")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Identification of a Novel VPS13B Mutation in a Chinese Patient with Cohen Syndrome by Whole-Exome Sequencing
Authors Hu X, Huang T, Liu Y, Zhang L, Zhu L, Peng X, Zhang S
Received 2 July 2021
Accepted for publication 10 November 2021
Published 4 December 2021 Volume 2021:14 Pages 1583—1589
DOI https://doi.org/10.2147/PGPM.S327252
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Xiaoyun Hu, Tao Huang, Yun Liu, Lina Zhang, Li Zhu, Xiaohong Peng, Sufang Zhang
Department of Pediatrics, The First Affiliated Hospital of Nanchang University, Nanchang, People’s Republic of China
Correspondence: Tao Huang Tel +86 13970012590
Email [email protected]
Objective: The present study aims to investigate the clinical features and diagnostic characteristics of children with Cohen syndrome caused by the vacuolar protein sorting 13 homolog B (VPS13B) gene mutation and to review the relevant literature to provide a reference for genetic counseling and the diagnosis of Cohen syndrome.
Methods: The clinical data and molecular genetic test results of a child with Cohen syndrome were retrospectively analyzed and a review of the relevant literature was conducted.
Results: A two-year-and-four-month-old boy was referred to the hospital for recurrent fever and shortness of breath. On physical examination, the boy was found to have growth retardation, thick bushy hair, microcephaly, hypertelorism, down-slanting palpebral fissures, and hypotonia. Genetic testing was performed, and the results suggested the presence of exon 20– 32 heterozygous deletion and c.8275 delC (p.R2759 fs*18) heterozygous variant on the VPS13B gene from phenotypically normal parents. These two mutation loci have not been reported in the literature, and they were predicted by relevant software to be pathogenic variants.
Conclusion: We identified two novel variants in the VPS13B gene (exon 20– 32 heterozygous deletion and c.8275 delC heterozygous variant) in a boy with Cohen syndrome, thus extending the spectrum of VPS13B gene variants in patients with Cohen syndrome.
Keywords: Cohen syndrome, VPS13B gene, heterozygous mutation, heterozygous deletion
Cohen syndrome is a rare autosomal recessive hereditary disease, also known as “brain-obesity-eye-bone” syndrome, which is characterized by characteristic facial features, mental–motor retardation, hypotonia, neutropenia, and abnormal eye and limb development.1–4 Cohen syndrome is caused by a variant or a massive genomic rearrangement of the autosomal 8q22.2 vacuolar protein sorting 13 homolog B (VPS13B) gene, and the VPS13B gene is the only gene responsible for Cohen syndrome.2,5 So far, approximately 1000 cases of Cohen syndrome have been reported at home and abroad of which five cases have been genetically diagnosed in China.6–10 In the present study, the clinical data of a child with Cohen syndrome diagnosed using genetic diagnosis were reviewed to retrieve the relevant literature to explore the clinical and genetic variation characteristics of Cohen syndrome.
Clinical Data
A two-year-and-four-month-old boy was referred to the hospital with a history of recurrent fever and shortness of breath for more than 10 days. The boy was gravida 1 para 1 and had been delivered by cesarean section at 40+5 weeks of gestation, with a birth mass of 1.98 kg (full-term, small, low birth weight infant), with no history of asphyxia or hypoxia but with intrauterine growth restriction on prenatal care. At the time of the visit, the boy could not walk independently and could only say simple words, such as dad and mom. The boy had a history of wheezing more than 10 times, multiple episodes of pneumonia, head lifting at 2–3 months, sitting at 6–7 months, feeding difficulties until 8 months, and crawling at 1 year and 3 months. The boy was diagnosed with growth retardation at another hospital, and cranial magnetic resonance imaging showed underdevelopment of the prefrontal temporal lobe and a slightly widened brain gap (see Figure 1), and a significant abnormality was identified on the electroencephalogram. The boy was treated with rehabilitation and intramuscular injections of rat nerve growth factor for more than three months. Subsequently, the boy was able to stand with the support of others, starting at one year and six months of age, and had no history of convulsions. There were no other family members with similar diseases, and the parents were not consanguineous. The results of the physical examination were as follows: body mass 12 kg, height 88 cm, and head circumference 44 cm with a closed anterior fontanel. The boy had clear consciousness with a blunt response and characteristic facial features (thick hair and eyebrows, hypertelorism, down-slanting palpebral fissures, short philtrum, and short upper lip; see Figure 2 and Table 1). There was no Tong-Guan palmprint (a special type of palmprint in which two lines combine into a single line across the entire palm) and no abnormalities of the heart or abdomen. He had coarse breathing sounds in both lungs, with wet rales and wheezing sounds. He had joint hyperextension and hypotonia of the four extremities. The eye examination was normal. The results of the laboratory examination were as follows: The routine blood test showed the white blood cell count was 3.86 × 109/L, the neutrophil count was 0.85 × 109/L, and the percentage of neutrophils was 22.1%. The result of karyotype analysis was 46, XY. The blood biochemistry was nearly normal, with negative blood culture and antibodies against Mycoplasma pneumonia. The electrocardiogram was normal. There was no abnormality in the phenotype of the parents. This study was conducted in accordance with the declaration of Helsinki, and written informed consent was obtained from all participants and guardians.
 |
Table 1 Clinical Features of This Case |
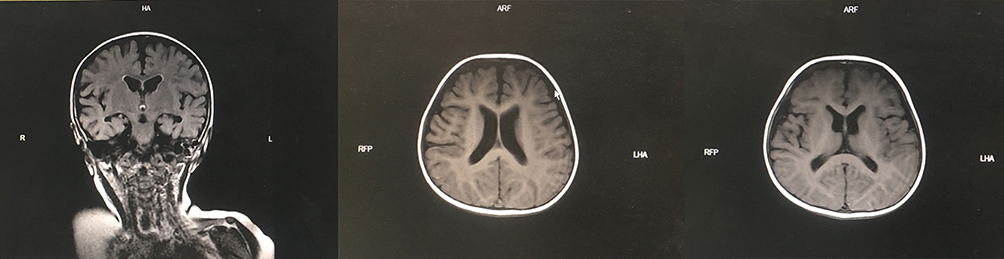 |
Figure 1 Cranial magnetic resonance imaging showed underdevelopment of the prefrontal temporal lobe and a slightly widened brain gap. |
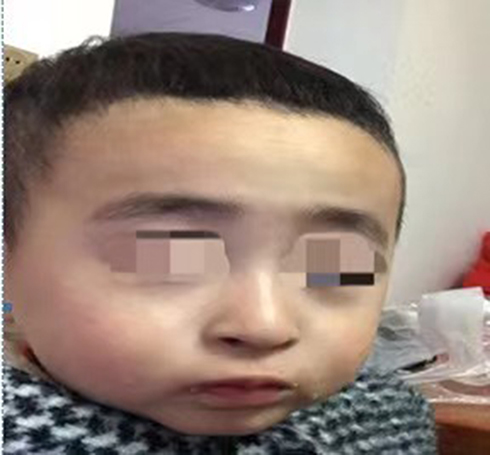 |
Figure 2 The characteristic facial features of the present case (thick hair and eyebrows, hypertelorism, down-slanting palpebral fissures, short philtrum, and short upper lip). |
The boy was diagnosed with pneumonia and wheezing, but he also had characteristic facial features, significant growth retardation, and feeding difficulties in infancy. Therefore, the possibility of Prader–Willi syndrome or Cohen syndrome was considered. To further clarify the diagnosis, after obtaining informed consent from the parents, high-throughput whole-exome sequencing was conducted on the boy and his parents, copy number variation analysis was performed on the father, and Sanger validation was performed on the mother.
Method
DNA Extraction
A sample of 5 mL of venous blood was collected from the boy and the parents using ethylenediaminetetraacetic acid anticoagulation tubes.
Whole-Exome Sequencing
The genomic DNA was randomly broken into fragments and linked with an Illumina PE splice oligonucleotide mixture. The products were subjected to amplification by the ligation-mediated polymerase chain reaction (PCR). A DNA library was obtained after purification, and quality testing was conducted. The above PCR products were hybridized with the target region capture chip to enrich the target region sequences, and the captured sequences were sequenced using the Illumina NextSeq 500 sequencing platform. The raw data were initially processed, corrected, and standardized. The contamination and splice sequences were removed from the raw sequencing data, and the filtered sequences were aligned to the human genome reference sequence in the National Center for Biotechnology Information database using the Burrows–Wheeler Alignment software. The information related to single-nucleotide polymorphism (SNP) and insertion/deletion (INDEL) mutations were analyzed using the genome analysis toolkit, and all SNPs and INDELs were then annotated using the ANNOVAR software.
Result
The mutation loci with frequency less than 0.05 in the normal human database were screened out, and finally, the I gene variants were verified by Sanger sequencing and next-generation sequencing. The results revealed a heterozygous variant and a heterozygous deletion on the VPS13B gene, constituting a compound heterozygous variant. The c.8275 delC variant was located in Chr 8: 100829869–100829870, exon 45, resulting in amino acid change p.R 2759 fs*18, a code-shifting mutation (see Figure 3A). The exon 20–32 deletion variant was located in Chr 8: 100396415–100588032, leading to exon deletion and resulting in a loss of gene function (see Figure 3B). The pedigree verification showed that the mother and father were carriers of the corresponding variants, with a c.8275 delC heterozygous mutation in the mother (see Figure 3C) and no variant at this locus in the father (see Figure 3D), and an exon 20–32 heterozygous deletion in the father (see Figure 3E). These two variants were not recorded in the Human Gene Mutation Database or the ClinVar database, and the frequencies of the two loci in the general population were not recorded. The above information was not recorded in the East Asian population in the Genome Aggregation Database. These two mutations were graded in relevant mutation interpretation guidelines by the American College of Medical Genetics as likely pathogenic. Thus, the boy was finally diagnosed with Cohen syndrome. Pneumonia and wheezing were cured using a comprehensive therapy of anti-infection and nebulization.
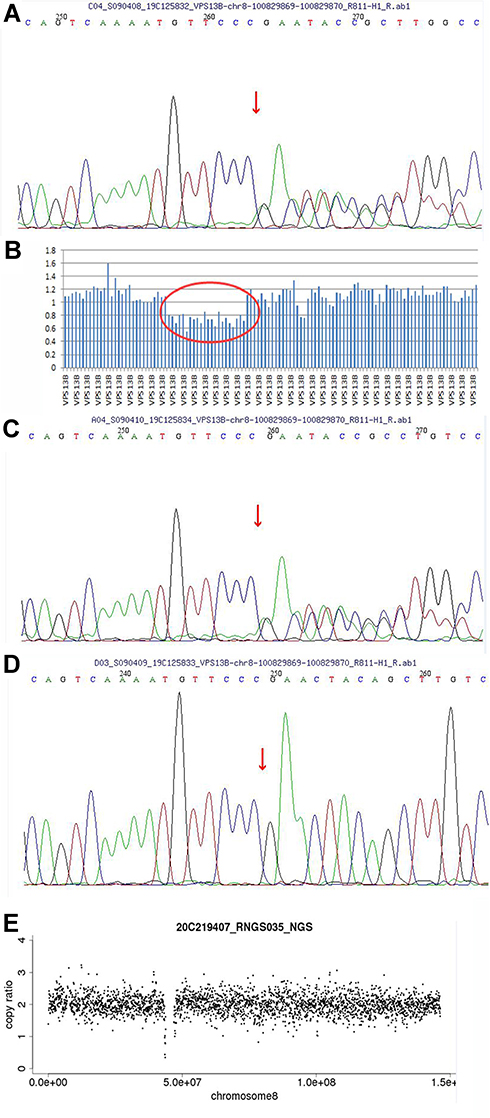 |
Figure 3 Results of the whole-exome sequencing of the VPS13B gene in the pedigree of the present case. (A) The heterozygous mutation c.8275 delC in exon 45 of the child. (B) The heterozygous deletion in exon 20–32 of the child. (C) The heterozygous mutation c.8275 delC in exon 45 of the mother. (D) The heterozygous mutation c.8275 delC in exon 45 absent in the father. (E) The heterozygous deletion in exon 20–32 of the father. |
Discussion
Cohen syndrome is a rare autosomal recessive hereditary disease that can involve multiple organs and systems throughout the body and was first reported by Cohen et al in 1973.11 Cohen syndrome is found worldwide, mostly in Finns and the Amish, with consanguineous marriage being the greatest risk factor. The keywords “Cohen syndrome” and “VPS13B” were used to search the literature, including the VIP database, WanFang Database, and China National Knowledge Infrastructure from establishment to November 2020. Six articles constituted the literature related to Cohen syndrome caused by VPS13B in these databases. Using “VPS13B” and “Cohen syndrome” as keywords, the PubMed and Web of Science databases were searched from establishment to November 2020, and more than 200 articles were retrieved, reporting on more than 1000 patients.
The clinical manifestations of patients with Cohen syndrome vary widely. Most patients do not show typical clinical manifestations until school age or later, making early diagnosis difficult,7,12–14 and therefore, early diagnosis relies on genetic testing. Currently, Finns have been reported to exhibit similar phenotypes.15 Cohen syndrome is characterized by myopia, retinal dystrophy, neutropenia, short stature, microcephaly, persistent hypotonia, developmental retardation, truncated obesity, joint hyperextension, characteristic facial features, and overly friendly behavior.1–3,16 Approximately half of patients with Cohen syndrome exhibit decreased fetal movement during gestation and hypotonia during infancy, which can sometimes lead to breathing and feeding difficulties.1 Almost all patients have varying degrees of language and motor retardation, and most patients are cheerful and friendly, but some studies have reported that patients may also have a combination of autism spectrum disorders.1–3,17 Patients with Cohen syndrome may have visual abnormalities, with progressive myopia and pigmentary retinopathy being the most common, while strabismus, ptosis, and lens clouding occur occasionally, with some studies suggesting that cataracts may develop in later stages.18 Neutropenia is a common feature of Cohen syndrome and may be associated with increased spontaneous apoptosis of neutrophils due to abnormalities in the VPS13B protein.1,19 Children with Cohen syndrome may have recurrent infections, such as pneumonia and oral ulcers, due to neutropenia,1,2 which is usually present from birth and is typically mild to moderate, intermittent, and nonfatal. Patients with Cohen syndrome often have hypotonia, hypermobile joints, and, later, muscle spasms, and some studies have reported cubitus valgus, genu valgum, slender fingers and/or toes, multi-line palm prints, and Guan-Tong palm prints.1 Patients with Cohen syndrome may develop cardiac lesions later in life, such as decreased left ventricular function, valve defects, and dilatation in the descending aorta.1 Some researchers20 have suggested that the combination of intellectual disability and neutropenia may be a risk factor for Cohen syndrome. In the present study, the patient had a history of recurrent infections (pneumonia), feeding difficulties before eight months of age, motor–intellectual retardation, hypotonia, joint hyperextension, microcephaly, short stature, neutropenia in the neonatal period, and characteristic facial features, such as hypertelorism, thick hair, short philtrum, and short upper lip, all of which are consistent with the clinical features of Cohen syndrome.
The pathogenic gene for Cohen syndrome is the VPS13B gene on chromosome 8q22.2.21 The VPS13B gene is approximately 864 kb in length with 62 exons, and the translated protein VPS13B has a relative molecular weight of 44,800 and consists of 4022 amino acids. It has been reported22 that VPS13B may be correlated with autophagy in Cohen syndrome, but the exact pathogenesis is not well understood. To date, more than 200 different VPS13B pathogenic gene variants have been identified in patients with Cohen syndrome. Boschann et al13 reported in 2020 on the presence of a c.3446–23T> G heterozygous variant and multiple exon (18–23) deletions in a child with Cohen syndrome who was from a phenotypically normal mother and father. Momtazmanesh et al21 reported a novel pure shift variant in VPS13B (LRG_351t1: c.7095del;p.Ser2366AlafsTer49) leading to loss of function in a four-year-old girl with Cohen syndrome born in a consanguineous heterozygous family of Iranian ancestry. Alipour1 et al23 reported on three Iranian families with Cohen syndrome and identified mutations in each family as c.4608_4609del (p.E1537Rfs * 7), c.11486dupG (p.L3830Tfs * 13), and c.10360dupC (p. L3454fs * 7), respectively. Further in-depth study is necessary to clarify the relationship between genotypes and phenotypes in Cohen syndrome. A compound heterozygous variant was found on the VPS13B gene in the present case, the c.8275 delC (p.R2759fs*18) variant caused deletion of base C in exon 45 at 8275, resulting in a shift variant at amino acid aspartate at position 2759 of the corresponding protein sequence, and the exon 20–32 deletion resulted in an exon deletion, leading to a loss of protein function. Bioinformatics analysis suggested that the above mutations were pathogenic, leading to a confirmation of the diagnosis of Cohen syndrome. The above two variants have not been reported internationally, and the results of the present study further expand the spectrum of VPS13B gene variants in patients with Cohen syndrome.
Currently, the exact pathogenesis of Cohen syndrome due to VPS13B variants is not well understood. It has been shown that VPS13B plays an essential role in a variety of cellular functions, which may maintain the integrity and function of the Golgi body, and in protein glycosylation and endosomal transportation.1,24,25 It has also been reported26 that the loss of function of the VPS13B protein may be a potential mechanism for the disease. VPS13B is an RAB 6 effector protein, and the reduced brain volume in patients with Cohen syndrome may be due to impaired VPS13B function in the Golgi complex, resulting in reduced nerve fiber production.25 Cohen syndrome has complex clinical manifestations, and some symptoms may progress with age, so it is difficult to diagnose. At present, the diagnosis of Cohen syndrome is mainly based on clinical manifestations combined with molecular gene examination, and a diagnosis can be achieved by confirming the presence of the VPS13B gene variant.1,2 Currently, there are no internationally agreed diagnostic criteria for Cohen syndrome. The most widely accepted clinical diagnostic criteria were proposed by Kolehmainen et al27 in 2004 who suggested that Cohen syndrome can be clinically diagnosed by meeting six of the following eight diagnostic criteria: developmental delay, microcephaly, typical facial features (thick bushy hair, thick eyebrows and eyelashes, wave-shaped eyelids and down-slanting palpebral fissures, prominent root of nose and bulbous nasal tip, and short and upturned philtrum), truncal obesity with slender extremities, overly social behavior, joint hypermobility, high myopia and/or retinal dystrophy, and neutropenia. Those fulfilling five or fewer criteria were considered to have a Cohen-like syndrome. At the same time, patients with Cohen syndrome should be differentiated from those with the microdeletion syndrome 12q14. The two syndromes are similar, with short stature, craniofacial dysmorphism, small stature for gestational age, delayed intellectual and linguistic development, and feeding difficulties, but microdeletion syndrome 12q14 is mainly characterized by precocious puberty, with microdeletions on chromosome 12, q14. Genes that may be associated with the clinical phenotype are HMGA2, LEMD3, IRAK3, and GRIP.28 In contrast, patients with Cohen syndrome mostly present with dysplasia and abnormalities on the VPS13B gene on chromosome 8, and chromosome microarray analysis is feasible to assist in the differentiation. In the present case, the patient had developmental delays, microcephaly, characteristic facial features, and neutropenia. Therefore, the diagnosis of Cohen syndrome was confirmed with a combination of genetic tests.
At present, there is no specific treatment for Cohen syndrome. The primary treatment is symptomatic, which can be conducted according to the patient’s clinical manifestations and individualized rehabilitation training. The management of Cohen syndrome includes regular surveillance and recovery. Recombinant human granulocyte colony-stimulating factor (rhG-CSF) can be used to treat neutropenia.20 Motor and speech delay in patients with Cohen syndrome require linguistic and physical therapy to improve the developmental delay, hypotonia, joint hyperextension, and clumsiness.1,7,20 Regular ophthalmologic examinations should be conducted to assess refractive error and retinal dystrophy, and early visual correction is necessary for patients with refractive error.1 However, there is no effective way to stop the progression of retinopathy. Growth and body mass gain should be monitored. Children with Cohen syndrome should have a scientific diet and moderate exercise during growth and development to improve physical fitness and prevent obesity. Short stature may be correlated with a concomitant growth hormone deficiency, which should be identified and treated with growth hormone.29
Conclusion
In summary, Cohen syndrome is a rare autosomal recessive hereditary disease caused by mutations on the VPS13B gene. Patients with Cohen syndrome have complex and diverse clinical presentations, and it should be noted when a child with psychomotor retardation has characteristic facial manifestations with neutropenia and hyperextension of joints. The gene assay of the VPS13B gene will help to clarify the diagnosis. Patients need to be followed up closely, and if necessary, rhG-CSF should be used to improve neutropenia. In the present case, the boy was discharged after one week of anti-infective treatment and nebulization, and the lung infection was cured. Regular follow-up and rehabilitation were recommended. However, the family did not undertake rehabilitation due to financial constraints. The boy was admitted to the hospital with coughing and wheezing more than a month later and was discharged after one week of anti-infective treatment and human immunoglobulin immune support. Currently, the boy is undergoing regular follow-ups. For the treatment of children with Cohen syndrome, active and standardized rehabilitation training measures should be taken, which can effectively improve the retarded motor development, and long-term follow-up and tracking of the complications are needed for early intervention to improve the quality of life of these patients.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of First Affiliated Hospital of Nanchang University. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from guardian.
Consent to Publish
Patient’s guardian signed a document of informed consent.
Funding
No external funding received to conduct this study.
Disclosure
The authors declare that they have no competing interests.
References
1. Rodrigues JM, Fernandes HD, Caruthers C, et al. Cohensyndrome: review of the literature. Cureus. 2018;10(9):e3330. doi:10.7759/cureus.3330
2. Rejeb I, Jilani H, Elaribi Y, et al. First case report of Cohen syndrome in the Tunisian population caused by VPS13B mutations. BMC Med Genet. 2017;18(1):134. doi:10.1186/s12881-017-0493-5
3. Zhao S, Luo Z, Xiao Z, et al. Case report: two novel VPS13B mutations in a Chinese family with Cohen syndrome and hyperlinear palms. BMC Med Genet. 2019;20(1):187. doi:10.1186/s12881-019-0920-x
4. Yang C, Hou M, Li Y, et al. Gene analysis: arare gene disease of intellectual deficiency-Cohen syndrome. Int J Dev Neurosci. 2018;68:83–88. doi:10.1016/j.ijdevneu.2018.05.004
5. Karimzadeh MR, Omidi F, Sahebalzamani A, Saeidi K. A novel VPS13B mutation identified by whole-exome sequencing in Iranian patients with Cohen syndrome. J Mol Neurosci. 2021;71(12):2566–2574. doi:10.1007/s12031-021-01852-4
6. Zhang F, Shi XY, Liu LY, et al. [Children’s psychomotor retardation with neutropenia for more than one year.]. Zhong Guo Dang Dai Er Ke Za Zhi. 2018;20(6):497–500. Chinese.
7. Yin LH, Cheng MM, Wang WX, et al. [Cohen syndrome: a case report and literature review.]. Zhong Hua Shi Yong Er Ke Lin Chuang Za Zhi. 2016;31(19):1498–1499. Chinese.
8. Liao XY, Qiu KY, Tan LJ, et al. [Cohen syndrome and literature review.]. Zhong Hua Fu You Lin Chuang Yi Xue Za Zh. 2020;16(1):59–66. Chinese.
9. Cao YH, ZHang GY, Cao KF, et al. [A case report of Cohen syndrome caused by complex heterozygosity variation of VPS 13b gene and review of literature]. Lin Chuang Er Ke Za ZHi. 2020;38(10):777–780. Chinese.
10. Zhou JL, HUang HL, Wen FY, et al. [Severe congenital neutropenia: a case report and literature review]. Lin Chuang Er Ke Za Zhi. 2020;38(1):61–64. Chinese.
11. Cohen MM
12. Koehler K, Schuelke M, Hell AK, et al. A novel homozygous nonsense mutation of VPS13B associated with previously unreported features of Cohen syndrome. Am J Med Genet A. 2020;182(3):570–575. doi:10.1002/ajmg.a.61435
13. Boschann F, Fischer-Zirnsak B, Wienker TF, et al. An intronic splice site alteration in combination with a large deletion affecting VPS13B (COH1) causes Cohen syndrome. Eur J Med Genet. 2020;63:103973. doi:10.1016/j.ejmg.2020.103973
14. Enomoto Y, Tsurusaki Y, Yokoi T, et al. CNV analysis using whole exome sequencing identified biallelic CNVs of VPS13B in siblings with intellectual disability. Eur J Med Genet. 2020;63:103610. doi:10.1016/j.ejmg.2018.12.015
15. Jones KLJM, Del Campo MG. Cohen Syndrome. Smith’s Recognizable Patterns of Human Malformation.
16. Kaushik P, Mahajan N, Girimaji SC, et al. Whole exome sequencing identifies a novel homozygous duplication mutation in the VPS13B gene in an Indian family with Cohen syndrome. J Mol Neurosci. 2020;70:1225–1228. doi:10.1007/s12031-020-01530-x
17. Rafiq MA, Leblond CS, Saqib MA, et al. Novel VPS13B mutations in three large Pakistani Cohen syndrome families suggests a Baloch variant with autistic-like features. BMC Med Genet. 2015;16:41. doi:10.1186/s12881-015-0183-0
18. Lhussiez V, Dubus E, Cesar Q, et al. Cohen Syndrome-associated cataract is explained by VPS13B functions in lens homeostasis and is modified by additional genetic factors. Invest Ophthalmol Vis Sci. 2020;61(11):18. doi:10.1167/iovs.61.11.18
19. Duplomb L, Rivière J, Jego G, et al. Serpin B1 defect and increased apoptosis of neutrophils in Cohen syndrome neutropenia. J Mol Med (Berl). 2019;97(5):633–645. doi:10.1007/s00109-019-01754-4
20. Momtazmanesh S, Rayzan E, Shahkarami S, et al. A novel VPS13B mutation in Cohen syndrome: a case report and review of literature. MC Med Genet. 2020;21:140. doi:10.1186/s12881-020-01075-1
21. Kolehmainen J, Black GC, Saarinen A, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72(6):1359–1369. doi:10.1086/375454
22. Lee Y-K, Lee S-K, Choi S, et al. Autophagy pathway upregulation in a human iPSC-derived neuronal model of Cohen syndrome with VPS13B missense mutations. Mol Brain. 2020;13:69. doi:10.1186/s13041-020-00611-7
23. Alipour N, Salehpour S, Tonekaboni SH, et al. Mutations in the VPS13B gene in Iranian patients with different phenotypes of Cohen syndrome. J Mol Neurosci. 2020;70:21–25. doi:10.1007/s12031-019-01394-w
24. Duplomb L, Duvet S, Picot D, et al. Cohen syndrome is associated with major glycosylation defects. Hum Mol Genet. 2014;23(9):2391–2399. doi:10.1093/hmg/ddt630
25. Seifert W, Kühnisch J, Maritzen T, et al. Cohen syndrome associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J Biol Chem. 2015;290(6):3349–3358. doi:10.1074/jbc.M114.608174
26. Seifert W, Kühnisch J, Maritzen T, et al. Cohen syndrome associated protein, COH1 is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem. 2011;286(43):37665–37675. doi:10.1074/jbc.M111.267971
27. Kolehmainen J, Wilkinson R, Lehesjoki AE, et al. Delineation of Cohen syndrome following a large-scale genotypephenotype screen. Am J Hum Genet. 2004;75(1):122–127. doi:10.1086/422197
28. Chen J, Meng Y, Wang B, et al. [12q14 microdeletion syndrome: a case report and literature review]. Lin Chuang Er Ke Za Zhi. 2020;38(11):872–876. Chinese.
29. Massa G, Dooms L, Vanderschueren-Lodeweyckx M. Growth hormone deficiency in a girl with the Cohen syndrome. J Med Genet. 1991;28(1):48–50. doi:10.1136/jmg.28.1.48
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.