical squamous cell carcinoma by an integrated bioinformatic analysis. J Cell Physiol. 2019;234(11):21260–21273. doi:10.1002/jcp.28731
29. Wen X, Liu S, Cui M. Effect of BRCA1 on the concurrent chemoradiotherapy resistance of cervical squamous cell carcinoma based on transcriptome sequencing analysis. Biomed Res Int. 2020;2020:3598417. doi:10.1155/2020/3598417
30. Zheng M, Zhou Y, Yang X, et al. High GINS2 transcript level predicts poor prognosis and correlates with high histological grade and endocrine therapy resistance through mammary cancer stem cells in breast cancer patients. Breast Cancer Res Treat. 2014;148(2):423–436. doi:10.1007/s10549-014-3172-7
31. Peng L, Song Z, Chen D, et al. GINS2 regulates matrix metallopeptidase 9 expression and cancer stem cell property in human triple negative Breast cancer. Biomed Pharmacother. 2016;84:1568–1574. doi:10.1016/j.biopha.2016.10.032
32. Ye Y, Song YN, He SF, Zhuang JH, Wang GY, Xia W. GINS2 promotes cell proliferation and inhibits cell apoptosis in thyroid cancer by regulating CITED2 and LOXL2. Cancer Gene Ther. 2019;26(3–4):103–113. doi:10.1038/s41417-018-0045-y
33. Shen YL, Li HZ, Hu YW, Zheng L, Wang Q. Loss of GINS2 inhibits cell proliferation and tumorigenesis in human gliomas. CNS Neurosci Ther. 2019;25(2):273–287. doi:10.1111/cns.13064
34. Huang L, Chen S, Fan H, Ji D, Chen C, Sheng W. GINS2 promotes EMT in pancreatic cancer via specifically stimulating ERK/MAPK signaling. Cancer Gene Ther. 2020. doi:10.1038/s41417-020-0206-7
35. Chen T, Yang S, Xu J, Lu W, Xie X. Transcriptome sequencing profiles of cervical cancer tissues and SiHa cells. Funct Integr Genomics. 2020;20(2):211–221. doi:10.1007/s10142-019-00706-y
36. Branca M, Ciotti M, Giorgi C, et al. Up-regulation of proliferating cell nuclear antigen (PCNA) is closely associated with high-risk human papillomavirus (HPV) and progression of cervical intraepithelial neoplasia (CIN), but does not predict disease outcome in cervical cancer. Eur J Obstet Gynecol Reprod Biol. 2007;130(2):223–231. doi:10.1016/j.ejogrb.2006.10.007
37. Kim TH, Han JH, Shin E, Noh JH, Kim HS, Song YS. Clinical implication of p16, ki-67, and proliferating cell nuclear antigen expression in cervical neoplasia: improvement of diagnostic accuracy for high-grade squamous intraepithelial lesion and prediction of resection margin involvement on conization specimen. J Cancer Prevention. 2015;20(1):70–77.
38. Madhumati G, Kavita S, Anju M, Uma S, Raj M. Immunohistochemical expression of cell proliferating nuclear antigen (PCNA) and p53 protein in cervical cancer. J Obstet Gynaecol India. 2012;62(5):557–561. doi:10.1007/s13224-012-0180-6
39. Issac MSM, Yousef E, Tahir MR, Gaboury LA. MCM2, MCM4, and MCM6 in breast cancer: clinical utility in diagnosis and prognosis. Neoplasia. 2019;21(10):1015–1035. doi:10.1016/j.neo.2019.07.011
40. Giaginis C, Georgiadou M, Dimakopoulou K, et al. Clinical significance of MCM-2 and MCM-5 expression in colon cancer: association with clinicopathological parameters and tumor proliferative capacity. Dig Dis Sci. 2009;54(2):282–291. doi:10.1007/s10620-008-0305-z
41. Kuku S, Proctor I, Loddo M, et al. Do cell-cycle phase-specific markers predict disease grade, stage, and outcome in cervical carcinoma? Int J Gynecological Cancer. 2015;25(6):1066–1072. doi:10.1097/IGC.0000000000000356
42. Amaro Filho SM, Nuovo GJ, Cunha CB, et al. Correlation of MCM2 detection with stage and virology of cervical cancer. Int J Biol Markers. 2014;29(4):e363–371. doi:10.5301/jbm.5000081
43. Das M, Prasad SB, Yadav SS, et al. Over expression of minichromosome maintenance genes is clinically correlated to cervical carcinogenesis. PLoS One. 2013;8(7):e69607. doi:10.1371/journal.pone.0069607
44. Kaur G, Balasubramaniam SD, Lee YJ, Balakrishnan V, Oon CE. Minichromosome maintenance complex (MCM) genes profiling and MCM2 protein expression in cervical cancer development. Asian Pacific J Cancer Prevention. 2019;20(10):3043–3049. doi:10.31557/APJCP.2019.20.10.3043
45. Ishimi Y, Okayasu I, Kato C, et al. Enhanced expression of Mcm proteins in cancer cells derived from uterine cervix. European J Biochem. 2003;270(6):1089–1101. doi:10.1046/j.1432-1033.2003.03440.x
46. Deng M, Sun J, Xie S, et al. Inhibition of MCM2 enhances the sensitivity of ovarian cancer cell to carboplatin. Mol Med Rep. 2019;20(3):2258–2266.
47. Wu W, Wang X, Shan C, Li Y, Li F. Minichromosome maintenance protein 2 correlates with the malignant status and regulates proliferation and cell cycle in lung squamous cell carcinoma. Onco Targets Ther. 2018;11:5025–5034. doi:10.2147/OTT.S169002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.