Back to Journals » Drug Design, Development and Therapy » Volume 18
Identification of Coumarin-Chalcone and Coumarin-Pyrazoline Derivatives as Novel Anti-Toxoplasma gondii Agents
Authors Ebaid MS ![]() , Chyb M, Furlan V, Ibrahim HAA
, Chyb M, Furlan V, Ibrahim HAA ![]() , Bren U, Gatkowska J, Dziadek J
, Bren U, Gatkowska J, Dziadek J ![]() , Eldehna WM
, Eldehna WM ![]() , Sabt A
, Sabt A ![]()
Received 4 October 2024
Accepted for publication 14 November 2024
Published 4 December 2024 Volume 2024:18 Pages 5599—5614
DOI https://doi.org/10.2147/DDDT.S495089
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Manal S Ebaid,1,2 Maciej Chyb,3,4 Veronika Furlan,5 Hoda Atef Abdelsattar Ibrahim,6 Urban Bren,5,7,8 Justyna Gatkowska,3 Jarosław Dziadek,9 Wagdy M Eldehna,10,11 Ahmed Sabt2
1Department of Chemistry, College of Science, Northern Border University, Arar, Saudi Arabia; 2Chemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Center, Dokki, Cairo, Egypt; 3Department of Molecular Microbiology, Faculty of Biology and Environmental Protection, University of Lodz, Łódź, Poland; 4Bio-Med-Chem Doctoral School of the University of Lodz and Lodz Institutes of the Polish Academy of Sciences, Łódź, Poland; 5Faculty of Chemistry and Chemical Engineering, University of Maribor, Maribor, Slovenia; 6Pediatric Department, Faculty of Medicine, Cairo University, Cairo, Egypt; 7Faculty of Mathematics, Natural Sciences and Information Technologies, University of Primorska, Glagoljaška 8, Slovenia; 8Institute of Environmental Protection and Sensors, Beloruska Ulica 7, Maribor, Slovenia; 9Laboratory of Genetics and Physiology of Mycobacterium, Institute of Medical Biology of the Polish Academy of Sciences, Łódź, Poland; 10Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt; 11Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Pharos University in Alexandria, Alexandria, Egypt
Correspondence: Ahmed Sabt; Wagdy M Eldehna, Email [email protected]; [email protected]
Introduction: Toxoplasmosis, a zoonotic infection caused by the apicomplexan parasite Toxoplasma gondii, affects a significant portion of the global human population. This condition, particularly dangerous for pregnant women and immunocompromised individuals, currently lacks effective treatment options.
Methods: Eighteen coumarin-based derivatives were synthesized, comprising coumarin-chalcone hybrids (5a-i) and coumarin-pyrazoline hybrids (6a-i). Cytotoxicity was evaluated using L929 mouse fibroblasts and Hs27 human fibroblasts. Anti-T. gondii activity was assessed, and molecular docking studies were performed to predict binding modes with TgCDPK1.
Results: Pyrazoline hybrids (6a-i) showed lower toxicity than chalcone-bearing coumarins (5a-i), with CC30 values exceeding the highest tested concentration (500 μg/mL) for most compounds. The synthesized molecules demonstrated strong anti-T. gondii activity, with IC50 values ranging from 0.66 μg/mL to 9.05 μg/mL. Molecular docking studies provided insights into potential binding mechanisms.
Conclusion: This study highlights the potential of coumarin-based hybrids as anti-T. gondii agents. The findings should contribute to the growing arsenal of small molecules against T. gondii and underscore the value of molecular hybridization in drug design. Further studies to elucidate these compounds’ mechanism of action and in vivo efficacy are warranted to fully realize their potential as anti-parasitic agents.
Keywords: Coumarin sulfonamides, Synthesis, N-acetylpyrazoline, TgCDPK1 enzyme, molecular docking
Introduction
Toxoplasmosis is a parasitosis caused by the intracellular protozoan parasite Toxoplasma gondii (T. gondii), a member of the apicomplexan phylum.1 While the only known T. gondii definitive hosts are felids, the parasite can also infect various warm-blooded animals, such as birds, rodents, and humans, which serve as intermediate hosts.2 This parasite is considered highly successful as it can cross the blood–brain barrier and establish a persistent infection in a drug-resistant form known as bradyzoite.3 Toxoplasmosis can be acquired through contaminated water or food, organ transplants, or vertical transmission.4 It is estimated to infect 2 billion individuals globally, with South America having the highest infection rates, likely due to regional lifestyles and dietary habits.5,6 In mammals, ingestion of this parasite can lead to higher rates of abortion and stillbirth.7
There are three primary infectious stages of T. gondii: tachyzoites, tissue cysts containing bradyzoites, and mature oocysts with sporozoites.8 Although Toxoplasma is a single species, it has various clonal lineages that vary in their ability to cause disease.9 Among these, type I (RH strain in this study) is the most virulent and is fatal at any dose in all strains of mice during the acute phase of the illness.10 During this stage of infection, the fast-growing tachyzoites invade host cells by attaching their apical complex and can transform into bradyzoites to form tissue cysts in the chronic stage.11,12 Bradyzoites are released when these cysts rupture, particularly in individuals with weakened immune systems, leading to the reactivation of diseases such as tachyzoites. The progression and prolonged duration of the infection rely on the tachyzoite-bradyzoite conversion process.13,14 In acute toxoplasmosis, the rapid multiplication of tachyzoites causes necrotic changes and cell destruction, resulting in conditions like retinochoroiditis and meningoencephalitis in immunocompromised patients.15 Moreover, T. gondii is commonly associated with miscarriages and congenital infections.16
Current drugs used to treat toxoplasmosis include medications from the sulfonamides group, such as sulfadiazine I and sulfamethoxazole II,17 Figure 1. However, these drugs, when combined with others, can lead to adverse effects like thrombocytopenia, liver and kidney problems, and bone marrow toxicity.18,19 Due to the drawbacks of current treatments for toxoplasmosis, there is a critical need for the development of a medication that is more effective, less toxic, and has fewer side effects.
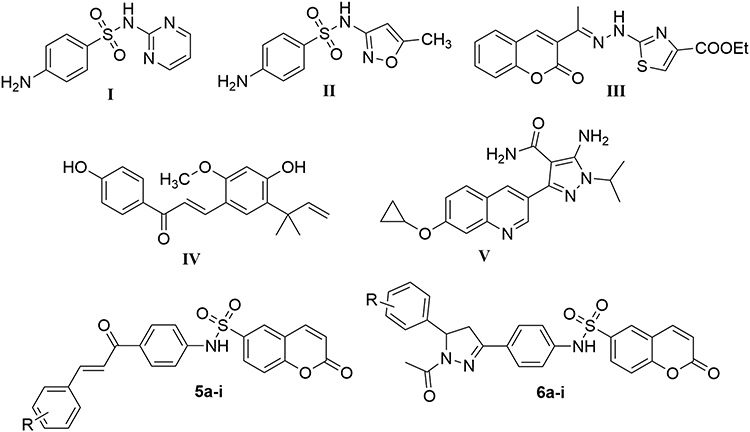 |
Figure 1 Structures of representative hybrids of bioactive cores I–V and our target coumarins 5a-i and 6a-i. |
Natural products, with their diverse molecular compositions and high bioavailability, play a crucial role in medicinal chemistry for the development of novel chemical structures.20 Extensive research has demonstrated that numerous natural products, particularly coumarins and chalcones, exhibit a broad spectrum of biological activities. These activities include anticancer, antioxidant, antimicrobial, antiviral, and antiparasitic properties.21–28
Recent investigations have elucidated the remarkable efficacy of both chalcone compounds and coumarin derivatives against Toxoplasma gondii.29,30 Of particular note is Compound III (Figure 1), which incorporates a coumarin scaffold, demonstrated good activity against T. gondii with an IC50 of 55 μM.31 Similarly, Licochalcone A (Compound IV, Figure 1), featuring a chalcone moiety in its molecular architecture, exhibited significant anti-T. gondii properties.32,33 Furthermore, nitrogen-containing heterocycles, exemplified by pyrazoline structures, have emerged as promising candidates in the fight against T. gondii.34–36 A striking example is Compound V (Figure 1), which boasts an exceptionally low EC50 of 0.42 μM against T. gondii, indicative of its formidable antiparasitic potential.37 Such findings underscore the diverse chemical scaffolds that can be utilized for the development of novel therapeutic molecules against T. gondii. The coumarin, chalcone, and pyrazoline moieties serve as auspicious starting points and building points for medicinal chemists in their quest to discover more potent and selective anti-Toxoplasma compounds. These advancements could potentially revolutionize the treatment landscape for toxoplasmosis, a disease of significant global health concern.
The concept of hybrid molecules represents a sophisticated approach in medicinal chemistry, wherein two or more pharmacologically active moieties are ingeniously integrated into a single molecular entity. This strategic amalgamation is designed to engender bioactive compounds that exhibit synergistic effects, thereby potentially surpassing the therapeutic efficacy of their individual constituents. This innovative methodology’s primary objectives are to augment overall efficacy, mitigate the development of drug resistance, and attenuate toxicity profiles compared to the progenitor compounds. Motivated by the aforementioned data, we have explored a diverse range of derivatives containing coumarin as the main scaffold as potential promising molecules against Toxoplasma gondii. Thereafter, this coumarin core has been judiciously conjugated with either a chalcone moiety or a pyrazoline ring. The conjugation process was facilitated by the incorporation of a sulfonamide group, which served as a molecular linker. This meticulous design strategy culminated in the synthesis of two distinct sets of small molecules: the coumarin-chalcone hybrids 5a-i and the coumarin-pyrazoline hybrids 6a-i (Figure 1).
The cytotoxicity of both compound sets (5a-i and 6a-i) was evaluated using L929 mouse fibroblasts and Hs27 human fibroblasts to assess their safety profiles, providing a comprehensive view of potential toxicity across species. Following this assessment, the compounds were investigated for their efficacy against Toxoplasma gondii. Additionally, a molecular docking study was conducted to predict the binding mode of the most promising candidate within the active site of a potential target enzyme, offering insights into the structural basis of the compound’s activity and guiding future modifications to enhance binding affinity and selectivity. This integrated approach ensures identifying and optimizing novel therapeutic molecules against Toxoplasma gondii while maintaining an acceptable safety profile.
Results and Discussion
Chemistry
The synthesis of chalcone-bearing coumarins 5a-i and N-acetyl pyrazoline-bearing coumarins 6a-i is outlined in Schemes 1 and 2. The key 4-amino-chalcone intermediates 3a-i were prepared via Claisen-Schmidt condensation between 4-aminoacetophenone 1 and various aryl aldehydes 2a-i, following a previously reported procedure.38 The target chalcone-bearing coumarins 5a-i were then obtained by reacting coumarin-6-sulfonyl chloride 4 with the 4-amino-chalcone derivatives 3a-i in methylene chloride and pyridine as an acid binder at room temperature (Scheme 1).
 |
Scheme 1 Synthesis of target chalcone-based molecules 5a-i. |
The 1H NMR and 13C NMR spectral data were used to determine the structure of derivatives 5a-i. The 1H NMR spectrum showed characteristic signals ranging from 7.60 to 8.08 ppm, corresponding to hydrogens of the olefinic double bond. The observed signals manifested as doublets, occurring in pairs, with coupling constants spanning from 15.6 to 16.0 hz, thereby suggesting a trans configuration. In the 13C NMR spectrum, the most deshielded signals, exhibiting chemical shifts in the 187 to 189 ppm range, were attributed to the carbonyl group of the trans-enone bridge.
Furthermore, the corresponding set of N-acetyl pyrazolines 6a-i was obtained by cyclo-addition reaction of chalcone derivatives 5a-i in glacial acetic acid with hydrazine hydrate (Scheme 2). The target compounds 6a-i was confirmed by using the spectral data of 1H-NMR and 13C-NMR. 1H-NMR analysis showed the disappearance of signals related to the olefinic double bond. Instead, three significant signals were observed. The first signal was a singlet ranging from 2.23 to 2.28 ppm, corresponding to CH3 (CO-CH3). The second signal consisted of two doublets of doublets ranging between 2.81–3.05 and 3.62–3.78 ppm, corresponding to CH2-pyrazoline. The third signal was a doublet of doublets ranging from 5.31 to 5.61 ppm, corresponding to CH pyrazoline. The 13C-NMR analysis showed three signals at approximately 20, 40, and 60 ppm related to CH3, CH2-pyrazoline, and CH-pyrazoline, respectively.
 |
Scheme 2 Synthesis of target N-acetyl pyrazolines-based molecules 6a-i. |
Biological Evaluations
Evaluation of the Potential Cytotoxicity
To assess the cytotoxicity of the newly synthesized chalcone-bearing coumarins 5a-i and N-acetyl pyrazoline-bearing coumarins 6a-i, a methyl thiazole tetrazolium (MTT) reduction method was used on both L929 mouse and Hs27 human fibroblasts. CC30 values, are presented in Table 1, along with the standard error of the mean (SEM) and the coefficient of determination R2 and plotted in Figure 2.
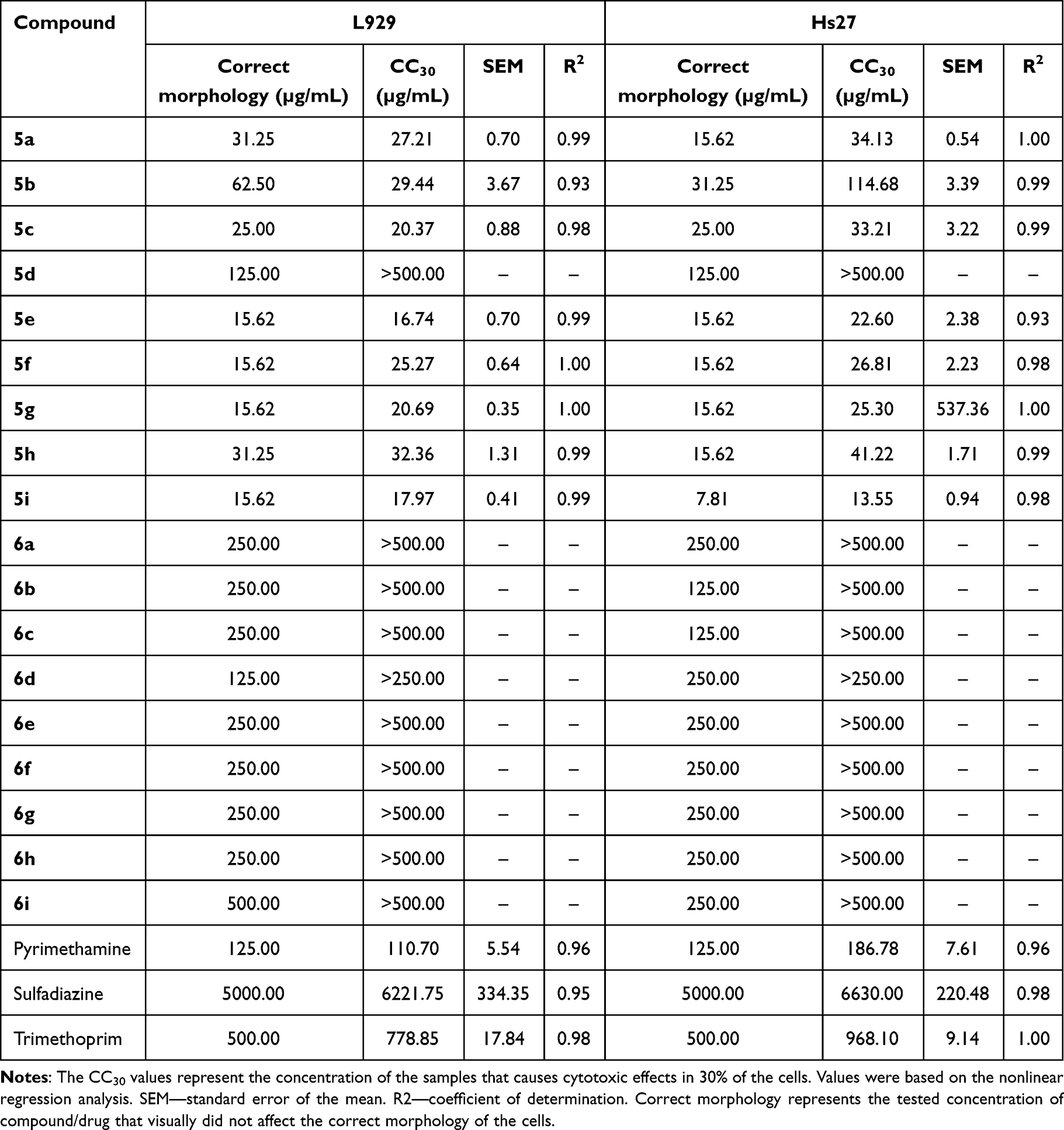 |
Table 1 Cytotoxicity Profile of Chalcone Derivatives (5a-i) and N-Acetyl Pyrazolines Derivatives (6a-i) Against Mouse L929 and Human Hs27 Fibroblasts |
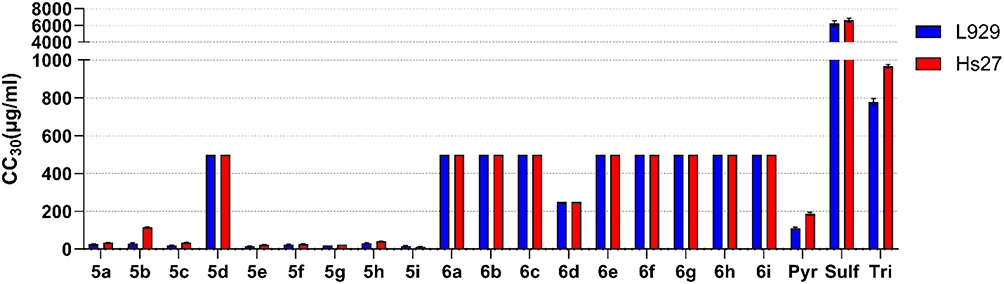 |
Figure 2 The cytotoxic effects of chalcone-bearing coumarins 5a-i and N-acetyl pyrazoline-bearing coumarins 6a-i against mouse L929 (blue) and human Hs27 (red) fibroblasts. The CC30 values represent the concentration of the samples to cause cytotoxic effects in 30% of the cells. CC30 and SEM values were based on the non-linear regression analysis. Reference drugs: Pyr: pyrimethamine; Tri: trimethoprim; Sulf: sulfadiazine. |
Evaluation of cytotoxicity showed that N-acetyl pyrazolines (6a-i) are far less toxic than chalcone-bearing coumarins (5a-i), with CC30 higher than the highest tested concentration 500/250 µg/mL in the case of all derivatives. The solubility of the compounds limited the testing of higher concentrations. In the case of 5a-i compounds, the exception is compound 5d, whose CC30 values were also higher than 500 µg/mL. 5d compound contains 2.4-dimethoxy substituents, which seem to positively affect the cytotoxicity of compounds, resulting in even 40-fold higher CC30 values compared to other chalcone derivatives. Cytotoxicity did not differ between cell lines, with the exception of the 5b compound. However, Hs27 cells tend to be more sturdy, as in the case of pyrimethamine and trimethoprim CC30 values. Compound 5g had comparable CC30 values for both cell lines; the SEM of Hs27 CC30 value is quite high due to the strong non-linearity of the toxicity of this compound for this cell line, which consequently showed a high hillslope of 22.27. Due to these reasons, the SEM of Hs27 CC30 value of compound 5g was not plotted in Figure 2. Compounds 5a-b and 5d-h showed relatively high toxicity, with CC30 values ranging from 13.55 to 114.68 µg/mL in both cell lines, compared to the reference drugs of which pyrimethamine has the lowest value of 110.7 µg/mL (L929) and 186.78 µg/mL (Hs27). The tested compounds did not significantly affect the correct morphology of the cells, which recovered completely at a concentration of CC30 or two-fold lower, comparable to the reference antibiotics; thus, these compounds may be safe for use and further testing.
Inhibition of Toxoplasma gondii in vitro
To assess the anti-T. gondii activity of the target chalcone-bearing coumarins 5a-i and N-acetyl pyrazoline-bearing coumarins 6a-i, highly virulent RH T. gondii strain expressing green fluorescent protein was used. IC50 and the selectivity values index are presented in Table 2, along with the standard error of the mean (SEM) and the coefficient of determination R2, and plotted in Figure 3.
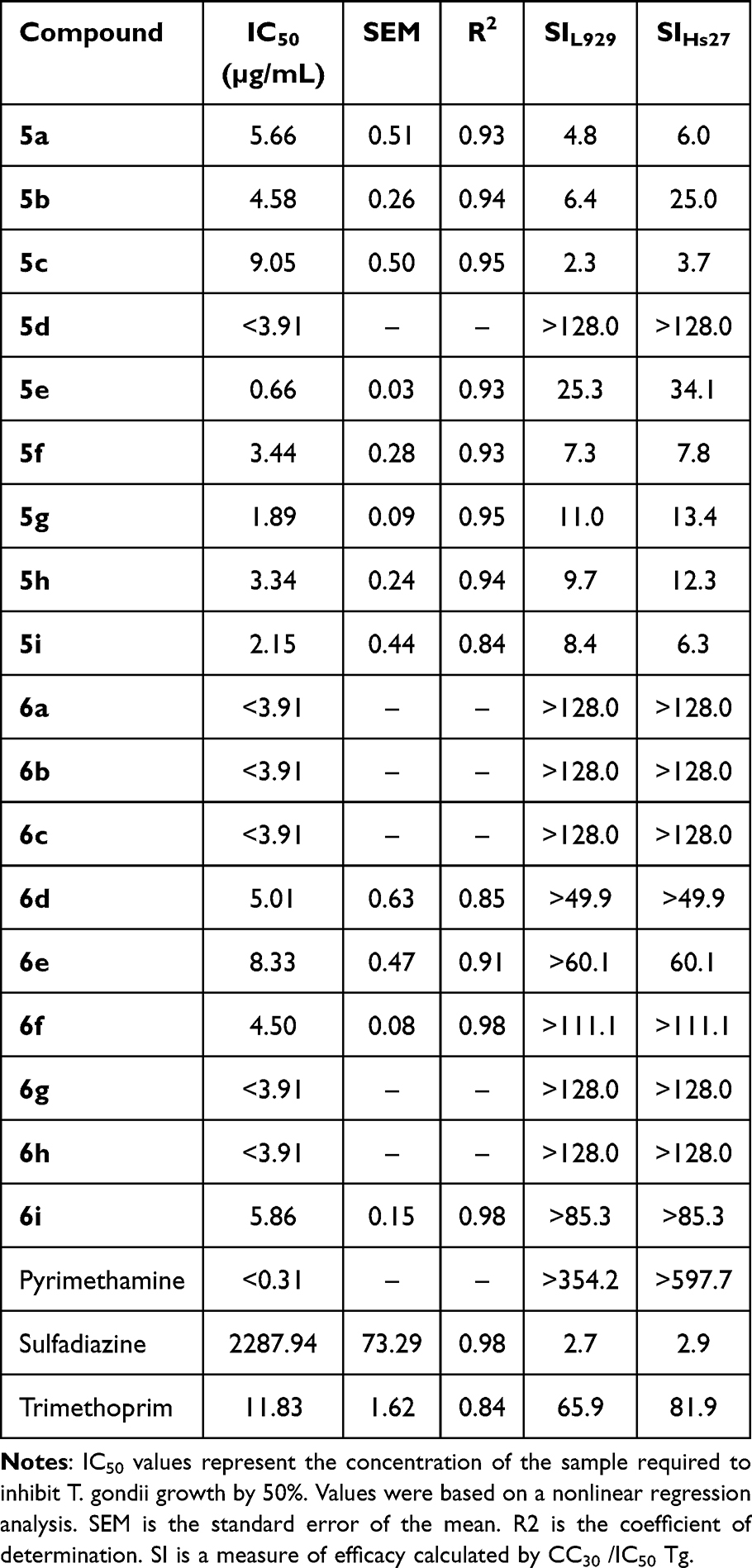 |
Table 2 Profile of Anti-T. Gondii Activity of the Target Chalcone-Bearing Coumarins 5a-I and N-Acetyl Pyrazoline-Bearing Coumarins 6a-I |
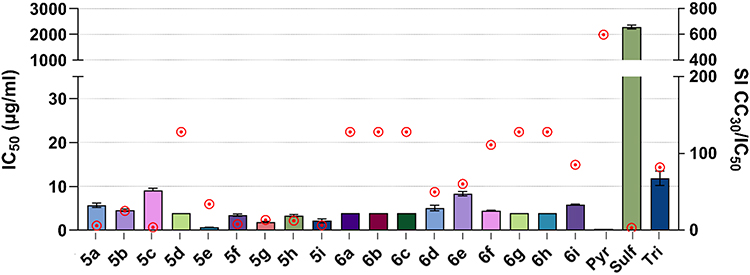 |
Figure 3 Anti-T. gondii activity of the coumarin-6-sulfonamide bearing chalcone derivatives (5a-i) and N-acetyl pyrazolines (6a-i). IC50 values with SEM are shown on the left y-axis, representing the concentration of the sample to cause inhibition of T. gondii growth by 50%. Values were based on the non-linear regression analysis. The right, y-axis (red dots above bars) represents the selectivity index SI, a measure of efficacy calculated by CC30 Hs27/IC50 Tg. Reference drugs: Pyr: pyrimethamine; Tri: trimethoprim; Sulf: sulfadiazine. |
All of the compounds tested showed strong anti-T. gondii activity, reaching IC50 values with good accuracy, comparable to trimethoprim 11.83 µg/mL, higher than pyrimethamine <0.31 µg/mL and significantly lower than sulfadiazine 2287.94 µg/mL. In this case, the set of N-acetyl pyrazoline-bearing coumarins 6a-i did not show better activity than derivatives 5a-i. The lowest IC50 value of 0.66 µg/mL was obtained for compound 5e (SIHs27 34.1) and the highest of 9.05 µg/mL for 5c (SIHs27 3.7). The selectivity index of the compounds tested was higher than 1 in each case and was in the ranges of 2.3–128 for line L929 and 3.7–128 for line Hs27 (Table 2). The lowest result was obtained for compound 5c, whose index was 2.3 and 3.7 for lines L929 and Hs27, respectively. Nevertheless, this is still close to the SI value of sulfadiazine (SIHs27 2.9; SIL929 2.7), the drug used for treatment. Coumarins 6a-c, 6f-i, and 5d, on the other hand, showed a particularly high selectivity index, above 128. This is a very promising result, suggesting a potentially safe and effective activity of these molecules due to the possible use of a large multiplication range of IC50 values, eg, 5 × IC50, which will still be safe and should enable eradication of the tachyzoites. These SI values are much higher than sulfadiazine and better than trimethoprim, while still much lower than pyrimethamine, providing opportunities for further improvements (Table 2).
Molecular Docking
Calcium-dependent protein kinase 1 (TgCDPK1) plays a crucial role in regulating various physiological functions of T. gondii, including adhesion, secretion, parasite motility, host-cell invasion, and egress.39 The crystal structure analysis of TgCDPK1 reveals a distinct ATP binding site that is more extensive compared to a homologous human kinase. This structural characteristic suggests that TgCDPK1 features a gatekeeper residue of small glycine, creating a favorable binding site for enzyme inhibitors to target a hydrophobic cavity adjacent to the ATP binding site. Consequently, TgCDPK1 emerges as a promising candidate for the development of inhibitors aimed at combating T. gondii infections.
To reveal the binding patterns of the seven most potent inhibitors, namely 6a, 6b, 6c, 6f, 6g, 6h, and 5d to TgCDPK1, molecular docking was performed using the CANDOCK algorithm with RMR6 scoring function.40 After visual inspection, the binding modes of seven studied inhibitors at the active site of TgCDPK1 with the lowest docking score values were selected for further analysis. As can be observed from Table 3, the docking score values of all seven inhibitors were below −53 arb. units, indicating their strong binding affinity to TgCDPK1. The halogenated inhibitors 6f, 6g, and 6h exhibited slightly lower docking score values, indicating a higher affinity for TgCDPK1, than the nonhalogenated inhibitors 6a, 6b, and 6c with the same molecular scaffold. From Table 3, it is also evident that compound 5d demonstrated the highest docking score value, indicating the lowest affinity to TgCDPK1 among the tested inhibitors, whereas compound 6d exhibited the lowest docking score value and, therefore, the highest affinity. From Table 3, it can also be observed that the differences between the docking score values are small, which confirms the inhibitory activity of all seven studied compounds against TgCDPK1 as well as corresponds to their similar experimental IC50 and CC30 values.
 |
Table 3 The Lowest Docking Score Values of the Seven Studied Inhibitors in Complex with TgCDPK1 |
To evaluate important intermolecular interactions between TgCDPK1 active site amino-acid residues and the seven studied inhibitors, the Protein–Ligand Interaction Profiler (PLIP) was utilized.41 The most important interactions of 6a, 6b, 6c, 6f, 6g, 6h, and 5d with amino-acid residues at the active site of TgCDPK1 are presented in Figure 4.
 |
Figure 4 Binding modes of a) 6a, b) 6b, c) 6c, d) 6f, e) 6g, f) 6h, and g) 5d at the active site of TgCDPK1. The carbon atoms of the studied inhibitors are presented in blue, while the carbon atoms of TgCDPK1 amino-acid residues are depicted in Orange. Oxygen atoms are red, nitrogen atoms are dark blue, and sulfur atoms are yellow. Hydrophobic interactions are presented with dark gray dashed lines, hydrogen bonds with dark blue lines, and salt bridges with yellow dashed lines. Hydrogen atoms were omitted for clarity reasons. |
From the binding poses, it can be observed that all seven inhibitors form non-bonded interactions with amino-acid residues Leu-57, Lys-59, Glu-135, and Asp-210. More specifically, all seven inhibitors 6a, 6b, 6c, 6f, 6g, 6h, and 5d form hydrophobic interactions with Leu-57 at distances 3.4, 4.0, 3.4, 3.5, 3.5, 3.5, 3.5, 3.6, 3.1, 3.4, and 3.6 Å, respectively. Inhibitors 6a, 6b, 6c, 6f, 6g, and 6h are additionally stabilized through a salt bridge interaction with Lys-59 at distances 5.0, 4.9, 4.1, 4.6, 5.1, and 4.0 Å, respectively, while compound 5d forms a hydrophobic interaction with Leu-57 at a distance of 3.4 Å and a salt bridge with Leu-185 at a distance 4.0 Å. Hydrophobic interactions between amino-acid residue Glu-135 and all seven inhibitors 6a, 6b, 6c, 6f, 6g, 6h, and 5d can also be observed at distances of 3.9, 3.7, 3.5, 3.8, 3.8, 3.6, and 3.8 Å, respectively. In addition, all inhibitors 6a, 6b, 6c, 6f, 6g, 6h, and 5d are stabilized by a hydrogen bond with amino-acid residue Asp-210 at distances 2.5, 2.2, 2.2, 2.0, 2.6, 1.9, and 3.2 Å, respectively. Inhibitors 6a, 6c, 6f, and 6h also form hydrogen bonds with residues Gln-341 (at distances of 4.0, 3.8, 4.0, and 3.8 Å, respectively) and Lys-338 (at distances of 3.5, 3.8, 3.9, and 4.1 Å, respectively), while hydrogen bond with Glu-178 is observed in the case of 6a, 6b, 6c and 6h (at distances of 3.8, 4.0, 4.1, and 3.9 Å, respectively).
Moreover, inhibitors 6a, 6b, 6c, 6f, 6g, and 6f are additionally stabilized by forming a hydrogen bond with Tyr-131 through their methyl carboxylate group at distances of 3.1, 2.7, 3.0, 3.4, 2.7, and 3.4 Å, respectively, which is not present in the case of 5d with a different molecular scaffold. Hydrophobic interactions with residue Leu-181 were also observed in the case of six inhibitors 6a, 6b, 6c, 6f, 6h, and 5d at distances of 3.4, 3.5, 3.5, 3.1, 3.6, and 3.5 Å, respectively, four inhibitors 6a, 6f, 6g, and 5d established hydrophobic interactions with residue Thr-132 at distances of 3.9, 3.7, 3.9, and 3.2 Å, respectively, while inhibitors 6g and 5d also formed additional hydrophobic interactions with Val-130 at distances of 3.9, and 4.0 Å, respectively.
Hydrophobic interactions and hydrogen bonds, therefore, play the most important role in the strong inhibitory activity of compounds 6a, 6b, 6c, 6f, 6g, 6h, and 5d, while salt bridges also significantly contribute to their inhibitory activity against TgCDPK1. Intermolecular interactions with similar amino-acid residues indicate similar binding modes for all seven investigated inhibitors at the hydrophobic pocket of the ATP-binding site of TgCDPK1, which also agrees with their similar experimental IC50 and CC30 values.
Conclusions
In conclusion, this study has unveiled a promising new class of anti-Toxoplasma gondii agents based on coumarin derivatives. We successfully synthesized and evaluated eighteen novel compounds, comprising coumarin-chalcone hybrids (5a-i) and coumarin-pyrazoline hybrids (6a-i), designed by conjugating the coumarin scaffold with either a chalcone moiety or a pyrazoline ring through a sulfonamide linker. The cytotoxicity assessment using L929 mouse fibroblasts and Hs27 human fibroblasts revealed a favorable safety profile for the pyrazoline-bearing compounds (6a-i), with CC30 values exceeding the highest tested concentration. Notably, compound 5d from the chalcone series also demonstrated low toxicity, with CC30 values above 500 µg/mL. Most importantly, these novel coumarin derivatives exhibited potent anti-T. gondii activity, with IC50 values ranging from 0.66 µg/mL to 9.05 µg/mL. This remarkable potency, coupled with the low toxicity profile of several molecules, particularly in the pyrazoline series, highlights the potential of these coumarin-based hybrids as promising leads for developing new anti-toxoplasmosis therapeutics. Molecular docking was performed for the seven most active compounds against the active site of TgCDPK1. The results indicated a favorable fit within the active site, characterized by the formation of non-covalent interactions with the amino acid residues Leu-57, Lys-59, Glu-135, and Asp-210. Notably, all seven inhibitors exhibited hydrophobic interactions with Leu-57 and were further stabilized by a salt bridge interaction with Lys-59. These findings should contribute to the growing arsenal of small molecules against T. gondii and underscore the value of molecular hybridization in drug design. Further studies to elucidate these compounds’ mechanism of action and in vivo efficacy are warranted to fully realize their potential as anti-parasitic agents.
Experimental Section
Chemistry
Melting points were determined using an Electrothermal IA 9000 apparatus and were uncorrected. Elemental analyses were performed at the Micro-Analytical Central Services Laboratory, Faculty of Science, Cairo University, Egypt. 1H-NMR and 13C-NMR spectra were measured using Bruker Avance II™ 400 MHz spectrometers (Bruker Biospin AG, Fällanden, Switzerland) in Prague, Czech Republic. The reactions were followed by TLC (silica gel, aluminium sheets 60 F254, Merck) using chloroform/methanol (9.5:0.5 v/v) as eluent and sprayed with iodine-potassium. Compounds 3a-i and 5a-i were reported in our previous work.42 The NMR spectra for all the newly synthesized compounds are available in the supplementary information (Figures S1–S18).
General Procedure for the Preparation of Coumarin-6-Sulfonamides-N-Acetylpyrazoline 6a-i
To a stirred solution of the appropriate chalcone derivative 5a-i (1 mmol) in glacial acetic acid (5 mL), hydrazine hydrated 95% (2 mL) was added. The reaction mixture was heated under reflux temperature for 6 hours, then cooled and poured onto ice water. The resulting precipitate was then filtered, dried, and crystallized with acetonitrile to obtain the target coumarins 6a-i.
N-(4-(1-Acetyl-5-Phenyl-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6a)
White powder; yield 79%, m.p. 223–225°C. 1H NMR (400 MHz, DMSO-d6): δ 2.26 (s, 3H, CH3), 2.99 (dd, 1Ha, J = 4.4 and 17.6 hz, CH2-pyrazoline), 3.72 (dd, 1Hb, J = 12 and 18 hz, CH2-pyrazoline), 5.47 (dd, 1Hb, J = 4.4 and 11.6 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.12 (d, 2H, J = 7.2 hz, H-Ar), 7.19–7.31 (m, 5H, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.64 (d, 2H, J = 8.4 hz, H-Ar), 7.93 (dd, 1H, J = 2.0 and 8.8 hz, H-Ar), 8.16 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.4 hz, H5 of coumarin), 10.76 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.13 (CH3), 42.53 (CH2-pyrazoline), 59.80 (CH-pyrazoline), 118.34, 119.44, 119.86, 125.82, 127.62, 127.17, 128.24, 128.29, 129.08, 130.01, 135.65, 139.64, 142.84, 143.82, 153.96 (C=N-pyrazoline), 156.41 (C9 of Coumarin), 159.52 (C2 of Coumarin), 167.71 (C=O). Anal. Calcd for C26H21N3O5S (487.12) C, 64.05; H, 4.34; N, 8.62; Found: C, 64.23; H, 4.41; N, 8.51.
N-(4-(1-Acetyl-5-(4-Nitrophenyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6b)
Pale yellow powder; yield 56%, m.p. 124–126°C. 1H NMR (400 MHz, DMSO-d6): δ 2.28 (s, 3H, CH3), 3.05 (dd, 1Ha, J = 4.8 and 18.0 hz, CH2-pyrazoline), 3.78 (dd, 1Hb, J = 12 and 18 hz, CH2-pyrazoline), 5.61 (dd, 1Hb, J = 4.8 and 12 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.19 (d, 2H, J = 8.8 hz, H-Ar), 7.42 (d, 2H, J = 8.8 hz, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.64 (d, 2H, J = 8.8 hz, H-Ar), 7.93 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.16 (d, 2H, J = 8.8 hz, NO2-2H-Ar), 8.18(d, 1H, J = 9.2 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.0 hz, H5 of coumarin), 10.77 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 19.60 (CH3), 42.14(CH2-pyrazoline), 59.81 (CH-pyrazoline), 118.35, 119.16, 119.47, 119.81, 124.39, 124.42, 125.75, 126.89, 127.42, 127.44, 127.95, 128.26, 128.39, 130.07, 135.63, 139.84, 141.90, 143.81, 147.10, 154.12 (C=N-pyrazoline), 156.46 (C9 of Coumarin), 159.49 (C2 of Coumarin), 167.99 (C=O). Anal. Calcd for C26H20N4O7S (532.53) C, 58.64; H, 3.79; N, 10.52; Found: C, 58.82; H, 3.76; N, 10.61.
N-(4-(1-Acetyl-5-(4-Fluorophenyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6c)
White powder; yield 61%, m.p. 167–169°C. 1H NMR (400 MHz, DMSO-d6): δ 2.25 (s, 3H, CH3), 3.00 (dd, 1Ha, J = 4.4 and 18.0 hz, CH2-pyrazoline), 3.71 (dd, 1Hb, J = 11.6 and 18 hz, CH2-pyrazoline), 5.47(dd, 1Hb, J = 4.4 and 11.6 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.09–7.13(m, 2H, H-Ar), 7.16–7.21(m, 4H, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.63 (d, 2H, J = 8.4 hz, H-Ar), 7.93 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.16 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.0 hz, H5 of coumarin), 10.77 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.13 (CH3), 42.40 (CH2-pyrazoline), 59.16 (CH-pyrazoline), 115.68, 115.89, 118.33, 119.44, 119.84, 127.11, 128.04, 128.30, 130.02, 135.66, 138.97, 139.68, 143.82, 153.98 (C=N-pyrazoline), 156.40 (C9 of Coumarin), 159.62 (C2 of Coumarin), 160.47(q, J = 243.41 hz, C-F), 167.77 (C=O). Anal. Calcd for C26H20FN3O5S (505.11) C, 61.78; H, 3.99; N, 8.31; Found: C, 61.99; H, 4.04; N, 8.40.
N-(4-(1-Acetyl-5-(4-Chlorophenyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6d)
White powder; yield 45%, m.p. 198–200°C. 1H NMR (400 MHz, DMSO-d6): δ 2.25 (s, 3H, CH3), 3.00 (dd, 1Ha, J = 4.4 and 18.0 hz, CH2-pyrazoline), 3.71 (dd, 1Hb, J = 12.0 and 18.0 hz, CH2-pyrazoline), 5.46 (dd, 1Hb, J = 4.8 and 12.0 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.15 (d, 2H, J = 8.4 hz, H-Ar), 7.19 (d, 2H, J = 8.8 hz, H-Ar), 7.34 (d, 2H, J = 8.4 hz, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.63 (d, 2H, J = 8.8 hz, H-Ar), 7.93 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.16 (d, 1H, J = 10.0 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.0 hz, H5 of coumarin), 10.79 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.10 (CH3), 42.30 (CH2-pyrazoline), 59.23 (CH-pyrazoline), 118.33, 119.43, 119.84, 127.05, 127.92, 128.25, 128.31, 129.02, 130.02, 132.13, 135.66, 139.71, 141.74, 143.90, 154.00 (C=N-pyrazoline), 156.47 (C9 of Coumarin), 159.52 (C2 of Coumarin), 167.83 (C=O). Anal. Calcd for C26H20ClN3O5S (521.08) C, 59.83; H, 3.86; N, 8.05; Found: C, 60.01; H, 3.84; N, 8.11.
N-(4-(1-Acetyl-5-(4-Methoxyphenyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6e)
White powder; yield 66%, m.p. 183–184°C. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 2.98 (dd, 1Ha, J = 4.4 and 18.0 hz, CH2-pyrazoline), 3.68 (dd, 1Hb, J = 11.6 and 17.6 hz, CH2-pyrazoline), 3.70 (s, 3H, OCH3), 5.41 (dd, 1Hb, J = 4.4 and 12.0 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 6.83 (d, 2H, J = 8.8 hz, H-Ar), 7.04 (d, 2H, J = 8.4 hz, H-Ar), 7.19 (d, 2H, J = 8.8 hz, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.64 (d, 2H, J = 8.8 hz, H-Ar), 7.93 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.16 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.4 hz, H5 of coumarin), 10.75 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.21(CH3), 42.51(CH2-pyrazoline), 55.58 (OCH3), 59.27 (CH-pyrazoline), 114.39, 118.34, 119.45, 119.86, 127.22, 128.38, 130.00, 134.99, 135.66, 139.72, 143.91, 154.08 (C=N-pyrazoline), 156.41 (C9 of Coumarin), 158.81 (C-OCH3), 159.61 (C2 of Coumarin), 167.64 (C=O). Anal. Calcd for C27H23N3O6S (517.13) C, 62.66; H, 4.48; N, 8.12; Found: C, 62.84; H, 4.43; N, 8.04.
N-(4-(1-Acetyl-5-(2,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-oxo-2H-chromene-6-sulfonamide (6f)
White powder; yield 77%, m.p. 156–157°C. 1H NMR (400 MHz, DMSO-d6): δ 2.27 (s, 3H, CH3), 2.81 (dd, 1Ha, J = 4.0 and 17.6 hz, CH2-pyrazoline), 3.62 (dd, 1Hb, J = 11.2 and 17.6 hz, CH2-pyrazoline), 3.71 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 5.51 (dd, 1Hb, J = 4.4 and 11.6 hz, CH-pyrazoline), 6.38 (dd, 1H, J = 2.4 and 8.4 hz, H-Ar), 6.55 (d, 1H, J = 2.4 hz, H-Ar), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 6.72 (d, 1H, J = 8.4 hz, H-Ar), 7.16 (d, 2H, J = 8.8 hz, H-Ar), 7.54 (d, 1H, J = 8.8 hz, H-Ar), 7.61 (d, 2H, J = 8.4 hz, H-Ar), 7.91 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.15 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.24 (d, 1H, J = 2.4 hz, H5 of coumarin), 10.72 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.21 (CH3), 41.61 (CH2-pyrazoline), 55.39 (OCH3), 55.67 (OCH3), 56.05 (CH-pyrazoline), 99.27, 104.94, 118.34, 119.48, 119.89, 122.09, 126.50, 127.42, 128.17, 130.00, 135.68, 139.51, 143.90, 154.47 (C=N-pyrazoline), 156.40 (C9 of Coumarin), 157.23 (C-OCH3), 159.53 (C2 of Coumarin), 160.24 (C-OCH3), 167.50 (C=O). Anal. Calcd for C28H25N3O7S (547.14) C, 61.42; H, 4.60; N, 7.67; Found: C, 61.59; H, 4.63; N, 7.59.
N-(4-(1-Acetyl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-oxo-2H-chromene-6-sulfonamide (6g)
Buff powder; yield 50%, m.p. 227–229°C. 1H NMR (400 MHz, DMSO-d6): δ 2.27 (s, 3H, CH3), 3.01 (dd, 1Ha, J = 4.8 and 18.0 hz, CH2-pyrazoline), 3.60 (s, 3H, OCH3), 3.67 (dd, 1Hb, J = 12.0 and 18.0 hz, CH2-pyrazoline), 3.69 (s, 6H, 2OCH3), 5.39 (dd, 1Hb, J = 4.8 and 12.0 hz, CH-pyrazoline), 6.40 (s, 2H, H-Ar), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.18 (d, 2H, J = 8.8 hz, H-Ar), 7.55 (d, 1H, J = 8.8 hz, H-Ar), 7.63 (d, 2H, J = 8.4 hz, H-Ar), 7.92 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.15 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.25 (d, 1H, J = 2.0 hz, H5 of coumarin), 10.74 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.12 (CH3), 42.60 (CH2-pyrazoline), 56.28 (2OCH3), 60.03 (CH-pyrazoline), 60.34 (OCH3), 102.84, 118.33, 119.43, 119.83, 127.21, 128.23, 128.31, 130.00, 135.66, 136.89, 138.63, 139.61, 143.80, 153.47 (C-OCH3), 153.99 (C=N-pyrazoline), 156.40 (C9 of Coumarin), 159.51 (C2 of Coumarin), 167.93 (C=O). Anal. Calcd for C29H27N3O8S (577.15) C, 60.30; H, 4.71; N, 7.27; Found: C, 60.18; H, 4.70; N, 7.35.
N-(4-(1-Acetyl-5-(4-Bromophenyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6h)
White powder; yield 49%, m.p. 180–182°C. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 3.00 (dd, 1Ha, J = 4.4 and 18.0 hz, CH2-pyrazoline), 3.71 (dd, 1Hb, J = 11.6 and 18.0 hz, CH2-pyrazoline), 5.44 (dd, 1Hb, J = 4.4 and 11.6 hz, CH-pyrazoline), 6.59 (d, 1H, J = 9.6 hz, H3 of coumarin), 7.08 (d, 2H, J = 8.4 hz, H-Ar), 7.15–7.20 (m, 3H, H-Ar), 7.47 (d, 2H, J = 8.4 hz, H-Ar), 7.54 (d, 1H, J = 8.8 hz, H-Ar), 7.62 (d, 2H, J = 8.8 hz, H-Ar), 7.92 (dd, 1H, J = 2.0 and 8.8 hz, H-Ar), 8.15 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.26 (d, 1H, J = 2.4 hz, H5 of coumarin), 10.75 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 22.09 (CH3), 42.24 (CH2-pyrazoline), 59.29 (CH-pyrazoline), 118.34, 119.43, 119.82, 120.61, 127.04, 127.92, 128.24, 128.27, 128.31, 129.02, 130.00, 131.94, 135.65, 139.70, 141.74, 142.17, 143.81, 153.99 (C=N-pyrazoline), 156.40 (C9 of Coumarin), 159.51 (C2 of Coumarin), 167.77 (C=O). Anal. Calcd for C26H20BrN3O5S (565.03) C, 55.13; H, 3.56; N, 7.42; Found: C, 55.01; H, 3.55; N, 7.51.
N-(4-(1-Acetyl-5-(p-Tolyl)-4,5-Dihydro-1H-Pyrazol-3-Yl)phenyl)-2-Oxo-2H-Chromene-6-Sulfonamide (6i)
White powder; yield 81%, m.p. 294–296°C. 1H NMR (400 MHz, DMSO-d6): δ 2.23 (s, 3H, CH3), 2.97 (dd, 1Ha, J = 4.4 and 18.0 hz, CH2-pyrazoline), 3.67 (dd, 1Hb, J = 11.6 and 18.0 hz, CH2-pyrazoline), 3.69(s, 3H, CH3), 5.40 (dd, 1Hb, J = 4.4 and 11.6 hz, CH-pyrazoline), 6.58 (d, 1H, J = 9.6 hz, H3 of coumarin), 6.82 (d, 2H, J = 8.8 hz, H-Ar), 7.03 (d, 2H, J = 8.8 hz, H-Ar), 7.18 (d, 2H, J = 8.8 hz, H-Ar), 7.54 (d, 1H, J = 8.8 hz, H-Ar), 7.63 (d, 2H, J = 8.8 hz, H-Ar), 7.92 (dd, 1H, J = 2.4 and 8.8 hz, H-Ar), 8.15 (d, 1H, J = 9.6 hz, H4 of coumarin), 8.25 (d, 1H, J = 2.4 hz, H5 of coumarin), 10.75 (s, 1H, NH), 13C NMR (101 MHz, DMSO-d6): δ 20.91(CH3), 22.16 (CH3), 42.51 (CH2-pyrazoline), 59.26 (CH-pyrazoline), 114.37, 118.32, 119.43, 119.86, 127.16, 127.21, 128.24, 130.00, 130.31, 134.88, 135.69, 139.66, 143.81, 153.94 (C=N-pyrazoline), 156.39 (C9 of Coumarin), 159.52 (C2 of Coumarin), 167.59 (C=O). Anal. Calcd for C27H23N3O5S (501.14) C, 64.66; H, 4.62; N, 8.38; Found: C, 64.43; H, 4.69; N, 8.45.
Biological Assessments
All compounds were dissolved in dimethyl sulfoxide suitable for cell cultures (DMSO, Sigma-Aldrich, St. Louis, MO) to a maximum of 25–50 mg/mL concentration, depending on each compound’s individual solubility. The reference anti-Toxoplasma gondii drugs, used as controls in the tests, were dissolved in DMSO (trimethoprim – TRI, pyrimethamine – PYR) or PBS (sulphadiazine – SULF) to the concentrations of 50 mg/mL for TRI and PYR and 200 mg/mL SULF. The final concentration of DMSO in final compound dilutions used in the cell tests was below 1.00%, which was determined as non-cytotoxic in preliminary tests.
Cell Culture
The L929 mouse fibroblasts (ATTC CCL-1, Manassas, VA, USA) were maintained in high glucose DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Biowest, Nuaille, France), 100 I.U./mL penicillin and 100 mg/mL streptomycin (Merck KGaA, Darmstadt, Germany). The Hs27 human fibroblasts (Hs27, CRL-1634, ATCC, Manassas, VA, USA), used as host cells for T. gondii, were maintained in high glucose DMEM (ATCC, Manassas, VA, USA), supplemented with 10% fetal bovine serum (ATCC, Manassas, VA, USA), 100 I.U./mL penicillin and 100 mg/mL streptomycin (Merck KGaA, Darmstadt, Germany). All used cells were grown to full confluency at 37°C and 5% of CO2, then detached from the culture flasks using 0.25% trypsin solution (Merck KGaA, Darmstadt, Germany) and transferred to new cell flasks twice a week.
Cytotoxicity Assessment
Cytotoxicity of compounds was determined by cell viability according to international standards (ISO 10993–5:2009) on L929 cells. The cytotoxicity towards T. gondii host cells Hs27 was also evaluated. Cells were seeded in their respective culture media in the 96-well plates at an initial density of 1 × 104 or 2 × 104 cells/well. After 24h (L929) or 72h (Hs27) of incubation the medium was removed, and compounds or drugs diluted (7 two-fold dilutions) in high glucose DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) medium without phenol red supplemented with 10% heat-inactivated FBS (Biowest, Nuaille, France) and 100 I.U./mL penicillin, 100 mg/mL streptomycin were added to the wells.
After 24h incubation, the medium was again replaced with 50 µL of MTT (Merck KGaA, Darmstadt, Germany) solution in a complete DMEM medium without phenol red at 1 mg/mL concentration. After 4h incubation, the MTT solution was removed, and resulting formazan crystals were dissolved in 150 µL of DMSO, and 25 mL 0.05 M glycine buffer (pH 10.5) (Merck KGaA, Darmstadt, Germany) was added to each well. The optical density was read at 570 nm using a microplate absorbance reader Multiskan EX (Thermo Fisher Scientific, Waltham, MA, USA). Optical density values were used to calculate the percent of cell viability regarding untreated cells. Cell viability values were changed to cytotoxicity values. Values of each compound/drug dilutions were plotted on graph, and least squares fit non-linear regression analysis was performed to calculate CC30. Compounds alone were tested for their ability to reduce MTT. Graphs and regression analysis were performed using GraphPad Prism 10.0.0 for Windows (Dotmatics, GraphPad Software, San Diego, California, USA). All experiments were performed in triplicate.
Parasite Culture
The highly virulent RH T. gondii strain expressing green fluorescent protein (ATCC 50940) was maintained on Hs27 human fibroblasts in high glucose DMEM medium (ATCC, Manassas, VA, USA) supplemented with 3% twice heat-inactivated FBS (Biowest, Nuaille, France) and 100 I.U./mL penicillin and 100 mg/mL streptomycin (Merck KGaA, Darmstadt, Germany). After the full lysis of infected host cells, released parasites were transferred to a fresh confluent monolayer of Hs27 cells at a density of 1 × 107 tachyzoites per 25cm2 culture flask and incubated at 37°C and 5% of CO2 for 3–4 days until the complete destruction of host cells.
Anti-T. gondii Activity Test
The influence of compounds on T. gondii tachyzoites was evaluated as follows: Hs27 cells were seeded in the 96-well plates at a density of 1 × 104 cells/ well and incubated at 37°C and 5% of CO2 for 72h to full confluency. Next, the medium was removed, and T. gondii tachyzoites were added at the density of 1 × 105 cells/well in 100 µL immediately followed by 100 µL of diluted compounds or drugs (7 two-fold dilutions). Both tachyzoites and tested compounds/drugs were prepared in high glucose DMEM medium without phenol red supplemented with 3% twice heat-inactivated FBS (Biowest, Nuaille, France) and 100 I.U./mL penicillin and 100 mg/mL streptomycin (Merck KGaA, Darmstadt, Germany). After incubation of cultures at 37°C and 5% of CO2 for 4 days, the mean fluorescence intensivity of 488/510 nm (MFI) was measured using SpectraMax i3 (Molecular Devices). MFI values were used to calculate the percent of T. gondii viability. Values of each compound/drug dilutions were plotted on graph and least squares fit non-linear regression analysis was performed to calculate IC50. Each compound was tested at each dilution used for its autofluorescence in the 488/510 nm channel. If present, it was subtracted from the test value. Selectivity values index was determined by dividing the CC30 concentration by the IC50 value to determine the actual efficacy of the compound. Graphs and regression analysis was performed using GraphPad Prism 10.0.0 for Windows (Dotmatics, GraphPad Software, San Diego, California, USA). All experiments were performed in four repeats.
Molecular Docking
The molecular docking was carried out by applying CANDOCK algorithm40 and radial-mean-reduced scoring function at a cutoff radius of 6 Å from each atom of the ligand (RMR6) to obtain the binding poses of the seven most potent inhibitors 6a, 6b, 6c, 6f, 6g, 6h, and 5d at the active site of TgCDPK1. The X-ray crystal structure of TgCDPK1 (PDB ID: 4TZR, chain A) was obtained from the Protein Data Bank (PDB). Avogadro was used to prepare 3D models of the seven studied inhibitors,43 which were subsequently geometrically optimized with the M06-2X method and 6–31G(d) basis set in Gaussian 16 program.44 The binding poses of the seven investigated inhibitors at the active site of TgCDPK1 with the lowest docking score values were chosen for analysis of intermolecular interactions, as the experimental crystal structures have not yet been experimentally determined.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2024-80-03”. JD was supported by the Ministry of Science and Higher Education, POL-OPENSCREEN, DIR/WK/2018/06 and 2024/WK/06.
Disclosure
Authors declare no conflict of interest. The authors alone are responsible for the content and writing of the paper.
References
1. Kochanowsky JA, Koshy AA. Toxoplasma gondii. Curr Biol. 2018;14:R770–R771. doi:10.1016/j.cub.2018.05.035
2. Mendez OA, Koshy AA. Toxoplasma gondii: entry, association, and physiological influence on the central nervous system. PLoS Pathog. 2017;7:e1006351. doi:10.1371/journal.ppat.1006351
3. Alday PH, Doggett JS. Drugs in development for toxoplasmosis: advances, challenges, and current status. Drug Des Dev Ther. 2017;11:273–293. doi:10.2147/DDDT.S60973
4. Innes EA, Hamilton C, Garcia JL, Chryssafidis A, Smith D. A one health approach to vaccines against Toxoplasma gondii. Food Waterborne Parasitol. 2019;15:e00053. doi:10.1016/j.fawpar.2019.e00053
5. Di Cristina M, Dou Z, Lunghi M, et al. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat Microbiol. 2017;2(8):17096. doi:10.1038/nmicrobiol.2017.96
6. Dubey JP, Jones JL. Toxoplasma gondii infection in humans and animals in the United States. Int J Parasitol. 2008;38:1257–1278. doi:10.1016/j.ijpara.2008.03.007
7. Rudin C, Boubaker K, Raeber PA, et al. Conge, Toxoplasmosis during pregnancy and infancy A new approach of Switzerland. Swiss Med Weekly. 2008;138:1–8. doi:10.4414/SMW.2008.w30271
8. Ozgonul C, Besirli CG. Recent developments in the diagnosis and treatment of ocular toxoplasmosis. Ophthalmic Res. 2017;57(1):1–12. doi:10.1159/000449169
9. Sanchez SG, Besteiro S. The pathogenicity and virulence of Toxoplasma gondii. Virulence. 2021;12(1):3095–3114. doi:10.1080/21505594.2021.2012346
10. Boyle JP, Saeij JP, Coller SP, Boothroyd JC. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Am J Trop Med Hyg. 2006;314(5806):1780–1783.
11. Jones EJ, Korcsmaros T, Carding SR. Mechanisms and pathways of Toxoplasma gondii transepithelial migration. Tissue Barriers. 2017;5(1):e1273865. doi:10.1080/21688370.2016.1273865
12. Paredes-Santos T, Martins-Duarte E, Vitor R, De Souza W, Attias M, Vommaro R. Spontaneous cystogenesis in vitro of a Brazilian strain of Toxoplasma gondii. Parasitol Int. 2013;62(2):1. doi:10.1016/j.parint.2012.12.003
13. Pan M, Ge CC, Fan YM, Jin QW, Shen B, Huang SY. The determinants regulating Toxoplasma gondii bradyzoite development. Front Microbiol. 2022;13:1027073. doi:10.3389/fmicb.2022.1027073
14. Skariah S, McIntyre MK, Mordue DG. Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol Res. 2010;107(2):253–260. doi:10.1007/s00436-010-1899-6
15. Park Y-H, Han J-H, Nam H-W. Clinical features of ocular toxoplasmosis in Korean patients. Korean J Parasitol. 2011;49(2):1. doi:10.3347/kjp.2011.49.2.167
16. Thebault A, Kooh P, Cadavez V, Gonzales-Barron U, Villena I. Risk factors for sporadic toxoplasmosis: a systematic review and meta-analysis. Microbial Risk Anal. 2021;17:100133. doi:10.1016/j.mran.2020.100133
17. E Schoondermark-van DV, Vree T, Melchers W, et al. In vitro effects of sulfadiazine and its metabolites alone and in combination with pyrimethamine on Toxoplasma gondii. Antimicrob Agents Chemother. 1995;39:763–765. doi:10.1128/AAC.39.3.763
18. Borkowski PK, Brydak-Godowska J, Basiak W, Olszynska-Krowicka M, Rabczenko D. Adverse Reactions in Antifolate-Treated Toxoplasmic Retinochoroiditis. Current Tren Immun Respiratory Infec. 2018;37–48.
19. Adeyemi OS, Murata Y, Sugi T, Kato K. Inorganic nanoparticles kill Toxoplasma gondii via changes in redox status and mitochondrial membrane potential. Int j Nanomed. 2017;12:1647–1661. doi:10.2147/IJN.S122178
20. Mouneir SM, Elhagrasi A, El-Shamy AM. A review on the chemical compositions of natural products and their role in setting current trends and future goals. Egypt. J. Chem. 2022;65(5):491–506.
21. Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM. Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg. Chem. 2020;103:104163. doi:10.1016/j.bioorg.2020.104163
22. Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhibition and Medicinal Chemi. 2018;33(1):1095–1107. doi:10.1080/14756366.2018.1477137
23. Eldehna WM, Taghour MS, Al-Warhi T, et al. Discovery of 2, 4-thiazolidinedione-tethered coumarins as novel selective inhibitors for carbonic anhydrase IX and XII isoforms. Jf Enzyme Inhibition and Medicinal Chem. 2022;37(1):531–541. doi:10.1080/14756366.2021.2024528
24. Tawfik HO, Shaldam MA, Nocentini A, et al. Novel 3-(6-methylpyridin-2-yl) coumarin-based chalcones as selective inhibitors of cancer-related carbonic anhydrases IX and XII endowed with anti-proliferative activity. Jf Enzyme Inhibition and Medicinal Chem. 2022;37(1):1043–1052. doi:10.1080/14756366.2022.2056734
25. Batran RZ, Sabt A, Khedr MA, Allayeh AK, Pannecouque C, Kassem AF. 4-Phenylcoumarin derivatives as new HIV-1 NNRTIs: design, synthesis, biological activities, and computational studies. Bioorg. Chem. 2023;141:106918. doi:10.1016/j.bioorg.2023.106918
26. Smyth T, Ramachandran VN, Smyth WF. A study of the antimicrobial activity of selected naturally occurring and synthetic coumarins. Int J Antimicrob Agents. 33(5):421–426. DOI:10.1016/j.ijantimicag.2008.10.022
27. Ibrahim HS, Abdelrahman MA, Nocentini A, et al. Insights into the effect of elaborating coumarin-based aryl enaminones with sulfonamide or carboxylic acid functionality on carbonic anhydrase inhibitory potency and selectivity. Bioorg. Chem. 2022;126(105888):105888. doi:10.1016/j.bioorg.2022.105888
28. González LA, Upegui YA, Rivas L, et al. Effect of substituents in the A and B rings of chalcones on antiparasite activity. Arch. Pharm. 2020;353(12):2000157. doi:10.1002/ardp.202000157
29. Dard C, Leforestier B, Francisco Hilário F, et al. Crossing of the cystic barriers of Toxoplasma gondii by the fluorescent coumarin tetra-cyclopeptide. Molecules. 2021;26(24):7506. doi:10.3390/molecules26247506
30. Mazzone F, Klischan MKT, Greb J, Smits SHJ, Pietruszka J, Pfeffer K. Synthesis and In vitro evaluation of bichalcones as novel anti-toxoplasma agents. Front Chem. 2024;12:1406307. doi:10.3389/fchem.2024.1406307
31. Chimenti F, Bizzarri B, Bolasco A, et al. Synthesis and evaluation of 4-acyl-2-thiazolylhydrazone derivatives for antiToxoplasma efficacy in vitro. J Med Chem. 2009;52(15):4574e4. doi:10.1021/jm9005862
32. Chen M, Theander TG, Christensen SB, et al. Licochalcone A, a new antimalarial agent, inhibits in vitro growth of the human malaria parasite Plasmodium falciparum and protects mice from P. yoelii infection, Antimicrob. Agents Chemother. 1994;38(7):1470e1475. doi:10.1128/AAC.38.7.1470
33. Chen M, Christensen SB, Blom J, et al. Licochalcone A, a novel antiparasitic agent with potent activity against human pathogenic protozoan species of Leishmania, Antimicrob. Agents Chemother. 1993;37(12):2550e2556. doi:10.1128/AAC.37.12.2550
34. Zhang Z, Ojo KK, Vidadala R, et al. Potent and selective inhibitors of CDPK1 from T. gondii and C. parvum based on a 5-aminopyrazole-4- carboxamide scaffold. ACS Med Chem Lett. 2014;5(1):40e44. doi:10.1021/ml400315s
35. Lourido S, Zhang C, Lopez MS, et al. Optimizing small molecule inhibitors of calcium dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J Med Chem. 2013;56(7):3068e3077. doi:10.1021/jm4001314
36. Rutaganira FU, Barks J, Dhason MS, et al. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and reoccurrence of central nervous system disease by Toxoplasma gondii. J Med Chem. 2017;60(24):9976e9989. doi:10.1021/acs.jmedchem.7b01192
37. Huang W, Ojo KK, Zhang Z, et al. SAR Studies of 5- aminopyrazole-4-carboxamide analogues as potent and selective inhibitors of Toxoplasma gondii CDPK1. ACS Med Chem Lett. 2015;6(12):1184e1189. doi:10.1021/acsmedchemlett.5b00319
38. Karthik R, Vimaladevi G, Chen SM, Elangovan A, Jeyaprabha B, Prakash P. Corrosion Inhibition and Adsorption Behavior of 4–Amino Acetophenone Pyridine 2-Aldehyde in 1 M Hydrochloric Acid. Int J Electrochem Sci. 2015;10:4666–4681. doi:10.1016/S1452-3981(23)06654-3
39. Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxo-plasma. Nature. 2010;465(7296):359–362. doi:10.1038/nature09022
40. Fine J, Konc J, Samudrala R, Chopra G. CANDOCK: chemical atomic network-based hierarchical flexible docking algorithm using generalized statistical potentials. J Chem Inf Model. 2020;60:1509–1527. doi:10.1021/acs.jcim.9b00686
41. Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi:10.1093/nar/gkv315
42. Sabt A, Kitsos S, Ebaid MS, et al. Novel coumarin-6-sulfonamide-chalcone hybrids as glutathione transferase P1-1 inhibitors. PLoS One. 2024;19(8):e0306124. doi:10.1371/journal.pone.0306124
43. Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4:17. doi:10.1186/1758-2946-4-17
44. Frisch M, Trucks G, Schlegel H, et al. Gaussian 16, Revision C. 01. Gaussian, Inc Wallingford CT; 2016. Google Scholar There is no corresponding record for this reference 2020.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.