Back to Journals » Cancer Management and Research » Volume 12
Identification of High-Frequency Methylation Sites in RNF180 Promoter Region Affecting Expression and Their Relationship with Prognosis of Gastric Cancer
Authors Han F, Liu S, Jing J ![]() , Li H, Yuan Y
, Li H, Yuan Y ![]() , Sun LP
, Sun LP ![]()
Received 22 January 2020
Accepted for publication 14 April 2020
Published 13 May 2020 Volume 2020:12 Pages 3389—3399
DOI https://doi.org/10.2147/CMAR.S246995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Fang Han,1,2,* Shuang Liu,1,3,* Jingjing Jing,1 Hao Li,1 Yuan Yuan,1 Li-Ping Sun1
1Tumor Etiology and Screening Department of Cancer Institute and General Surgery, The First Hospital of China Medical University, and Key Laboratory of Cancer Etiology and Prevention in Liaoning Education Department, Shenyang 110001, People’s Republic of China; 2Hepatobiliary and Pancreatic Surgery, Minimal Invasive Surgery, Zhejiang Provincial People’s Hospital, Hangzhou Medical College, Hangzhou 310014, People’s Republic of China; 3Department of Oncology, Shanxi Provincial Tumor Hospital, Xi’an 710076, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li-Ping Sun; Yuan Yuan Email [email protected]; [email protected]
Background: Ring finger protein 180 (RNF180) is a tumor suppressor gene regulated by promoter methylation. We previously demonstrated that the RNF180 promoter methylation could be a risk factor for gastric cancer (GC); and eight high-frequency hypermethylated CpG sites were associated with GC. However, it is not clear whether these key sites can affect gene expression and involve in prognosis. The aim of this study was to investigate the effects of above CpG sites on the gene expression and prognosis of GC.
Patients and Methods: A total of 164 GC tissues were enrolled and followed up. Tissue samples were used for DNA and RNA isolation. Methylation status of RNF180 was detected using bisulfite sequencing PCR (BSP). Expression levels of RNF180 were detected using quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR). JASPAR and PROMO databases were used to predict the transcription factors (TFs) binding to the CpG site.
Results: The methylation in RNF180 promoter region increased and mRNA expression decreased in GC tissue. Correlation analysis revealed that the average methylation rate (AMR) and four CpG sites methylation rate were negatively related to RNF180 expression, including M3(− 165)(Chr5:64165942), M5(− 148)(Chr5:64,165,959), M7(− 133)(Chr5:64,165,974) and M8(− 130)(Chr5:64,165,977). Furthermore, the methylation rate of M5(− 148)(Chr5:64,165,959) and M27(− 26)(Chr5:64,166,081) above 0.3 indicated poor prognosis (PM5 = 0.008, PM27 = 0.003, HRM5(− 148) = 2.000 (1.201,3.332), HRM27(− 26)=2.389 (1.336,4.271)), which could be independent factors of prognosis.
Conclusion: By focusing on the methylation sites in the RNF180 promoter region, we identified two high-frequency methylation sites, M5(− 148)(Chr5:64,165,959) and M27(− 26)(Chr5:64,166,081), which could affect gene expression and predict the prognosis of GC. In the future, the possible molecular mechanism involved needs to be further studied.
Keywords: gastric cancer, RNF180, methylated CpG sites, prognosis, methylation
Introduction
DNA methylation, an important regulation mechanism of gene expression, is a critical epigenetic modification which is not based on DNA sequence changes. DNA aberrant methylation commonly occurs in the CpG island region of the tumor suppressor gene.1 It has been found that the abnormal methylation of tumor suppressor genes affects the various malignant biological characteristics.2,3 Moreover, the CpG island of promoter methylation is often related to prognosis.4 As an important tumor suppressor gene family in homo sapiens, RNF (Ring Finger Protein) genes are involved in the construction of E3 ubiquitin protein ligase in ubiquitin proteasome system (UPS), which are also involved in ubiquitin degradation.5 It has been found that RNFs participate in a variety of cellular processes including DNA repair, gene transcription and apoptosis.6 The disorder of UPS often occurs in various tumors, which leads to tumorigenesis.7 RNF180 is a tumor suppressor gene that encodes the ubiquitin ligase E3 RING structure protein.8 Its function covers various biological behaviors such as cell proliferation, apoptosis, differentiation and tumor formation by affecting the protein ubiquitination degradation.8 Inactivation of RNF180 is related to apoptosis, tumor invasion and metastasis.9
Gastric cancer (GC) is a malignant tumor that seriously threatens human health. The incidence rate of gastric cancer ranks fifth in malignant tumor worldwide. And the third leading cause of cancer-related death.10 Our previous study found that the methylation level of RNF180 promoter region was related to gastric tumorigenesis.11 According to the results of our previous work, a series of highly frequent-methylated CpG sites were considered to be highly associated with atrophy gastritis (AG) and GC, which are M3(−165)(Chr5:64,165,942), M5(−148)(Chr5:64,165,959), M7(−133)(Chr5:64,165,974), M8(−130)(Chr5:64,165,977), M20(−57)(Chr5:64,166,050), M25(−34)(Chr5:64,166,073), M27(−26)(Chr5:64,166,081) and M30(+5)(Chr5:64,166,112). Risk analysis indicated that increased methylation rate at multiple CpG sites could significantly increase the risk of GC, suggesting that methylation of RNF180 CpG sites in the promoter region could be used as a molecular marker for prediction and early diagnosis of GC. However, it is not clear whether these key sites could affect gene expression and involve in prognosis. This study focused on the methylation of specific CpG sites in RNF180 promoter region to evaluate the correlation of gene expression, and combined with clinical and prognostic information to discuss the feasibility of methylation as a molecular biomarker for GC prognosis.
Patients and Methods
Patients and Tissue Samples
A total of 164 GC patients were enrolled in the First Hospital of China Medical University, Shenyang, China, from 2011 to 2015, including 112 males and 52 females with average ages of 60.2±10.7 and 56.9±12.6 years, respectively. The tumor tissues were collected from patients who underwent surgical resection within 30 mins. Among them, tumor-adjacent non-cancerous tissues in 80 cases were also collected meanwhile. Tissue samples were frozen at –80°C until DNA and RNA extraction. Two gastrointestinal pathologists were invited to evaluate the pathologic diagnosis for all cases independently in accordance with the WHO classification and updated Sydney System. Epidemiological information and clinical-pathological data were collected by questionnaires and computerized medical records. After surgery, all subjects were followed up every 6 months at the outpatient department or by telephone until the patient died. The follow-up period was from August 2012 to March 2018. Median follow-up time was 35.5 months. This study was approved by the Human Ethics Review Committee of the First Hospital of China Medical University (Shenyang, China). Written informed consent was provided to all patients. All consent progress was conducted in accordance with the Declaration of Helsinki. The Ethical Review Committee number of this study was 2016 No.2016-155-2.
DNA Extraction
Tissue DNA was extracted according to chloroform/phenol extraction procedure as follows: take 50 mg fresh tissue, cryogenic grinding, add lysate (400 ul Tris-EDTA (Cat. PH0620, Phygene China) + 25 μL, 10% Sodium dodecyl sulfate (SDS, Sigma US)+10μL Proteinase K(Cat. P2308 Sigma US)), mix and digest overnight in 37°C. On the next day, phenol and chloroform were added to extract diluted DNA. Absolute ethanol was used to precipitate DNA. At last genomic DNA was diluted by using Tris-EDTA and stored at −20°C during the experiment.
Bisulfite Sequencing PCR
Bisulfite sequencing PCR (BSP) was used as a quantitatively analyzed method for detecting the methylation rate of the RNF180 promoter area. The gene sequence for this study was obtained from Genebank. The MethylKIT package (http://www.bioconductor.org/packages/release/bioc/html/methylKit.html) of R software was used to analyze methylation data, all CpG sites and transcription initiation site (TSS). Genomic DNA sodium bisulfite modification was performed by Zymo DNA Methylation-Gold kit (Cat. Nos. D5005 or D5006, Zymo Research, US) according to the manufacturers’ instructions. Polymerase chain reaction (PCR) was performed by the enzyme TaKaRa Taq™ Hot Start Version (Code No. R007A Takara, Japan). The primers were as follows: F:5′-GTGGTTTTGGTAAGGGGATGAT-3′; R: 5′-AACAACCAAACTCTAAAAACTC-3′.11 The condition was as follows: 94°C for 3 mins for initial denaturation, run 45 cycles (94°C 20 s, 58.5°C 30 s, 72°C 45 s); after the end of the cycles followed by an extension 10 mins at 72°C, a final termination at 4°C. PCR purification products were used for forward sequence analysis. Chromas version 2.6.6 (Technelysium Pty Ltd., Australia) was used to analyze sequencing results. According to the theory of bisulfite modification, methylation rate calculation formula was as follows: Meth%=C/(C+T)*100%.12
RNA Extraction
Tissue RNA was extracted with the Animal Tissue Total RNA Extraction Kit (TIANGEN, DP431, CHINA). A 1μg total RNA was reversed transcribed into cDNA using the Fast Quant cDNA First-Strand Synthesis Kit (TIANGEN, KR106). RNA extraction and reverse transcription were performed according to the instructions.
Quantitation Real-Time Polymerase Chain Reaction (RT-qPCR)
The expression level of RNF180 mRNA was detected by RT-qPCR. The primers were as follows: Forward: 5ʹ-TCTGACTTTCCTGATGGACCTG-3ʹ, Reverse: 5ʹ-CCTGAGTATTTACCCTGCTTCTGT-3ʹ. GAPDH was used as a control. The primers were as follows: Forward: 5ʹ-TGCACCACCAACTGCTTAG-3ʹ, Reverse: 5ʹ-GGATGCAGGGATGATGTTC-3ʹ. PCR reaction mix is 20 μL in total. PCR reaction system contains SYBR® Premix Ex TaqTMII 10 ul (TaKaRa, Japan), primers (0.8 μL each) and cDNA template (2 μL). PCR cycling conditions were as follows: initial denaturation at 95°C for 30 s, followed by 40 cycles (95°C for 15 s, 55°C for 30 s, 72°C for 30 s). Using 2-ΔΔCT for data statistics.
Transcription Factors (TFs) Binding Sites Prediction
JASPAR (http://jaspar.genereg.net/) and PROMO (http://alggen.lsi.upc.es) databases were used to analyze protein and DNA binding sites. Promoter sequence of RNF180 was import into JASPAR and PROMO datasets. The intersection of two datasets is the predicted TFs.
Statistical Analysis
Mann–Whitney U-test was used to compare the difference of methylation between GC and control. Student’s T test, two-tailed, was used to compare the difference of expression between GC and control. Spearman correlation test was used to analyze the correlation between methylation and expression. Survival analysis was performed using Kaplan-Meier. Multi-factor regression survival analysis was performed using COX regression. Log-rank test was used to determine significance. Hazard ratios (HR) and 95% CI were generated at the same time. All analyses were performed using SPSS software (version 19.0 Chicago III) and R software (version 3.3.1 2016 R). Survival package and survminer package were used to perform the cox regression and plot the survival curve. P < 0.05 was considered statistically significant.
Results
Methylation Sequencing Results of RNF180
A 318 bp fragment was amplified covering −224 to +94 in RNF180 promoter region. Two samples, which were GC tissue and adjacent normal tissue, are listed in Figure 1A and B. Four pairs of methylation rate analysis results are shown in Figure 1C (N1 to N4, T1 to T4).
 |
Figure 1 (A) A case of gastric cancer hypermethylation sequencing results. (B) A case of normal hypomethylation sequencing results. (C) Methylation detection results of RNF180 promoter region in GC and normal. TSS: transcription initiation site. For the reason of readable area, we amplified 318bp from −224 to +94 but only shown readable sequencing with CpG sites from −207 to 94. |
Methylation Level of RNF180 Promoter Region Significantly Increased in GC
In 80 pairs of GC and adjacent normal tissues, we analyzed the differences in the methylation rate of RNF180. The average methylation rate (AMR) of RNF180 in GC tissues was significantly higher than that in adjacent non-cancerous tissues, with AMRmedian-GC=0.30(0.22,0.48) and AMRmedian-CON=0.21(0.07, 0.33)(P <0.001). Eight high frequent-hypermethylated CpG sites as follows, which were identified in our previous study: M3(−165)(Chr5:64,165,942), M5(−148)(Chr5:64,165,959), M7(−133)(Chr5:64,165,974), M8(−130)(Chr5:64,165,977), M20(−57)(Chr5:64,166,050), M25(−34)(Chr5:64,166,073), M27(−26)(Chr5:64,166,081), M30(+5)(Chr5:64,166,112). (Table 1).
 |
Table 1 Methylation Rate Difference Between GC and Control |
The Expression Level of RNF180 mRNA Decreased in GC
In 80 pairs of GC and adjacent normal tissues, we analyzed the differences of RNF180 mRNA expression between GC and adjacent normal tissues. The expression of RNF180 was significanly downregulated in GC tissues compared to adjacent non-cancerous tissues. (P=3.35E-4, Figure 2).
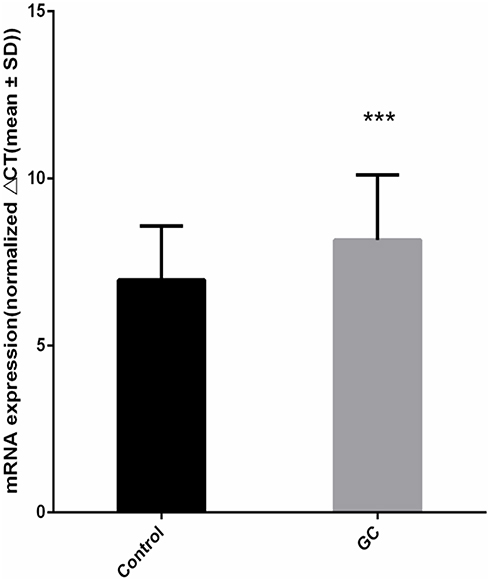 |
Figure 2 RNF180 mRNA expression difference (Nnormal=80 Ncancer=80 P value=3.35E-04 ***p < 0.001 Mann–Whitney U-test by using Normalized 2−ΔΔCt). |
The Methylation of RNF180 Was Negatively Correlated with Gene Expression
In order to investigate the effect of aberrant methylation on RNF180 expression, we analyzed the correlation between methylation rate and mRNA expression. There was negative correlation between AMR and mRNA expression in GC tissues (rAMR-GC = −0.238, PAMR-GC = 0.033), but not in adjacent non-cancerous tissues. Further we found 4 CpG sites related to gene expression, including M3(−165)(Chr5:64165942), M5(−148)(Chr5:64165959), M7(−133)(Chr5:64165974), M8(−130)(Chr5:64165977), the trends of which were consistent with AMR. Curiously, for M27(−26)(Chr5:64166081), there was large negative correlation coefficient between methylation and expression in GC (r=−0.941), but with no statistical significance (P=0.084) (Table 2). Considering that statistical differences may be affected by total sample size, we still consider recruiting M27(−26)(Chr5:64166081) in our further survival analysis.
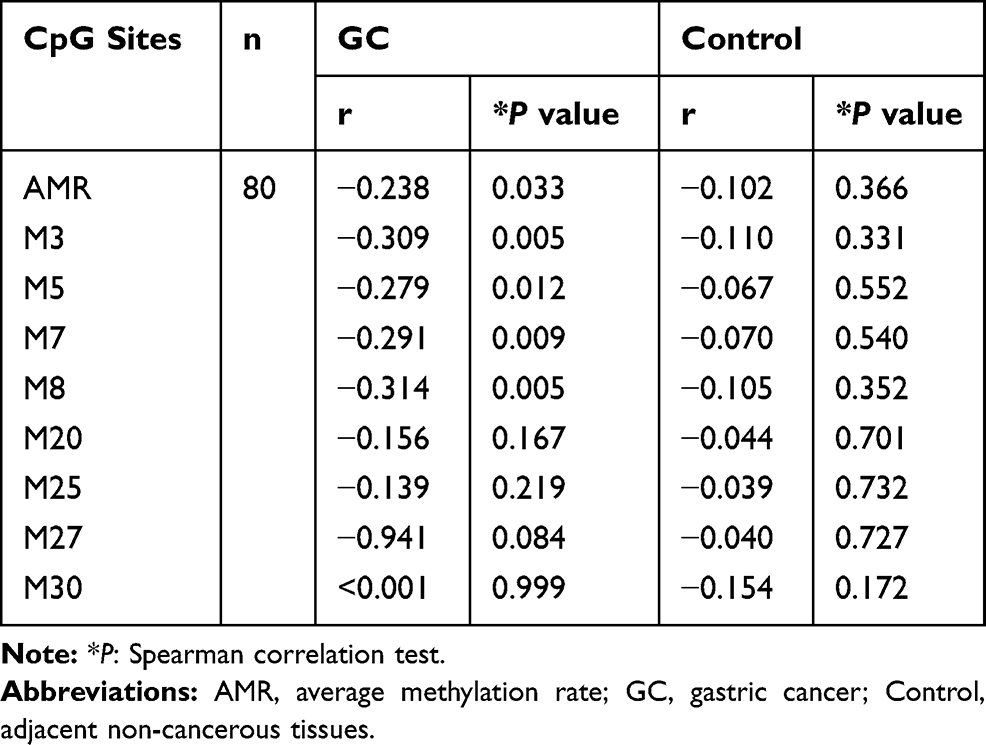 |
Table 2 Correlation Between RNF180 Expression and Methylation Rate |
No Significant Correlation Between RNF180 Methylation and Clinical Information
We analyzed the correlation between RNF180 methylation and clinical information in 164 GC cases, including gender, age, smoking, lymph node metastasis, histologic differentiation, vascular cancer embolus, lauren’s classification, growth pattern, invasioin depth, metastasis and TNM stages. For AMR and M27(−26)(Chr5:64166081), the methylation rate was different in TNM staging groups, but it was not statistically significant(P=0.06,P=0.054). No significant relationships were found with other sites or clinical parameters. (Table 3).
Methylation Rate Greater Than 0.3 Could Predict the Prognosis of GC
We used R packages to analyze the best cut-off value of the methylation rate for GC survival. The results showed that methylation rate >0.3 could be a best prognosis indicator in M5(−148)(Chr5:64,165,959), M27(−26)(Chr5:64,166,081) (PM5(−148) = 0.012;PM27(−26) = 0.002). In other words, a methylation rate greater than 0.3 suggested a poor prognosis (supplementary Table 1). Although a methylation rate >0.8 demonstrated poor prognosis in M3(−165)(Chr5:64,165,942), M7(−133)(Chr5:64,165,974), M8(−130)(Chr5:64,165,977), the positive cases were few. The practical significance was not obvious. We did not include these sites in the subsequent analysis.
Methylation Rate Greater Than 0.3 Could Be an Independent Factor to Predict the Prognosis of Gastric Cancer
In order to evaluate the efficacy of RNF180 methylation in predicting prognosis of GC, we further analyzed the impact of methylation rate on survival for AMR, M3(−165)(Chr5:64,165,942), M5(−148)(Chr5:64,165,959), M7(−133)(Chr5:64,165,974), M8(−130)(Chr5:64,165,977), M27(−26)(Chr5:64,166,081). The following clinical factors were used in univariate survival analysis and multivariate COX regression sequentially, including smoke, lymph node metastasis, histological differentiation, growth pattern, vascular cancer embolus, invasion depth, TNM stage and metastasis were included. The results showed methylation rate of M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081) >0.3 could be independent risk factors for poor prognosis (PM5(−148) = 0.008, PM27(−26) = 0.003, HRM5(−148) = 2.000(1.201,3.332), HRM27(−26)=2.389(1.336,4.271)).(Table 4, Figure 3).
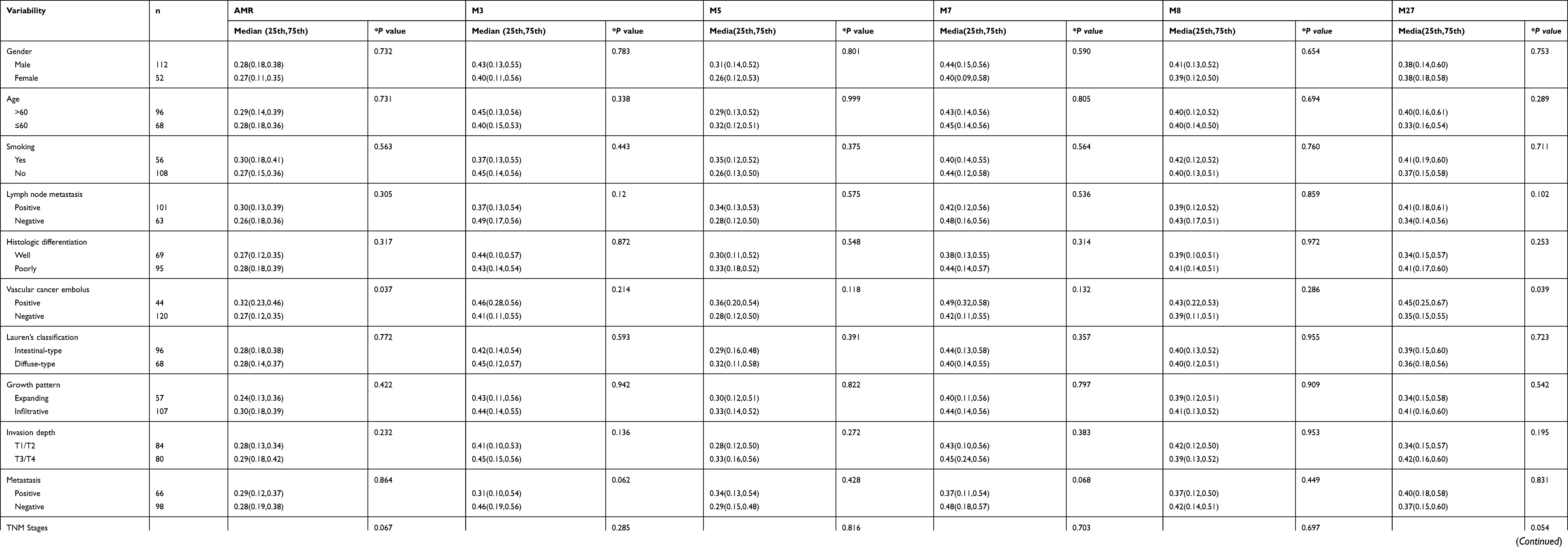 |
Table 3 Association Between Methylation Rate of RNF180 and Clinicopathological Parameters in GC |
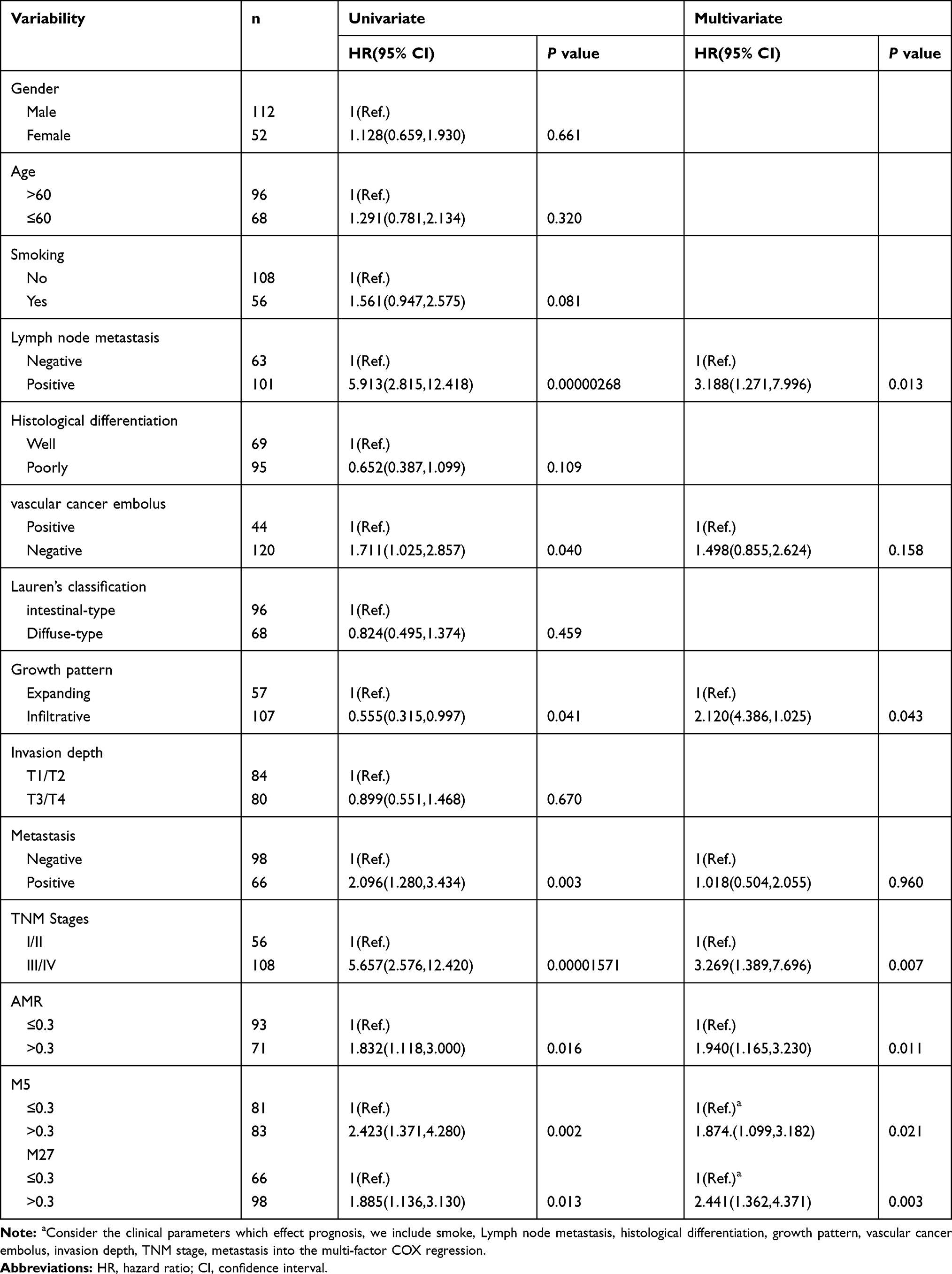 |
Table 4 Survival Analysis Combined with Clinical Parameter and Methylation |
 |
Figure 3 Methylation rate (MR) of specific sites show differential clinical outcomes. A total of 164 cases recruit in our study. (A) Cox Regression of AMR >0.3 shows poor prognosis (P = 0.011). (B) Cox Regression of M5(−148)MR >0.3 shows poor prognosis (P = 0.021). (C) Cox Regression of M27(−26)MR > 0.3 shows poor prognosis (P = 0.003). |
Transcription Factors Binding Sites Prediction
We used JASPAR and PROMO datasets to predict the binding sites with transcript factors. M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081) bind to many transcription factors (TFs), such as FOXA1, FOXC1, FOXC2, FOXF2, FOXP1, EGR1, EGR2 and EGR3, etc. (supplementary Table 2).
Discussion
It is generally believed that the malignant behavior of molecular biology precedes the appearance of the pathological phenotype of malignant tumors.13 DNA methylation plays an important role in tumorigenesis.14 They believe that methylation has broad application prospects as a biomarker. As decades passed, many researchers have made on the feasibility of DNA methylation as a molecular biomarker.15 It has been clarified that promoter methylation of the tumor suppressor gene is often negatively correlated with its expression.16 Demethylation treatment can significantly inhibit the malignant biological behavior of tumors.17 The methylation degree of specific gene region is directly related to lymph node metastasis, TNM stage, tumor pathological classification and prognosis.18 These findings open the door to molecular diagnosis for cancers. We can detect specific methylation sites as molecular biological indicators to predict tumor prognosis.19,20
RNF180 plays an important role, as a tumor suppressor gene involved in various cellular biological processes, such as DNA repair, gene transcription, apoptosis, etc.5 In our previous study, we found that the methylation rate of RNF180 promoter gradually increased with the severity of gastric mucosal lesions from gastritis, atrophy gastritis to GC. Moreover, we located eight CpG sites in RNF180 promoter with high methylation distribution frequency. However, it is not clear whether specific methylated sites affect the expression of RNF180 in these sites, resulting in a poor prognosis. In this study, we explicitly locate the high-specific methylation sites in the RNF180 promoter region and clarify that the M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081) locus methylation rate can independently determine the prognosis.
We firstly examined the correlation between methylation and mRNA level with GC and adjacent tissue samples. The results showed that compared with the adjacent tissues, the AMR of RNF180 in GC tissue was significantly increased. Moreover, there was a negative correlation between mRNA expression and AMR. In addition, there were five CpG sites: M3(−165)(Chr5:64,165,942), M5(−148)(Chr5:64,165,959), M7(−133)(Chr5:64,165,974), M8(−130)(Chr5:64,165,977), M27(−26)(Chr5:64,166,081), which were negatively correlated with mRNA expression. As an important tumor suppressor gene, RNF180 is an essential RING protein with the function of E3 ubiquitin ligase, the most important protein complex in UPS.8,21 The down-expression of RNF180 would lead to dysfunction of E3 and disorder of UPS.9 It is well known that abnormal UPS can terminate the normal protein degradation process, cause the disorder of tumor metabolism, and promote multiple malignant biological behaviors of tumor cells eventually.7 Therefore, we supposed that hypermethylation in RNF180 promoter region promoted the occurrence and development of GC by down-regulating gene expression level.
Methylation can affect transcription through multiple mechanisms.22 It is generally known that promoter methylation alters the structure of chromatin,23 and has a negative effect in a variety of diseases such as metabolic abnormalities, Alzheimer,24 aging and tumor.25 Gene expression depends on the binding of TFs (TFs) to promoter-specific regions.26,27 CpG sites methylation in these regions interfere with gene expression and affect the binding of TFs to these regions.28 Hypermethylation of promoter of the tumor suppressor gene leads to the down-regulation of gene expression,29 which further leads to tumor development and progression.30 For the above reasons, hypermethylation of tumor suppressor genes can lead to poor prognosis for tumor patients.31
In order to analyze the methylation and prognosis, we analyzed the relationship between the methylation rate of the above five sites and the prognosis of GC. The results showed that the methylation rate of M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081) could indicate poor prognosis by setting a cut-off value of 0.3. Furtherly, considering the effect of clinical-pathological parameters, we comprehensively analyzed the clinical data of GC, including gender, age, smoking, lymph node metastasis, histological differentiation, vascular cancer embolus, Lauren’s classification, growth pattern, invasion depth, distant metastasis and TNM stage. After adjusting for clinical factors, the methylation rates of M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081), with a cut-off value of 0.3, were still independent prognostic factors of GC. Many researchers have found that specific methylation sites can be biomarkers for various tumors.32 STEP1 promoter methylation rate could be a sensitive and specific biomarker for colorectal cancer.33,34 Another research has implied that −1415 CpG site in the promoter of RUNX3 contributes to poor prognosis. This specific site could be a prognostic biomarker in GC.18 In this study, we located two sites as prognostic factors of GC, which may also be binding sites of various transcript factors.
Studies have reported that TFs can bind to specific sequences of DNA, especially some important functional CpG sites.35,36 Abnormal methylation sites can affect TFs binding and lead to abnormal gene function.37 In this study, using JASPAR and PROMO database, we found some TFs, such as FOXA1, FOXC1, FOXC2, FOXF2, FOXP1, EGR1, EGR2 and EGR3 may bind to the methylation sites above. Therefore, we speculated that abnormal methylation status of these CpGs might alter the structure of the transcription factor binding region, and thereby change the expression level of RNF180. This speculation coincides with the molecular mechanism by which methylation affects transcription. Future researches are needed to elucidate the effect of methylation of RNF180-specific CpG sites on gene function and relevant mechanism.
In conclusion, for the first time, we located the functional two CpG sites in the RNF180 promoter region by combining the relationship between methylation rate and expression. It is further clarified that the methylation rate of M5(−148)(Chr5:64,165,959) and M27(−26)(Chr5:64,166,081) site can be independent biomarkers for prognosis of GC, with the optimal methylation rate threshold at 0.3 as a cut-off value. If the conclusions of this study can be verified in a large population, the molecular biology test kit can be developed targeting these two sites in clinic. In terms of precision medicine for GC, it will be helpful to guide clinical treatment and more advanced diagnosis through accurate detection and analysis.
Acknowledgments
This work was supported by the National Key R&D Program, Grant 2016YFC1303202, and the Key Laboratory of Cancer Intervention in Liaoning Province, Grant LS201605.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Deng G, Chen A, Hong J, et al. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59(9):2029–2033.
2. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi:10.1016/j.cell.2007.01.029
3. Liu M, Thomas SL, DeWitt AK, et al. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 2018. doi:10.1158/0008-5472.CAN-17-3953
4. Wang C, Liu Y, Guo W, et al. MAPT promoter CpG island hypermethylation is associated with poor prognosis in patients with stage II colorectal cancer. Cancer Manag Res. 2019;11:7337–7343. doi:10.2147/CMAR.S206731
5. Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;102(5):549–552. doi:10.1016/S0092-8674(00)00077-5
6. Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11(1):39–46. doi:10.1016/S0959-440X(00)00167-6
7. Shearer RF, Iconomou M, Watts CK, et al. Functional roles of the E3 ubiquitin ligase UBR5 in cancer. Mol Cancer Res. 2015;13(12):1523–1532. doi:10.1158/1541-7786.MCR-15-0383
8. Ogawa M, Mizugishi K, Ishiguro A, et al. Rines/RNF180, a novel RING finger gene-encoded product, is a membrane-bound ubiquitin ligase. Genes Cells. 2008;13(4):397–409. doi:10.1111/j.1365-2443.2008.01169.x
9. Cheung KF, Lam CN, Wu K, et al. Characterization of the gene structure, functional significance, and clinical application of RNF180, a novel gene in gastric cancer. Cancer. 2012;118(4):947–959. doi:10.1002/cncr.26189
10. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
11. Han F, Sun LP, Liu S, et al. Promoter methylation of RNF180 is associated with H.pylori infection and serves as a marker for gastric cancer and atrophic gastritis. Oncotarget. 2016;7(17):24800–24809. doi:10.18632/oncotarget.8523
12. Xie XM, Deng JY, Hou YC, et al. Evaluating the clinical feasibility: the direct bisulfite genomic sequencing for examination of methylated status of E3 ubiquitin ligase RNF180 DNA promoter to predict the survival of gastric cancer. Cancer Biomark. 2015;15(3):259–265. doi:10.3233/CBM-150466
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
14. Klutstein M, Nejman D, Greenfield R, et al. DNA methylation in cancer and aging. Cancer Res. 2016;76(12):3446–3450. doi:10.1158/0008-5472.CAN-15-3278
15. Tahara T, Arisawa T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics. 2015;7(3):475–486. doi:10.2217/epi.15.4
16. Rose M, Bringezu S, Godfrey L, et al. ITIH5 and ECRG4 DNA methylation biomarker test (ei-BLA) for urine-based non-invasive detection of bladder cancer. Int J Mol Sci. 2020;21:3. doi:10.3390/ijms21031117
17. Hong SY, Kao YR, Lee TC, et al. Upregulation of E3 ubiquitin ligase CBLC enhances EGFR dysregulation and signaling in lung adenocarcinoma. Cancer Res. 2018;78(17):4984–4996. doi:10.1158/0008-5472.CAN-17-3858
18. Wang N, Sui F, Ma J, et al. Site-specific hypermethylation of RUNX3 predicts poor prognosis in gastric cancer. Arch Med Res. 2016;47(4):285–292. doi:10.1016/j.arcmed.2016.07.011
19. Gomez-Zamudio JH, Mendoza-Zubieta V, Ferreira-Hermosillo A, et al. High thyroid-stimulating hormone levels increase proinflammatory and cardiovascular markers in patients with extreme obesity. Arch Med Res. 2016;47(6):476–482. doi:10.1016/j.arcmed.2016.10.007
20. Avincsal MO, Jimbo N, Fujikura K, et al. Epigenetic down-regulation of SOX2 is an independent poor prognostic factor for hypopharyngeal cancers. Histopathology. 2018;72(5):826–837. doi:10.1111/his.13436
21. Cetin Z, Yakut S, Clark OA, et al. A 5q12.1-5q12.3 microdeletion in a case with a balanced exceptional complex chromosomal rearrangement. Gene. 2013;516(1):176–180. doi:10.1016/j.gene.2012.12.013
22. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi:10.1038/npp.2012.112
23. Shokouhian M, Bagheri M, Poopak B, et al. Altering chromatin methylation patterns and the transcriptional network involved in regulation of hematopoietic stem cell fate. J Cell Physiol. 2020. doi:10.1002/jcp.29642
24. Qazi TJ, Quan Z, Mir A, et al. Epigenetics in Alzheimer’s disease: perspective of DNA methylation. Mol Neurobiol. 2018;55(2):1026–1044. doi:10.1007/s12035-016-0357-6
25. Morgan AE, Davies TJ, Mc Auley MT. The role of DNA methylation in ageing and cancer. Proc Nutr Soc. 2018;77(4):412–422. doi:10.1017/S0029665118000150
26. Bartlett A, O’Malley RC, Huang SC, et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc. 2017;12(8):1659–1672. doi:10.1038/nprot.2017.055
27. Deplancke B, Alpern D, Gardeux V. The genetics of transcription factor DNA binding variation. Cell. 2016;166(3):538–554. doi:10.1016/j.cell.2016.07.012
28. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17(9):551–565. doi:10.1038/nrg.2016.83
29. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–5440. doi:10.1038/sj.onc.1205600
30. Wang LH, Wu CF, Rajasekaran N, et al. Loss of tumor suppressor gene function in human cancer: an overview. Cell Physiol Biochem. 2018;51(6):2647–2693. doi:10.1159/000495956
31. Ruan J, Xu P, Fan W, et al. Quantitative assessment of aberrant P16(INK4a) methylation in ovarian cancer: a meta-analysis based on literature and TCGA datasets. Cancer Manag Res. 2018;10:3033–3046. doi:10.2147/CMAR.S170818
32. Ding W, Chen G, Shi T. Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis. Epigenetics. 2019;14(1):67–80. doi:10.1080/15592294.2019.1568178
33. Warren JD, Xiong W, Bunker AM, et al. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011;9(1):133. doi:10.1186/1741-7015-9-133
34. Gao C, Zhuang J, Zhou C, et al. Developing DNA methylation-based prognostic biomarkers of acute myeloid leukemia. J Cell Biochem. 2018;119(12):10041–10050. doi:10.1002/jcb.27336
35. Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288(48):34287–34294. doi:10.1074/jbc.R113.512517
36. Domcke S, Bardet AF, Adrian Ginno P, et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528(7583):575–579. doi:10.1038/nature16462
37. Zhang X, Yang R, Jia Y, et al. Hypermethylation of Sp1 binding site suppresses hypothalamic POMC in neonates and may contribute to metabolic disorders in adults: impact of maternal dietary CLAs. Diabetes. 2014;63(5):1475–1487. doi:10.2337/db13-1221
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.