Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Identification of a Novel Mutation of Extracellular Matrix Protein 1 Gene in a Chinese Family with Lipoid Proteinosis
Authors Xu M, Zhou J ![]() , Yan J, Wang J
, Yan J, Wang J
Received 4 April 2023
Accepted for publication 7 June 2023
Published 14 June 2023 Volume 2023:16 Pages 1515—1519
DOI https://doi.org/10.2147/CCID.S415682
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Mengjun Xu, Jiong Zhou, Jianliang Yan, Jianyou Wang
Department of Dermatology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
Correspondence: Jianyou Wang, Department of Dermatology, Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, 310009, People’s Republic of China, Email [email protected]
Abstract: Lipoid proteinosis (LP) is a rare autosomal recessive disorder caused by mutations in extracellular matrix protein 1 (ECM1), a glycoprotein expressed in skin. Whole-exome sequencing (WES) was used to investigate two Chinese siblings with suggestive clinical features of LP. They shared one known (c.960G>A) and one novel (c.1081G>T) pathogenic variant in ECM1 gene, inherited from their unaffected parents. The novel mutation (c.1081G>T) led to a termination codon at position 361 and caused nonsense-mediated mRNA decay and lost the function. Our finding expands the genetic etiology spectrum of LP.
Keywords: lipoid proteinosis, mutation, extracellular matrix protein 1, ECM1
Introduction
Lipoid proteinosis (LP), also known as hyalinosis cutis et mucosae or Urbach-Wiethe disease, is a rare autosomal recessive disorder characterized by skin or mucosa infiltration and hoarseness.1 Clinical manifestations differ in individuals with different degrees of organ involvement. Pathological feature of LP is widespread deposition of eosinophilic hyaline-like material in dermal-epidermal junction, blood vessels and eccrine sweat glands. The incidence is unknown, yet about 300 cases have been reported worldwide.
LP is caused by mutations in the gene encoding extracellular matrix protein 1 (ECM1), which is located on chromosome 1q21.2 ECM1 is a glycoprotein with a potential role in epidermal differentiation, basement membrane and extracellular matrix formation.3 To date, more than 50 mutations have been reported on the 1q21 chromosome in the ECM1 gene.4 Here, we reported a Chinese family with two siblings suffering from LP and uncovered a novel pathogenic ECM1 variant (c.1081G>T). The result provided genetic counseling for the LP family and extended the mutation spectrum of ECM1.
Methods
Patients and Ethics
The proband (II1) was a 6-year-old boy, experienced hoarseness and multiple skin-colored papules on both upper eyelids for 3 years. His elder sister (II2), a 10-year-old girl, also had a similar clinical manifestation since school age, without a hoarse voice. Clinical and lab examinations were performed. Their parents were not related and asymptomatic. No similar symptoms were reported from other distant relatives. Legal guardian of both probands provided written informed consent for the case details and images to be published. This study was approved by the Ethics Committee of the Second Affiliated hospital of Zhejiang University.
Whole-Exome Sequencing
Peripheral blood of all subjects was collected. Genomic DNA of all the family members was extracted using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Whole-exome sequencing (WES) was performed for the proband. WES data were processed using AfterQC to generate “clean reads” for further analysis. The “clean reads” were then aligned to the human genome reference (hg19) using the BWA (Burrows Wheeler Aligner) software. GATK (Genome Analysis Toolkit) software was used to detect copy number variants (SNVs) and indels. All SNVs and indels were filtered and estimated by 1000 Genomes (1000 human genome dataset), Genome AD (Genome Aggregation Database dataset) and ExAC (The Exome Aggregation Consortium dataset). The Human Gene Mutation Database (HGMD) and Clinvar Database were used to screen mutations reported in published studies. According to WES results, conventional Sanger sequencing was performed to validate the pathogenic mutations in all family members. Variants were classified following the American College of Medical Genetics and Genomics (ACMG) interpretation standards and guidelines.5
Results
Clinical Manifestations
Clinical features of both siblings are presented in Figure 1. The proband (II1) was a 6-year-old boy who had severe hoarseness since infancy. Laryngoscopy revealed vocal cord nodules 2 years ago. The growth and intellectual development were normal. Clinical examinations showed firm beaded papules on both upper eyelids (Figure 1A) and several isolated papules on the inside of the lower lip (Figure 1B). Neurological examinations were negative. His affected elderly sister (II2) also presented with similar clinical manifestations (Figure 1C–E) without history of hoarse voice. Laboratory tests for blood routine, liver and kidney function were normal in both two siblings. Their parents had no symptoms of lipoid proteinosis.
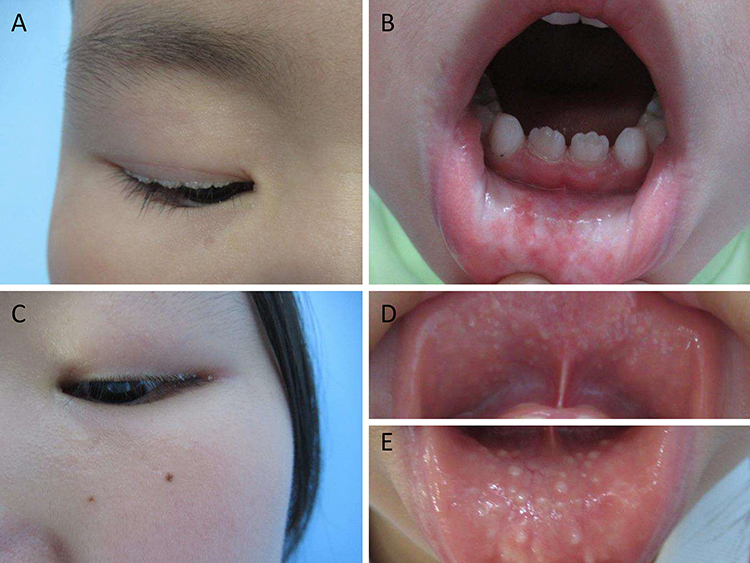 |
Figure 1 Clinical sign of the proband and his sister. (A) beaded papules on both upper eyelids and (B) isolated papules on the lip of the proband. (C) papules on the eyelids and (D and E) mucosa infiltration of his sister. |
Molecular Genetic Analysis
To identify the genetic lesion causing lipoid proteinosis in the family, gDNA samples of all family members were collected, and whole-exome sequencing was performed for the proband. Two candidate variants of the ECM1 gene were identified in the proband and his sister (Figure 2A and B). The first variant (c.960G>A) was inherited from the father, resulting in no amino acid change at position 320. This is a rare variant with 0.00002 in ExAC, 0.00001 in gnomAD and no referenced in 1000 Genomes. This variant has been previously reported to be associated with lipoid proteinosis in two Chinese families.6 This mutation site affected splicing.
 |
Figure 2 Pedigree and sequencing results. (A) Pedigree of the family with two affected siblings. (B) Sanger validation of the ECM1 variants. II1: proband, II2: sister, I1: father, I2: mother. |
The second variant (c.1081G>T) was inherited from the mother. It leads to a termination codon at position 361, which is expected to lead to a nonsense-mediated mRNA decay and lost the function. This variant was absent in population databases including 1000 Genomes, ExAC and gnomAD. According to the ACMG interpretation standards, c.1081G>T was classified as pathogenic variants (PVS1, PM2).
Discussion
Clinical manifestations of LP were highly variable, including a hoarseness of the voice, warty hyperkeratosis and scarring of the skin.7,8 The hoarseness of voice usually occurred during infancy caused by hyaline-like material deposited in the mucous membranes of the vocal cords. Mucosa infiltration of the pharynx, tongue, soft palate and lips can also be observed. In many cases, beaded papules on the eyelid margins were typical. These features could be found in our cases. Widespread hyaline-like material deposited in the papillary dermis and extending around sweat glands and blood vessels is the histologic feature of LP. Differential diagnoses include erythropoietic protoporphyria, lichen myxedematosus, and amyloidosis. The distribution of hyaline-like material and stain for PAS and amyloid were often used for differential diagnosis.
In 1997, Johnson et al9 first isolated human ECM1 gene and mapped it to chromosome 1q21. Three splice variants of ECM1 caused by two alternatively spliced exons (exon 5a and exon 7) were found, including ECM1a, ECM1b and ECM1c. All three variants were expressed in skin.10,11 However, the precise role of ECM1 in human skin is still not fully understood. The ECM1 protein is responsible for skin adhesion, epidermal differentiation, angiogenesis and basement membrane integrity in human skin.12 After secreting into the dermis, ECM1 acts as a “biological glue” binding to growth or differentiation factors and fibrillar proteins,13 and then regulates basement membrane function. Therefore, loss of function mutation of ECM1 gene leads to clinical features of scarring and skin infiltrations.
In the present study, we identified a known (c.960G>A) and a novel nonsense mutation (c.1081G>T) in the ECM1 gene in two Chinese siblings with LP. The new mutation 1081G>T in exon 7 leads to a premature termination codon which may cause severe phenotype of LP. Previous studies have shown that most pathologic mutations are located in exon 6 and 7. In a cohort of South African patients carrying a truncating mutation in exon 7 (Q276X), complete dermatological and neurological manifestation were exhibited.7 Meanwhile, in a Spanish family carrying nonsense mutation in exon 7 of ECM1 (c.1076G>A), hoarse voice, skin lesions and neurological symptoms were observed.14 However, it is difficult to study the genotype–phenotype correlation.
Conclusion
Our findings show a new pathogenic ECM1 mutations in a Chinese family with two siblings suspected with LP, which expand the mutation spectrum of the ECM1 gene.
Data Sharing Statement
Data are available on request from the corresponding author.
Consent Statement
Informed consent was provided by the patient for publication of the case.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82003332).
Disclosure
The authors declare that there is no conflict of interest.
References
1. Hofer PA. Urbach-wiethe disease (lipoglycoproteinosis; lipoid proteinosis; hyalinosis cutis et mucosae). A review. Acta Derm Venereol Suppl. 1973;53:1–52.
2. Hamada T, McLean WH, Ramsay M, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ecm1). Hum Mol Genet. 2002;11:833–840. doi:10.1093/hmg/11.7.833
3. Chan I, Liu L, Hamada T, Sethuraman G, McGrath JA. The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1. Exp Dermatol. 2007;16:881–890. doi:10.1177/2050313X19850359
4. Jahanimoghadam F, Hasheminejad J. Oral manifestations and dental management considerations of lipoid proteinosis: a case report and review of literature. J Dent. 2022;23:321–326. doi:10.30476/DENTJODS.2021.89748.1435
5. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi:10.1038/gim.2015.30
6. Yu W, Fu X, Sun L, Wang Z, Liu H, Zhang F. Mutation analysis of ECM1 gene in two families with lipoid proteinosis. China J Lepr Skin Dis. 2017;33(3):137–139.
7. Van Hougenhouck-Tulleken W, Chan I, Hamada T, et al. Clinical and molecular characterization of lipoid proteinosis in namaqualand, South Africa. Br J Dermatol. 2004;151:413–423. doi:10.1111/j.1365-2133.2004.06076.x
8. Nanda A, Alsaleh QA, Al-Sabah H, Ali AM, Anim JT. Lipoid proteinosis: report of four siblings and brief review of the literature. Pediatr Dermatol. 2001;18:21–26. doi:10.1046/j.1525-1470.2001.018001021.x
9. Johnson MR, Wilkin DJ, Vos HL, et al. Characterization of the human extracellular matrix protein 1 gene on chromosome 1q21. Matrix Biol. 1997;16:289–292. doi:10.1016/s0945-053x(97)90017-2
10. Mongiat M, Fu J, Oldershaw R, Greenhalgh R, Gown AM, Iozzo RV. Perlecan protein core interacts with extracellular matrix protein 1 (ecm1), a glycoprotein involved in bone formation and angiogenesis. J Biol Chem. 2003;278:17491–17499. doi:10.1074/jbc.M210529200
11. Smits P, Ni J, Feng P, et al. The human extracellular matrix gene 1 (ecm1): genomic structure, cDNA cloning, expression pattern, and chromosomal localization. Genomics. 1997;45:487–495. doi:10.1006/geno.1997.4918
12. Chan I. The role of extracellular matrix protein 1 in human skin. Clin Exp Dermatol. 2004;29:52–56. doi:10.1111/j.1365-2230.2004.01440.x
13. Smits P, Poumay Y, Karperien M, et al. Differentiation-dependent alternative splicing and expression of the extracellular matrix protein 1 gene in human keratinocytes. J Invest Dermatol. 2000;114:718–724. doi:10.1046/j.1523-1747.2000.00916.x
14. Mondejar R, Garcia-Moreno JM, Rubio R, et al. Clinical and molecular study of the extracellular matrix protein 1 gene in a Spanish family with lipoid proteinosis. J Clin Neurol. 2014;10:64–68. doi:10.3988/jcn.2014.10.1.64
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.