Back to Journals » International Journal of Nanomedicine » Volume 18
Identification and Characterization of a Novel Nanobody Against Human CTGF to Reveal Its Antifibrotic Effect in an in vitro Model of Liver Fibrosis
Authors Liu R, Zhu M, Chen J, Gai J, Huang J, Zhou Y, Wan Y, Tu C
Received 1 July 2023
Accepted for publication 14 September 2023
Published 21 September 2023 Volume 2023:18 Pages 5407—5422
DOI https://doi.org/10.2147/IJN.S428430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Rong Liu,1,* Min Zhu,2,* Jiaojiao Chen,3 Junwei Gai,2 Jing Huang,2 Yingqun Zhou,1 Yakun Wan,2 Chuantao Tu4
1Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, 200072, People’s Republic of China; 2Shanghai Novamab Biopharmaceuticals Co., Ltd, Shanghai, 201318, People’s Republic of China; 3Department of Gastroenterology, Shanghai Fourth People’s Hospital, Tongji University School of Medicine, Shanghai, 200434, People’s Republic of China; 4Department of Gastroenterology, Shanghai Public Health Clinical Center, Fudan University, Shanghai, 201508, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chuantao Tu; Yakun Wan, Email [email protected]; [email protected]
Background: No agents are currently available for the treatment or reversal of liver fibrosis. Novel antifibrotic therapies for chronic liver diseases are thus urgently needed. Connective tissue growth factor (CTGF) has been shown to contributes profoundly to liver fibrogenesis, which makes CTGF as a promising target for developing antifibrotic agents.
Methods: In this study, we identified a novel nanobody (Nb) against human CTGF (anti-CTGF Nb) by phage display using an immunized camel, which showed high affinity and specificity in vitro. LX-2 cells, the immortalized human hepatic stellate cells, were induced by transforming growth factor beta1 (TGFβ 1) as an in vitro model of liver fibrosis to verify the antifibrotic activity of the anti-CTGF Nb.
Results: Our data demonstrated that anti-CTGF Nb effectively alleviated TGFβ 1-induced LX-2 cell proliferation, activation, and migration, and promoted the apoptosis of activated LX-2 cells in response to TGFβ 1. Moreover, the anti-CTGF Nb remarkably reduced the levels of TGFβ 1, Smad2, and Smad3 expression in LX-2 stellate cells stimulated by TGFβ 1.
Conclusion: Taken together, we successfully identified a novel Nb against human CTGF, which exhibited antifibrotic effects in vitro by regulating the biological functions of human stellate cells LX-2.
Keywords: nanobody, connective tissue growth factor, liver fibrosis, hepatic stellate cells
Graphical Abstract:

Introduction
Chronic liver injury of various etiologies can lead to liver fibrosis characterized by excessive accumulation of extracellular matrix (ECM) in the liver.1–3 If remain unresolved, liver fibrosis may progress to cirrhosis, liver failure, and even hepatocellular carcinoma (HCC).1 Chronic fibrotic liver disease and cirrhosis represent a major global health challenge. However, there are currently no approved therapeutic agents to treat or reverse liver fibrosis.1,2 Therefore, a deep understanding of molecular mechanisms underlying hepatic fibrogenesis is needed to identify potential targets for antifibrotic therapies.
Hepatic stellate cells (HSCs) have been identified as the primary precursor cells of myofibroblasts (MFBs), which orchestrate the deposition of ECM in normal and fibrotic liver.1–3 Upon liver injury, hepatocytes and nonparenchymal cells, such as Kupffer cells, endothelial cells, and neutrophil granulocytes, release cytokines and chemokines, ultimately activate HSCs to release their vitamin A and retinoid stores, and undergo dramatic phenotypic alterations, and transdifferentiate into MFBs.1,3–5 Furthermore, the activated MFBs exhibit a variety of cellular processes such as proliferation, migration, contractility, and apoptosis resistance, accompanied by secretion of cytokines, chemokines, and ECM components.4 Thus, targeting the activation of MFBs are emerging as a high potential a strategy for treating liver fibrosis.1,6
Transforming growth factor beta (TGFβ) signaling is considered as a master profibrogenic pathway responsible for ECM production and plays a key role in the pathogenesis of liver fibrosis.7–9 TGFβ1 is the principal isoform of TGFβ which mediates SMAD2/3 dependent pathway, commonly activating HSCs and stimulating the production of ECM synthesis.7,8 Therefore, blocking TGFβ signaling represents an attractive antifibrotic therapy.5,7 However, TGFβ is a multifunctional cytokine and has important roles in liver physiology, and its inhibition results in undesirable side effects,5,7,10 which is a major challenge for translation into use of the clinic. Therefore, an improved understanding of the downstream regulatory mechanisms of TGF-β may help develop therapeutic strategies against liver fibrosis.7
Connective tissue growth factor (CTGF), also known as cellular communication network-2 (CCN2), is considered a downstream mediator of the effects of TGFβ on fibroblasts.7–9 A growing body of evidence has revealed that CTGF contributes to ECM production through several mechanisms including increasing cell proliferation, differentiation, adhesion, and migration, as well as directing interaction with matrix components.3,6,9 Moreover, CTGF acts synergistically with TGFβ to promote matrix protein deposition and fibrogenesis both in vitro and in vivo.11 Although the level of CTGF expression is low in healthy livers of human and mice, it is strikingly enhanced in fibrotic liver diseases of various etiologies.11–14 Thus, CTGF might be an attractive target for antifibrotic therapy in chronic liver diseases. Indeed, several animal model studies have shown that inhibition of CTGF limited liver fibrosis, such as pharmacological agents,11,14 targeted siRNA approaches,15 vaccination against CTGF,16 and neutralizing antibody.17 Notably, Pamrevlumab (FG-3019), a fully recombinant human monoclonal antibody against CTGF, has emerged as a potential agent for idiopathic pulmonary fibrosis.18 However, these strategies still face a number of obstacles for clinical application, such as off-target effects, safety, and tolerability.18,19 Meanwhile, the advantages of reduced size of therapeutic identities, in most cases, are offset by poor in vivo stability, which causes polymerization, lower binding affinity, and issues with mass manufacturing.20
Nanobodies (Nbs) have been used as imaging agents and therapeutics in preclinical and clinical studies.21–25 Nbs are novel type of single-domain antibody fragments (VHHs) derived from natural heavy chain-only antibodies in the Camelidae family of mammals.21,24 It possesses several advantages for therapeutic applications including tumor targeting, owing to low immunogenicity, high physicochemical stability and refolding capacity, and good tissue penetration. Moreover its antigen affinity is equal to that of conventional antibodies.22–25 Here, we constructed the a Nb against human CTGF (anti-CTGF Nb) which showed high affinity and specificity in vitro. Then, using human immortalized hepatic stellate cells LX-2, the feasibility of the anti-CTGF Nb as an antifibrotic agent was further confirmed.
Materials and Methods
Immunization and Library Construction
Human CTGF domain II (AA101-167) was optimized using a human codon and synthesized into the pFUSE vector (Invitrogen, Carlsbad, CA, USA), as previously described.21,24 CTGF domain II-Fc was expressed in HEK-293F cells (American Type Culture Collection) and purified by protein A affinity chromatography. The purified antigen was mixed with Freund’s adjuvant and immunized with two young camels. After seven injections, total RNA was isolated from peripheral blood lymphocytes (PBLs) of immunized camels. Reverse transcription was performed to acquire cDNA, and VHH was amplified using nested PCR. The pMECS phagemid vector and VHH were digested using restriction enzymes, and the digested vector was ligated with the target fragment. The ligation product was electrotransformed into competent Escherichia coli (E. coli) strain TG1 cells to construct a phage display library. Library capacity was measured by counting the number of monoclonal colonies grown using a gradient dilution. Twenty-four colonies were randomly selected from the library to estimate the correct insertion rate by PCR amplification.
Screening and Purification of Anti-CTGF Nb
Anti-CTGF Nb was screened through phage display bio-panning according to established methods.21,26,27 To identify positive colonies, individual colonies from the library were randomly selected for periplasmic extract ELISA verification. After sequencing of the screened colonies, the CTGF-specific Nb gene was amplified and subcloned into the pFUSE-mFc vector. VHH-mFc was expressed in HEK293F cells and purified by protein A affinity chromatography.
Affinity and Specificity Identification for Nanobodies
The affinity of anti-CTGF-mFc was detected by bio-layer interferometry (BLI) with a ForteBio Octet RED96 instrument (ForteBio, Menlo Park, CA, USA). The diluted biotinylated CTGF-Fc was coupled to SA biosensors and incubated with a series of diluted anti-CTGF Nb-mFc antibodies, followed by dissociation in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T). The binding curves were fitted to a 1:1 binding model using the Octet Data Analysis software 9.0. The association and dissociation rates were monitored and the equilibrium dissociation constant (Kd) was determined.
To determine the binding activities of anti-CTGF Nb, human and mouse recombinant CTGF-Fc (1 μg/mL in 100 μL per well) were captured in a 96-well plate overnight. After blocking with 1% BSA for 2 h and washing with TBS-T for four times, the biotinylated anti-CTGF Nb was diluted in gradient of 300 μL/mL and incubated in the relevant wells for 1 h at 37 °C, followed by SA-HRP for 1 h at 37 °C. Then, 100 μL 3,3′,5,5′-tetramethylbenzidine (TMB; Solarbio Biotechnology, Beijing, China) substrate was added to each well for 3–5 min at 37 °C. The reaction was stopped by the addition of H2SO4 (50 μL/well). Absorbance was measured at 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). For epitope competition assays, the procedure was the same as that mentioned above, except for mixing anti-CTGF Nb with the reference antibody pamrevlumab analog (FibroGen).
Immunohistochemistry
To test the binding of anti-CTGF Nb in liver sections, mouse liver samples from our recent study on nonalcoholic steatohepatitis (NASH) fibrosis were used.28 In brief, NASH fibrosis was induced by diets with a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) for 8 weeks, and a normal chow diet (NCD) as a control. Formalin-fixed, paraffin-embedded liver sections were pretreated with dewaxed, rehydrated, and heat-induced antigen retrieval, according to the standard protocol. After treatment with 3% hydrogen peroxide to block the endogenous peroxidase and 5% BSA to block the non-specific binding sites, staining for CTGF was performed overnight at 4 °C using the prepared anti-CTGF Nb (1:100). The sections were subsequently washed and incubated with HRP-conjugated goat anti-mouse IgG secondary antibodies (Jackson Immunoresearch Inc., USA; 1:500) for 1 h at room temperature (RT). DAB chromogenic reagent (AR1022; Boster; Wuhan, China) was added to visualize positive staining and hematoxylin was used to mark the nucleus. Images were captured under randomly non-overlapping fields (×10 and ×20) using a BX43 microscope (Olympus, Japan).
Cell Culture and in vitro Experimental Design
The human hepatic stellate cell line LX-2 was purchased from the Cell Bank of the Chinese Academy of Science (SCSP-527, Shanghai, China) and the cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U penicillin, and 100 μg streptomycin (all from Gibco BRL, CA) at 37 °C in a humidified atmosphere containing 95% O2 and 5% CO2. Cells were seeded into 6-well, 24-well, or 96-well plates for further studies after growth to the logarithmic stage. Recombinant TGFβ1 protein (YEASEN, Shanghai, China) is reconstituted in sterile 4 mM HCl to a concentration of 0.1 mg/mL and stored at −20 °C.
CCK-8 Cell Proliferation and Cytotoxicity Assay
Cell proliferation and cytotoxicity assays were conducted using Cell Counting Kit (CCK)-8-based spectrophotometric methods (Beyotime Institute Biotechnology, Shanghai, China) according to the manufacturer’s protocol. LX-2 cells were seeded in 96-well plates at a density of 2.0–5.0×104 cells/mL. TGFβ1 was diluted in a gradient (1, 2, 5, and 10 ng/mL) to determine a suitable concentration to induce LX-2 cell activation in our subsequent experiments, using different concentrations of anti-CTGF Nb (1, 5, and 10 μg/mL) or vehicle to treat LX-2 cells for another 24, 48, or 72 h. The effect of anti-CTGF Nb on LX-2 cell proliferation was explored and the cytotoxicity of anti-CTGF Nb was examined. 10 µL CCK-8 solution was added and the cells were incubated at 37 °C for 1 h before measuring the absorbance at 450 nm on a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Wound-Healing Assay
The effect of anti-CTGF Nb on the migratory behavior of LX-2 cells was identified using a wound-healing assay. The logarithmic phase LX-2 cells were seeded in 6-well plates (2×105 per well) and cultured with or without TGFβ1 (2 ng/mL) in a 37 °C incubator. After 24 h, anti-CTGF Nb (5 μg/mL) was added to the experimental group and incubated for another 24 h. Scratches were gently created with a 200 µL pipette tip, and the medium was replaced with serum-free medium after washing three times with PBS to remove non-adherent cells. Images of the wound areas were captured at 0 h and 24 h using a phase-contrast microscope. Cell migration was quantified by measuring the wound width. The migration rate was calculated as (%) = [W (24 h) − W (0 h)]/W (0 h).
Transwell Assay
The migration ability was further validated using Transwell chambers (Corning Costar, Cambridge, Massachusetts, pore size 8 μm). LX-2 cells were seeded in Transwell chambers in a 24-well plate (2×105/well) with or without TGFβ1 (2 ng/mL) and starved in DMEM for 24 h in advance. After reaching 80–90% confluence, the cells were digested, the cell suspension (200 μL) was added to the upper chamber, and DMEM containing 10% FBS (500 μL) was added to the lower chamber. The anti-CTGF Nb (5 μg/mL) or PBS was added to the upper chamber the following day. After incubation for another 24 h, the unmigrated cells were washed with a swab, fixed in 4% paraformaldehyde, and stained with 0.1% crystal violet for 15 min. Images of migrated cells were captured under a 100× microscope and counted in five randomly selected visual fields.
Flow Cytometric Analysis of Apoptosis
The extent of apoptosis was measured using an Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. After treatment with TGFβ1 (2 ng/mL) or vehicle for 24 h, anti-CTGF Nb (5 μg/mL) was added to the experimental group for another 24 h. LX-2 cells were harvested and resuspended in 195 μL Annexin V-FITC binding buffer, 5 μL Annexin V-FITC, and 10 μL propidium iodide (PI) solution, followed by incubation for 15 min in the dark at RT. Apoptosis of LX-2 cells was detected using a flow cytometer (ACEA Biosciences, San Diego, CA, USA). The fraction of the cell population in the different quadrants was analyzed using quadrant statistics. The lower left quadrant contained intact cells, the lower right quadrant contained apoptotic cells, and the upper right quadrant contained necrotic and post-apoptotic cells.
Quantitative Real-Time PCR
Total cellular RNA was extracted using an RNAeasyTM RNA isolation kit with a spin column (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. cDNA was prepared using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and quantitative real-time PCR was performed using a SYBR Green PCR Master Mix Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and a CFX Connect Real-Time PCR system (Bio-Rad, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard, and the relative expression of target gene mRNA was calculated using the 2−ΔΔCt method. Primer sequences used in this study are listed in Table 1.
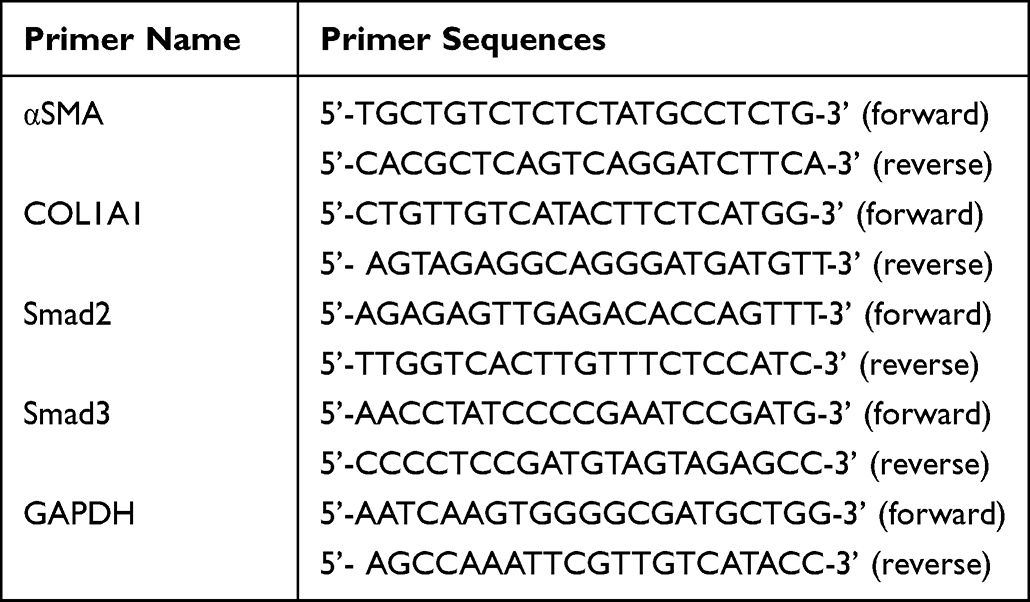 |
Table 1 Primer Sequences Used for Quantitative RT-PCR in This Study |
Western Blot Analysis
Cell lysates were extracted from LX-2 cells using a radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). The protein concentration was quantified using a modified bicinchoninic acid (BCA) protein concentration assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of protein (30 µg) were separated on a 7.5–12.5% SDS-PAGE gel and then transferred onto polyvinylidene (PVDF) membranes (Bio-Rad, California, USA). After blocking with 5% skim milk in TBS-T for 2 h, membranes were incubated with primary antibodies (1:1000 dilution) in the same buffer overnight at 4 °C. The primary antibodies used were anti-αSMA (Proteintech, Wuhan, China), anti-collagen I (ab6308, Abcam Inc., Cambridge, MA, USA), anti‑MMP9 (Servicebio Technology, Co., Ltd, Wuhan, China), anti‑Smad2 (Proteintech, Wuhan, China), anti‑Smad3 (ab4084, Abcam Inc., Cambridge, MA, USA), and anti-β-actin (Proteintech, Wuhan, China). After washing with TBST, the membranes were incubated with appropriate secondary antibodies for 1 h at RT: HRP-conjugated goat anti-rabbit (1:2000; Proteintech, Wuhan, China) or HRP-conjugated goat anti-mouse (1:3000; Proteintech, Wuhan, China). BeyoECL Plus (Cat #P0018, Beyotime) was used to develop images using a Tanon 5200 machine (Tanon Science and Technology Co., Ltd., Shanghai, China). For semi-quantitative analysis, images were acquired by microscopy and analyzed using the ImageJ V1.8.0 software (National Institutes of Health, Bethesda, MD, USA). Relative protein abundance in each sample was normalized to that of β-actin.
Immunofluorescence
For immunofluorescence staining, LX-2 cells treated with or without TGFβ1 (2 ng/mL) were seeded into 24-well plates and cultured with or without anti-CTGF Nb (5 μg/mL) for 24 h. The LX-2 cells were fixed in 4% paraformaldehyde for 30 min before treatment with 0.5% Triton X-100 for 15 min at RT and then blocked with 3% BSA for 1 h to avoid non-specific staining. Next, the cells were incubated with 1:500 diluted anti-CTGF Nb and anti-αSMA antibodies (Proteintech, Wuhan, China) at 4 °C overnight, followed by Alexa Fluor 488-labeled (1:500; ab234075, Abcam Inc., Cambridge, MA, USA) or Alexa Fluor 594-labeled (1:500; ab150116, Abcam Inc., Cambridge, MA, USA) goat anti-mouse secondary antibodies for 1 h at RT, and stained with 1 μg/mL DAPI (C1002; Beyotime; Shanghai, China) for 5 min. Images were obtained using a fluorescence microscope (Nikon).
Public Datasets Analysis
Four Gene Expression Omnibus (GEO) datasets, GSE68000, GSE155017, GSE68001, and GSE49301, were downloaded from the GEO website (https://www.ncbi.nlm.nih.gov/geo/). A p value less than 0.05, and |log2-fold change (FC)| > 1 were considered statistically significant. The results of differentially expressed genes (DEGs) are presented using heatmaps and volcano maps.
Statistical Analysis
Data are expressed as mean ± standard deviation (SD). Comparisons between two groups were performed using Student’s t-test (unpaired, two-tailed). Differences between multiple groups were determined by one-way analysis of variance (ANOVA), followed by Tukey’s post-hoc test. All analyses were performed using the GraphPad Prism 9.4.1 software (La Jolla, CA, USA). Differences were considered statistically significant at p < 0.05. All our Western blots and cell culture experiments were repeated independently at least 3 times with similar results.
Results
CTGF VHHs Were Screened from the High Quality Libraries by Phage Display Technology
CTGF domain II fused to Fc (CTGF domain II-Fc) was purified as an antigen using the HEK-293F eukaryotic expression system (Figure 1A). After immunization and RNA extraction from PBLs, the VHHs gene was amplified through the first and second PCR to produce ~700 bp fragments of VHH-h-CH2 (Figure 1B) and ~400 bp fragments of VHH (Figure 1C). Immunization and screening strategies are shown in Figure 1A. The capacity of libraries was determined by counting the number of colonies. The sizes of the library 1 and 2 were measured at 4.0×108 and 1.0×109 colony-forming units (CFU), respectively (Figure 1D). Individual colonies from libraries 1 and 2 were randomly selected for PCR analysis, and the results showed that the library insertion rates were 100% (Figure 1E), suggesting that high-quality phage display libraries for anti-CTGF VHHs were obtained.
 |
Figure 1 Construction and screening of VHH library. (A) Schematic depictions of the immunization and screening strategy used to isolate anti-CTGF Nbs. PBLs, peripheral blood lymphocytes. The cropping gel of purified CTGF-domain II-Fc antigen is displayed. (B) The segments containing VHH gene fragments were amplified by a first PCR. (C) The fragments were then amplified by a second PCR. (D) The capacity of library 1 and 2 was estimated by counting colony numbers after serial dilutions and plating on plates containing selective antibodies. (E) The correct insertion rates of library 1 and 2 were estimated by performing PCR on randomly selected 24 colonies. Clones were randomly selected to detect the percentage of clones with a phagemid containing an insert of a proper size for a VHH. (F) The enrichment for phage particles of library 1 and 2 was detected after three consecutive rounds of panning. Land M DNA marker. +: positive screened phages transformed into TG1 cells after panning with CTGF-Fc. -: negative screened phages panning with Fc were used as control. |
The CTGF-specific Nb library was identified by biopanning the phage-display library. To evaluate the screening effect, positively screened phage (phage specifically bound to antigen) and negatively screened phage (phage bound to Fc) were infected with E. coli TG1 cells in a logarithmic growth phase, the bacterial solution was cultured in serial gradient dilutions, and the library enrichment fold (+/−) was calculated as the ratio of the number of colonies. After three consecutive rounds of panning, the phage particles in the two libraries were gradually enriched 68 and 71 folds, respectively (Figure 1F). PE-ELISA was performed to detect positive clones in randomly selected colonies. Positive clones were sequenced and nanobodies from different families were fused with mouse Fc in the HEK-293F expression system.
Functional Activity Determination of the Anti-CTGF Nb
SDS-PAGE analysis showed that the Nbs had a single band of high purity (Figure 2A). In the affinity assay, we showed that the KD value of the anti-CTGF Nb was 1.86E-09, implying that the anti-CTGF Nb exhibits a high affinity towards CTGF (Figure 2B). To assess the function of the anti-CTGF Nb, the binding ability of CTGF in humans and mice was determined. The results showed that anti-CTGF Nb exhibited high binding activity to both human and mouse CTGF with EC50 values of 33.15 ng/mL and 29.18 ng/mL, respectively (Figure 2C). In the epitope competition assay (Figure 2D), the anti-CTGF Nb exhibited a comparable affinity and similar epitope to pamrevlumab (FG-3019), which has been established as a potential therapeutic option for patients with idiopathic pulmonary fibrosis in a Phase 2 trial.18,28
 |
Figure 2 Functional activity identification of anti-CTGF Nb. (A) SDS-PAGE gel analysis for the purified anti-CTGF Nb. Lane 1 was loaded with protein marker (M); lane 2 exhibited purified human CTGF-domain II-Fc antigen. The samples were separated by using 10% gradient PAGE gels and stained with Coomassie blue. (B) The affinity of CTGF nanobody was detected by Fortebio detection. The experiment was performed in triplicate and one representative experiment was shown. (C) The specificity of anti-CTGF Nb to human CTGF and mouse CTGF was detected by ELISA. (D) The epitope competition assays of CTGF nanobody with the reference antibody pamrevlumab analog. |
Effect of the Anti-CTGF Nb on Proliferation and Migration of LX-2 Cells Stimulated by TGFβ1
TGFβ1 is a master fibrogenic cytokine that contributes to the trans-differentiation of the LX-2 human HSC cell line, and TGFβ1 was introduced to establish an in vitro model of liver fibrosis.2,29 In the present study, cell viability was determined to verify whether the anti-CTGF Nb can limit the growth and proliferation of LX-2 cells in response to TGFβ1. First, a gradient concentration of TGFβ1 (1, 2, 5, and 10 ng/mL) was used to determine the optimal concentration in this experimental study. Compared with the vehicle-treated groups, the proliferation of TGFβ1-treated LX-2 cells increased in an approximately time-dependent manner and reached its maximum after treatment with 2 ng/mL TGFβ1 for 48 h. However, after inducing higher concentrations of TGFβ1 (5–10 ng/mL), the viability of LX-2 cells tended to decrease (Figure 3A), which may be attributed to the excessive cytotoxic effects of high-dose TGFβ1 stimulation. To assess the effect of the anti-CTGF Nb on the proliferation of LX-2 cells, different concentrations of the anti-CTGF Nb (1, 5, and 10 μg/mL) were added to LX-2 cells after treating with 2 ng/mL TGFβ1 for 24 h. Treatment with anti-CTGF Nb markedly alleviated the proliferation of LX-2 cells stimulated with TGFβ1 in a concentration-dependent manner (Figure 3B). In line with this result, microscopic observation showed that LX-2 cells tended to display quiescent morphological alterations after treatment with the anti-CTGF Nb compared to the vehicle-treated and TGFβ1-stimulated groups (Figure 3C).
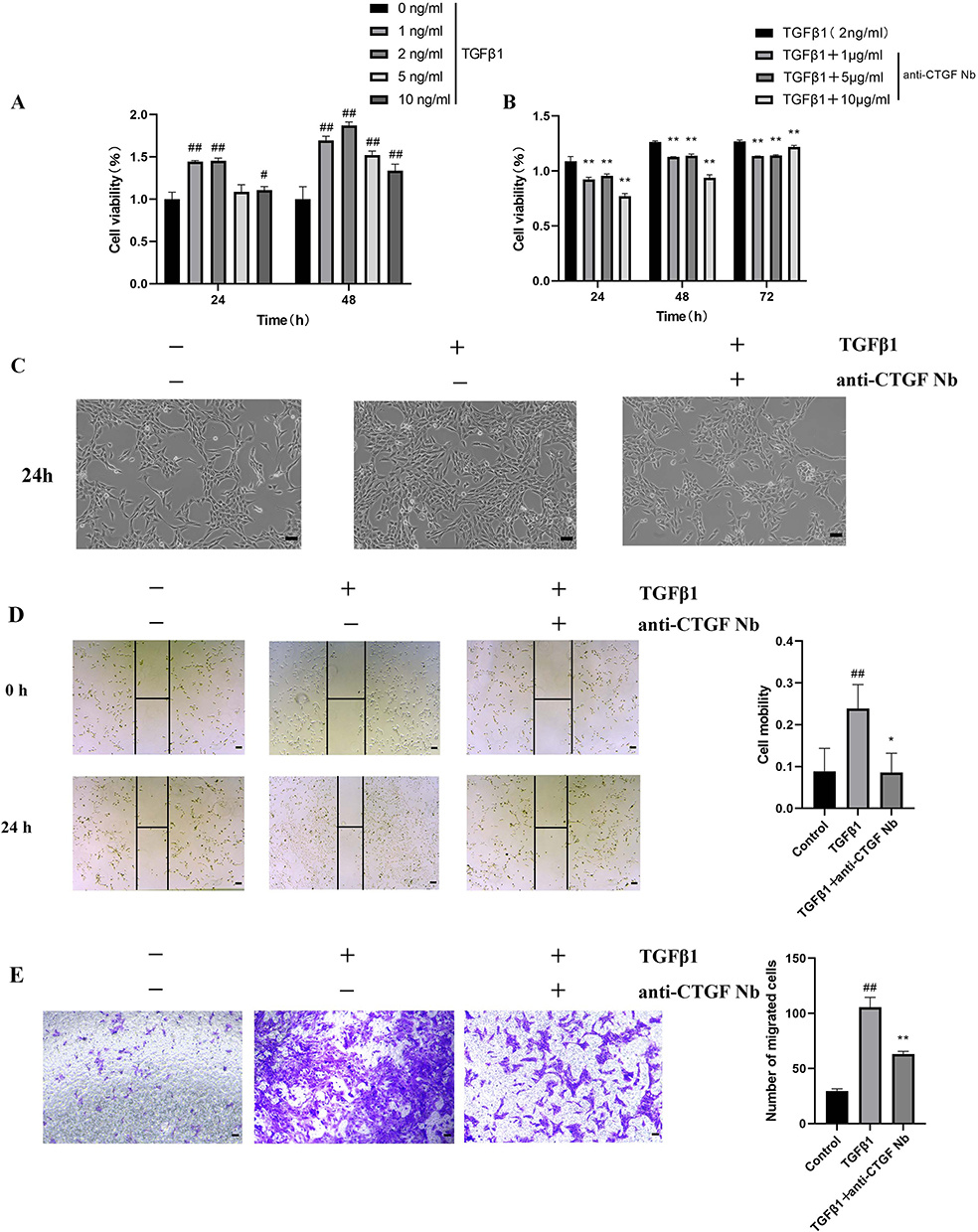 |
Figure 3 Effect of anti-CTGF Nb on proliferation and migration of LX-2 cells treated with TGFβ1. (A) CCK-8 assays showed proliferation change of cells. LX-2 cells were treated with the increased concentration of TGFβ1 (1, 2, 5, and 10 ng/mL) or an equal volume of PBS for 24 h. (B) Effect of anti-CTGF Nb at various concentrations (1, 5, and 10 μg/mL) on the proliferation of TGFβ1-treated LX-2 cells was determined by CCK-8 assay after 24, 48 or 72 h. (C) Morphology of LX-2 cells under different treatments for 24 h. (D) The migration rate of cells was detected by wound-healing test. Representative microphotographs showing the wound closure distance at 0h and 24 h post wounding. Dashed lines indicate initial wounds. Morphometrical analysis was performed to measure the remnant wounding area. The migration rate is shown as % of initial wounding area. (E) Cell mobility was detected by Transwell assay. Scale bars: 100 μm. All data are presented as mean ± SD from three independent experiments. #p < 0.05, ##p < 0.01 vs the control group; *p < 0.05, **p < 0.01 vs the TGFβ1 group. |
As activated HSCs acquire migratory ability, the potential effect of the anti-CTGF Nb on this parameter was assessed by wound healing and Transwell assays. The wound-healing assay demonstrated that the migration rate of LX-2 cells increased remarkably after stimulated with TGFβ1 (2 ng/mL) for 24 h compared to that of vehicle-treated cells, whereas a lower migration rate was observed when 5 μg/mL anti-CTGF Nb co-existed (Figure 3D). In parallel, the Transwell migration assay also confirmed the positive effect of the anti-CTGF Nb in reducing the migration of LX-2 cells stimulated by TGFβ1. But, a significant increase in the number of migrated cells was observed in the TGFβ1-stimulated groups, which remarkably decreased when the cells were subsequently incubated with the anti-CTGF Nb (5 μg/mL) (Figure 3E). These results suggest that anti-CTGF Nb limits the migration of LX-2 cells in response to TGFβ1.
Taken together, these results indicated that the anti-CTGF Nb strikingly inhibited the proliferation and migration of LX-2 cells treated with TGFβ1 in vitro.
Effects of the Anti-CTGF Nb on the Activation of LX-2 Cells and Fibrogenesis
To identify the antifibrotic activity of the anti-CTGF Nb in LX-2 cells in vitro, we measured the expression of fibrotic markers including collagen type 1 alpha 1 (COL1A1), αSMA, and MMP9. As expected, TGFβ1 (2 ng/mL) led to the activation of LX-2 cells and subsequently increased the protein levels of αSMA, COL1A1, and MMP9, which further triggered a positive feedback loop to accelerate the progression of fibrosis. Notably, the expression of these proteins failed to up-regulate or even decrease in TGFβ1-treated LX-2 cells when anti-CTGF Nb (5 µg/mL) was present (Figure 4A). In addition, TGFβ1 also enhanced the expression of Smad2 and Smad3 protein; however, treatment with the anti-CTGF Nb (5 µg/mL) resulted in the decreased expression of these two proteins (Figure 4B), indicating an inhibitory effect on the TGFβ1-induced Smad2/3 pathway by the anti-CTGF Nb. Similarly, TGFβ1-activated LX-2 cells showed up-regulation of mRNA transcript levels of COL1A1, αSMA, Smad2, and Smad3, which was abrogated following anti-CTGF Nb (5 µg/mL) treatment (Figure 4C). The antifibrotic effects of the anti-CTGF Nb (5 µg/mL) were also validated by immunofluorescence staining for CTGF and α-SMA (Figure 4D and E). Taken together, our results revealed that the anti-CTGF Nb had an inhibitory effect on the activation of LX-2 cells and subsequently limited the production of ECM.
 |
Figure 4 Effects of anti-CTGF Nb on the activation of LX-2 cells and fibrogenesis. (A and B) Western blot analysis for αSMA, COL1A1, MMP9, Smad2, and Smad3 in LX-2 cells; β-actin as loading control. (C) The mRNA levels of αSMA, COL1A1, Smad2, Smad3 were detected by qRT-PCR. Results were normalized to the GAPDH mRNA and expressed as fold change compared to control group. (D) Representative immunofluorescence staining of CTGF (green). Nuclei were stained with 4’, 6-diamidino-2-phenylindole (DAPI) (blue). Scale bars: 50 μm. (E) Representative immunofluorescence staining of αSMA (red). Nuclei were stained with DAPI (blue). Scale bars: 50 μm. ##p < 0.01 vs the control group; *p < 0.05, **p < 0.01 vs the TGFβ1 group. |
Effects of the Anti-CTGF Nb on Apoptosis of LX-2 Cells in Response to TGFβ1
To assess cellular tolerance, CCK-8 assay showed that pretreatment on unstimulated LX-2 cells with different dosage of anti-CTGF Nb (0, 1, 5 and 10 µg/mL) for 24 or 48 h did not significantly affect cells viability (Figure 5A). Then, the apoptosis of LX-2 cells was assessed by measuring the levels of cleaved caspase-3, the active form of caspase-3, using Western blot. Our results exhibited that the anti-CTGF Nb (5 µg/mL) treatment led to a higher expression of the apoptotic marker protein (cleaved caspase-3) (Figure 5B). Meanwhile, apoptotic LX-2 cells were also assessed by flow cytometry after cells stained with a combination of Annexin V/FITC and PI. Our results demonstrated that TGFβ1-stimulated LX-2 cells led to a relatively lower apoptosis rate compared with the vehicle-treated cells, which was significantly reversed and further induced after treatment with anti-CTGF Nb (5 µg/mL) (Figure 5C). Altogether, our results indicate that the anti-CTGF Nb promotes apoptosis of the activated LX-2 cells while free from cytotoxicity to LX-2 cells under quiescent conditions.
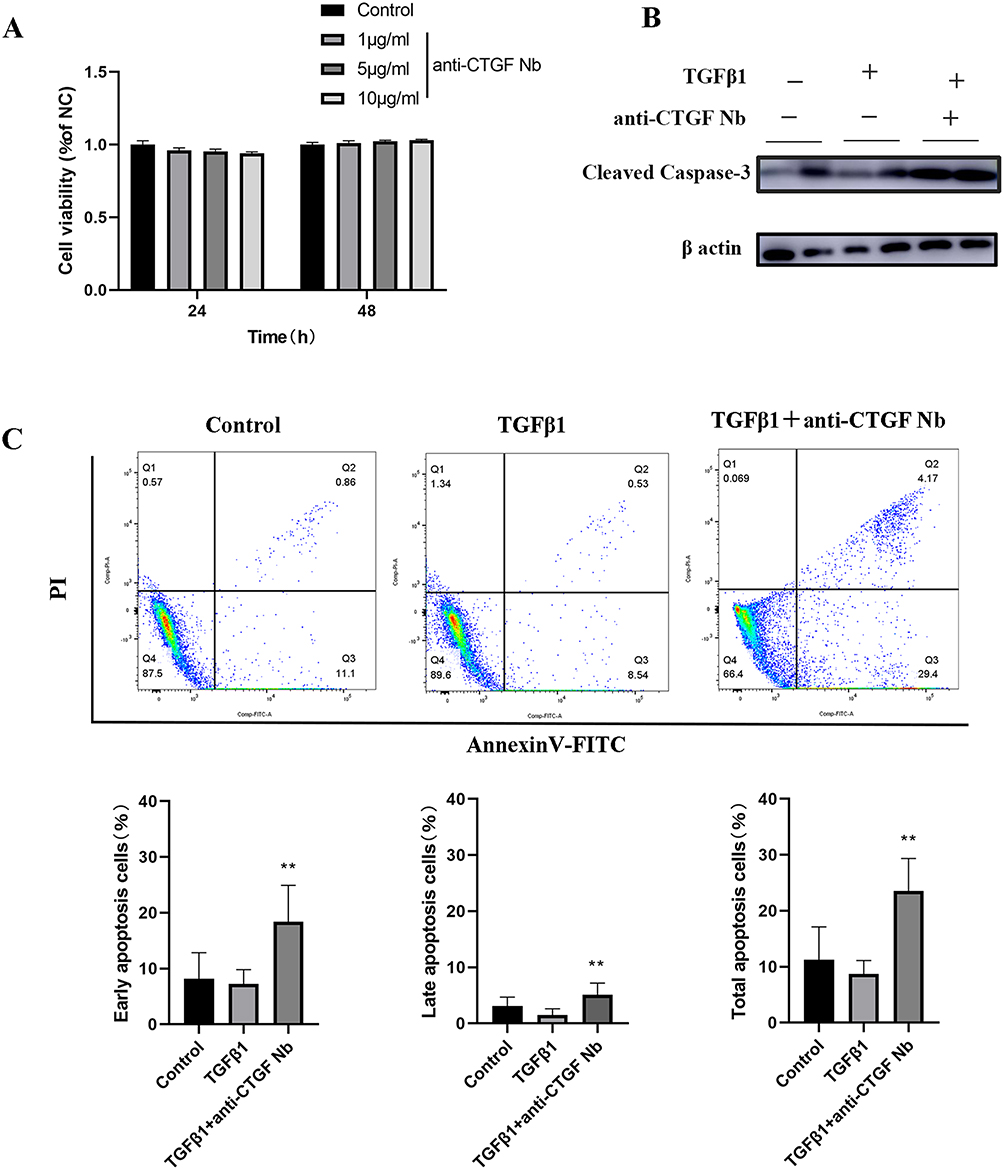 |
Figure 5 Effects of anti-CTGF Nb on apoptosis of LX-2 cells in response to TGFβ1. (A) The cytotoxic effect of anti-CTGF Nb in LX-2 cells was determined by CCK-8 assay at various concentrations (0, 1, 5, and 10 µg/mL) for 24 h and 48 h. (B) Representative Western blots showing cellular extracts cleaved caspase-3, and β-actin as loading control. (C) Anti-CTGF Nb (5 µg/mL) increased the rate of apoptosis in activated LX-2 cells in response to TGFβ1 (2 ng/mL) for 24 h. Cell apoptosis was detected using a Annexin V-FITC Apoptosis Detection Kit. The results are shown as the mean ± SD from three independent experiments. **p < 0.01 vs the TGFβ1 group. |
CTGF is Up-Regulated in Fibrotic Liver Samples from Patients and Mice
Histological alterations, collagen deposition, and CTGF expression in livers were detected through HE, Sirius red staining, and IHC. HE staining revealed that CDAHFD induced obvious hepatic steatosis, inflammation, and hepatocyte ballooning; Sirius red staining showed notable collagen accumulation (Figure 6A). Hepatic CTGF expression was significantly elevated and was predominantly distributed in the fibrogenic region around the portal area at the interface between the fibrous septa and liver parenchyma (Figure 6A). Of note, HSCs are considered the primary source of CTGF expression and are compressed by the endothelium and hepatocytes in the perisinusoidal space.
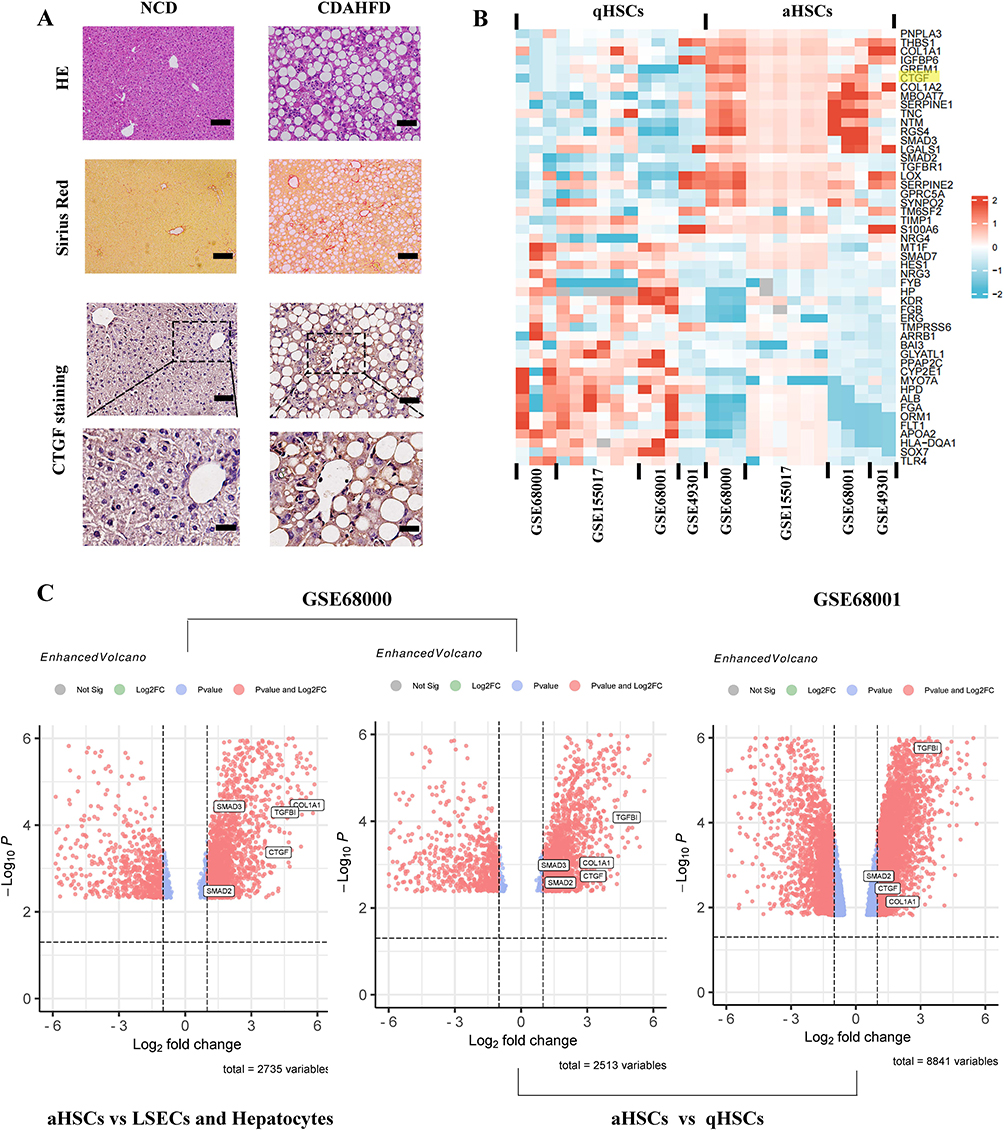 |
Figure 6 Expression and function identifications of CTGF in NASH liver fibrosis model of mice and human. (A) Histology of liver sections from NCD or CDAHFD-fed mice stained for CTGF, H&E, as well as Sirius red staining. Insert showing the typical CTGF-positive cells, lobular inflammation, and pericellular fibrosis. Scale bars: 100 μm (up 3 panels) and 200 μm (bottom panel). (B) Heatmap depicting the representative DEGs (sorted by fold change and p value) in the human quiescent HSCs (qHSCs) and human activated HSCs (aHSCs). CTGF is among the top DEGs and is highlighted in yellow. Database-based gene expression analysis was conducted using public data sets obtained from GEO at site the NCBI (http://www.ncbi.nlm.nih.gov/geo/). (C) Volcano plots of DEGs between human activated HSCs (aHSCs) and human hepatocytes, liver sinusoidal endothelial cells (LSECs) (from GSE68000), and DEGs between human activated HSCs (aHSCs) and human quiescent HSCs (qHSCs) (from GSE68000 and GSE68001 respectively). Representative fibrogenesis-related genes including CTGF are labeled in the plots. Abbreviations: NCD, normal chow diet; CDAHFD, choline-deficient, L-amino acid-defined, high-fat diet. |
We further excavated the GEO database to validate the overexpression of CTGF in activated HSCs from liver tissues of patients with NASH. The heatmap of the top DEGs indicated that CTGF was significantly up-regulated in human activated HSCs (aHSCs) compared with human quiescent HSCs (qHSCs) (p < 0.05) (Figure 6B). As shown in the volcano plots (Figure 6C), GSE68000 also indicated that CTGF was up-regulated in human aHSCs compared to human hepatocytes and liver sinusoidal endothelial cells (LSECs) (p < 0.05), which supports the notion that HSCs are the primary source of CTGF expression. Moreover, both GSE68000 and GSE68000 further validated the increased expression level of CTGF in human aHSCs compared with that in human qHSCs, the finding is consistent with the heatmap.
Discussion
Fibrotic liver disease and cirrhosis represent a massive global health concern with limited therapeutic agents.30 Consequently, there is an urgent need to develop curative options that can mitigate or reverse liver fibrosis.3,30 In this study, we successfully identified and screened stable nanobodies that specifically target human CTGF. Furthermore, the antifibrotic effect of the anti-CTGF Nb was characterized by regulation of the physiopathological function of LX-2 cells in an in vitro model of liver fibrosis.
CTGF/CCN2 has received considerable attention as a critical determinant of progressive fibrosis; and TGFβ is a master regulator of HSC activation and liver fibrogenesis and most TGFβ responses involved CTGF stimulation.9 Furthermore, CTGF functions broadly in liver fibrogenesis that mediates cell proliferation, differentiation, migration, adhesion, ECM production and remodeling.9 Therefore, blocking CTGF may provide a potential target for antifibrotic therapies.17–19
In the current study, VHHs genes were amplified from the mRNA of peripheral blood lymphocytes of immunized Bactrian camels. We obtained the anti-CTGF Nbs from a phage display library derived from camels immunized with human domain II of CTGF (von Willebrand type C repeat homology domain) (Figure 2A). Notably, CTGF/CCN2 is originally an inactive precursor that needs to undergo proteolytic processes to display the biological activity of signaling fragments.31,32 N-terminal CTGF is produced from CTGF by proteolytic cleavage in the linker region and is commonly observed in biological fluids such as plasma or urine.31,32 Indeed, FG-3019 targets domain II of CTGF, which has been validated for the inhibition of CTGF activities in experimental animal models of fibrotic diseases, as well as in clinical trials for kidney fibrosis and idiopathic pulmonary fibrosis.18,31,33 Previous studies have revealed that the C-terminal domain of CTGF is an attractive target for profibrotic disorders using neutralizing antibodies.32,34 Not surprisingly, this novel anti-CTGF Nb has a higher avidity binding to human CTGF, and camelid single-chain antibodies have exceptional properties, making them appropriate for treating fibrotic liver diseases. However, the combination of FG-3019 and CTGF in vivo have been demonstrated to be subject to highly rapid clearance, necessitating the administration of higher doses and/or more frequently.31 We further validated the high affinity and specificity of the identified nanobody targeting CTGF using Fortebio detection and ELISA, respectively (Figure 2B and 2C).
To characterize the antifibrotic effect of this novel anti-CTGF Nb, we used an in vitro liver fibrosis model to study.35 LX-2 cells consists of immortalized HSCs derived from normal human liver tissue, which are characterized by the expression of αSMA and the production of cytokines and collagen after activation.2,35 We found that nanobodies against CTGF inhibited the activation of LX-2 cells and limited the levels of fibrotic markers such as αSMA, COL1A1, and MMP9 as confirmed by Western blot (Figure 4). Thus, these results indicated that anti-CTGF Nb might exert striking effects against liver fibrosis in vitro. Furthermore, sustained activation of HSCs led to discrete alterations in cell behaviour, including proliferation, chemotaxis, fibrogenesis, contractility, matrix degradation, and chemoattractant/cytokine release.4 Our results exhibited that treatment with the anti-CTGF Nb inhibited TGFβ1-induced cellular proliferation and migration, supporting CTGF as a mitogenic regulator in LX-2 stellate cells (Figure 3). Since CTGF is believed to be responsible for the increased survival and resistance of MFBs to apoptotic stimuli in the state of persistent activation,4,19 blocking CTGF using this targeted-CTGF Nb led to the promotion of LX-2 stellate cell apoptosis, as confirmed by the up-regulation of the apoptotic marker cleaved caspase-3 and elevated Annexin V/PI staining (Figure 5). Interestingly, a previous study has revealed that pamrevlumab might down-regulate pro-survival signals in MFBs, thereby mitigating fibrotic and inflammatory responses.18
In this study, the levels of Smad2 and Smad3 were induced in TGFβ1 treated LX-2 cells but were dramatically repressed by anti-CTGF Nb (Figure 4B and C), suggesting that anti-CTGF Nb-mediated inhibition of liver fibrosis was associated with down-regulation of the TGFβ1/Smad2/3 pathway. It is well documented that the TGFβ1/Smads signaling pathway plays a crucial role in HSC activation during liver fibrogenesis.10 Upon phosphorylation by activated TGFβ1, Smad2 and Smad3 translocate into the nucleus and subsequently regulate the transcription of downstream fibrosis-related genes.36 In turn, CTGF has been shown to regulate TGFβ1 production and amplify the fibrogenic activity of TGFβ1, creating a positive feedback loop.37 Moreover, persistent liver inflammation led to this loop activation and uncontrolled, then subsequently induced a large amounts of ECM production.37 However, targeting CTGF with this nanobody could block the CTGF-TGFβ1 interaction by limiting Smad2 and Smad3 expression. Although we demonstrated that the anti-CTGF Nb had excellent antifibrotic effects in an in vitro experimental model using LX-2 stellate cells, further studies are needed to better characterize these Nbs and to assess their pharmacokinetics, efficacy, and safety in preclinical animal models.
Additionally, we used murine NASH fibrosis and healthy liver samples for immunostaining to visualize and assess the performance of our anti-CTGF nanobodies bound to liver sections. Our results showed that CTGF expression was remarkably higher in fibrotic livers than in normal livers (Figure 6A), and that CTGF staining was mainly located in the hepatic sinusoid. Moreover, animal model studies have confirmed that blocking CTGF using siRNA has a beneficial effect on liver fibrosis,15,38 indicating that CTGF functions as a critical profibrogenic cytokine in the liver. Although the cellular distribution of CTGF in fibrotic livers may depend on the etiology and time of the disease,39,40 several studies have shown that HSCs and fibroblasts are the major sources of CTGF in the liver during fibrogenesis.16–18 Here, we used the GEO database to validate the overexpression of CTGF in liver tissues of human NASH. Indeed, CTGF was distributed in the activated HSCs of fibrotic livers (Figure 6B and C).
Conclusion
In conclusion, we successfully identified Nbs against human CTGF, which exhibited the highest affinity and specificity in vitro. Using a cellular model of liver fibrosis, we confirmed the antifibrotic properties of the anti-CTGF Nb by regulating the profibrotic function of LX-2 stellate cells.
Ethics Statement
The normal and NASH liver specimens in this study was approved by the Animal Care and Use Committee of Shanghai Tongji University (Experimental Animal Center of Tongji University School of Medicine (No.TJBH06821101). All camels are carefully raised with consent from the owner in Jiaxiang County Aquaculture Farm, Shandong Province, China. All procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Acknowledgments
The authors thank the National Natural Science Foundation of China (grant number 81970531) and the Natural Science Foundation of Shanghai (22ZR1448500) for their financial support.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151–166. doi:10.1038/s41575-020-00372-7
2. Ding N, Yu RT, Subramaniam N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. doi:10.1016/j.cell.2013.03.028
3. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi:10.1038/nrgastro.2017.38
4. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. 2019;65:37–55. doi:10.1016/j.mam.2018.09.002
5. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587(7835):555–566. doi:10.1038/s41586-020-2938-9
6. Wiering L, Subramanian P, Hammerich L. Hepatic stellate cells: dictating outcome in nonalcoholic fatty liver disease. Cell Mol Gastroenterol Hepatol. 2023;15(6):1277–1292. doi:10.1016/j.jcmgh.2023.02.010
7. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8(11):1419. doi:10.3390/cells8111419
8. Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. doi:10.1038/nm1663
9. Ramazani Y, Knops N, Elmonem MA, et al. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018;68–69:44–66. doi:10.1016/j.matbio.2018.03.007
10. Fabregat I, Moreno-Caceres J, Sanchez A, et al. TGF-beta signalling and liver disease. FEBS J. 2016;283(12):2219–2232. doi:10.1111/febs.13665
11. Wang Q, Usinger W, Nichols B, et al. Cooperative interaction of CTGF and TGF-beta in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4(1):4. doi:10.1186/1755-1536-4-4
12. Paradis V, Dargere D, Vidaud M, et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology. 1999;30(4):968–976. doi:10.1002/hep.510300425
13. Williams EJ, Gaca MD, Brigstock DR, Arthur MJ, Benyon RC. Increased expression of connective tissue growth factor in fibrotic human liver and in activated hepatic stellate cells. J Hepatol. 2000;32(5):754–761. doi:10.1016/S0168-8278(00)80244-5
14. Pi L, Sun C, Jn-Simon N, et al. CCN2/CTGF promotes liver fibrosis through crosstalk with the Slit2/Robo signaling. J Cell Commun Signal. 2023;17(1):137–150. doi:10.1007/s12079-022-00713-y
15. George J, Tsutsumi M. siRNA-mediated knockdown of connective tissue growth factor prevents N-nitrosodimethylamine-induced hepatic fibrosis in rats. Gene Ther. 2007;14(10):790–803. doi:10.1038/sj.gt.3302929
16. Li S, Lv YF, Su HQ, Zhang QN, Wang LR, Hao ZM. A virus-like particle-based connective tissue growth factor vaccine suppresses carbon tetrachloride-induced hepatic fibrosis in mice. Sci Rep. 2016;6:32155. doi:10.1038/srep32155
17. Ren J, Wang X, Parry SN, et al. Targeting CCN2 protects against progressive non-alcoholic steatohepatitis in a preclinical model induced by high-fat feeding and type 2 diabetes. J Cell Commun Signal. 2022;16(3):447–460. doi:10.1007/s12079-022-00667-1
18. Sgalla G, Franciosa C, Simonetti J, Richeldi L. Pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2020;29(8):771–777. doi:10.1080/13543784.2020.1773790
19. Fu M, Peng D, Lan T, Wei Y, Wei X. Multifunctional regulatory protein connective tissue growth factor (CTGF): a potential therapeutic target for diverse diseases. Acta Pharm Sin B. 2022;12(4):1740–1760. doi:10.1016/j.apsb.2022.01.007
20. Sun S, Ding Z, Yang X, et al. Nanobody: a small antibody with big implications for tumor therapeutic strategy. Int J Nanomedicine. 2021;16:2337–2356. doi:10.2147/IJN.S297631
21. Ma L, Zhu M, Li G, et al. Preclinical development of a long-acting trivalent bispecific nanobody targeting IL-5 for the treatment of eosinophilic asthma. Respir Res. 2022;23(1):316. doi:10.1186/s12931-022-02240-1
22. Jovcevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs. 2020;34(1):11–26. doi:10.1007/s40259-019-00392-z
23. Wang W, Xu C, Wang H, Jiang C. Identification of nanobodies against hepatocellular carcinoma marker glypican-3. Mol Immunol. 2021;131:13–22. doi:10.1016/j.molimm.2021.01.010
24. Ma L, Zhu M, Gai J, et al. Preclinical development of a novel CD47 nanobody with less toxicity and enhanced anti-cancer therapeutic potential. J Nanobiotechnology. 2020;18(1):12. doi:10.1186/s12951-020-0571-2
25. van der Linden RH, Frenken LG, de Geus B, et al. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431(1):37–46. doi:10.1016/S0167-4838(99)00030-8
26. Papp KA, Weinberg MA, Morris A, Reich K. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: a multicentre, randomised, placebo-controlled, phase 2b study. Lancet. 2021;397(10284):1564–1575. doi:10.1016/S0140-6736(21)00440-2
27. Van Roy M, Ververken C, Beirnaert E, et al. The preclinical pharmacology of the high affinity anti-IL-6R nanobody(R) ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):135. doi:10.1186/s13075-015-0651-0
28. Li S, Li X, Chen F, Liu M, Ning L, Yan Y. Nobiletin mitigates hepatocytes death, liver inflammation, and fibrosis in a murine model of NASH through modulating hepatic oxidative stress and mitochondrial dysfunction. J Nutr Biochem. 2022;100:108888. doi:10.1016/j.jnutbio.2021.108888
29. Navarro-Corcuera A, Lopez-Zabalza MJ, Martinez-Irujo JJ, et al. Role of AGAP2 in the profibrogenic effects induced by TGFbeta in LX-2 hepatic stellate cells. Biochim Biophys Acta Mol Cell Res. 2019;1866(4):673–685. doi:10.1016/j.bbamcr.2019.01.008
30. Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75(2):473–488. doi:10.1002/hep.32285
31. Brenner MC, Krzyzanski W, Chou JZ, et al. FG-3019, a human monoclonal antibody recognizing connective tissue growth factor, is subject to target-mediated drug disposition. Pharm Res. 2016;33(8):1833–1849. doi:10.1007/s11095-016-1918-0
32. Kaasboll OJ, Gadicherla AK, Wang JH, et al. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J Biol Chem. 2018;293(46):17953–17970. doi:10.1074/jbc.RA118.004559
33. Richeldi L, Fernandez Perez ER, Costabel U, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2020;8(1):25–33. doi:10.1016/S2213-2600(19)30262-0
34. Xue X, Fan X, Qu Q, Wu G. Bioscreening and expression of a camel anti-CTGF VHH nanobody and its renaturation by a novel dialysis-dilution method. AMB Express. 2016;6(1):72. doi:10.1186/s13568-016-0249-1
35. Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54(1):142–151. doi:10.1136/gut.2004.042127
36. Lu ZN, Niu WX, Zhang N, et al. Pantoprazole ameliorates liver fibrosis and suppresses hepatic stellate cell activation in bile duct ligation rats by promoting YAP degradation. Acta Pharmacol Sin. 2021;42(11):1808–1820. doi:10.1038/s41401-021-00754-w
37. Adler SG, Schwartz S, Williams ME, et al. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol. 2010;5(8):1420–1428. doi:10.2215/CJN.09321209
38. Li G, Xie Q, Shi Y, et al. Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J Gene Med. 2006;8(7):889–900. doi:10.1002/jgm.894
39. Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34(4 Pt 1):738–744. doi:10.1053/jhep.2001.28055
40. Gressner OA, Gressner AM. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008;28(8):1065–1079. doi:10.1111/j.1478-3231.2008.01826.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.