Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Identification and Bioinformatic Analysis of Circular RNA Expression in Peripheral Blood Mononuclear Cells from Patients with Chronic Obstructive Pulmonary Disease
Authors Duan R ![]() , Niu H
, Niu H ![]() , Yu T, Cui H
, Yu T, Cui H ![]() , Yang T
, Yang T ![]() , Hao K, Wang C
, Hao K, Wang C
Received 11 March 2020
Accepted for publication 18 May 2020
Published 16 June 2020 Volume 2020:15 Pages 1391—1401
DOI https://doi.org/10.2147/COPD.S252896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chunxue Bai
Ruirui Duan,1– 4 Hongtao Niu,2– 4 Tao Yu,2– 5 Han Cui,2– 4,6 Ting Yang,1– 5 Ke Hao,7 Chen Wang1– 4,6
1Peking University China-Japan Friendship School of Clinical Medicine, Beijing, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 3National Clinical Research Center for Respiratory Diseases, Beijing, People’s Republic of China; 4Institute of Respiratory Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 5Institute of Clinical Medical Sciences, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 6Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, People’s Republic of China; 7Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Correspondence: Chen Wang
Peking University China-Japan Friendship School of Clinical Medicine, No. 2, East Yinghua Road, Chaoyang District, Beijing 100029, People’s Republic of China
Tel/ Fax +86-01-8420 6276
Email [email protected]
Purpose: Circular RNAs (circRNAs) regulate other RNA transcripts by competing for shared microRNAs, which play roles in the pathogenesis of many diseases, including chronic obstructive pulmonary disease (COPD). However, the role of circRNAs in COPD remains unknown. This study aimed to investigate the expression profile and the role of circRNAs in COPD.
Patients and Methods: Twenty-one COPD patients and twenty-one normal controls were recruited. Total RNAs were collected from peripheral blood mononuclear cells (PBMCs) of each participant. CircRNAs and protein-coding mRNAs were profiled by microarray and systematically compared between patients with COPD and control subjects. The top differentially expressed circRNAs and mRNAs were validated by quantitative real-time PCR (RT-qPCR). Functional analysis identified pathways relevant to the pathogenesis of COPD. Next, the circRNA target pathway network, the circRNA-miRNA-mRNA network (ceRNA network) and functional ceRNA regulatory modules were constructed.
Results: In total, 2132 circRNAs and 2734 protein-coding mRNAs were differentially expressed (|fold change| > 1.5 and P-value < 0.05) in COPD patients. Six out of nine selected RNAs were confirmed by RT-qPCR validation. Our functional analysis suggested that immune imbalances and inflammatory responses play roles in the pathogenesis of COPD. The ceRNA network highlighted the differentially expressed circRNAs and their related miRNAs and mRNAs in COPD. In the circRNA target pathway network and functional ceRNA regulatory modules, hsa_circRNA_0008672 appeared in the top three KEGG pathways (NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity and Th17 cell differentiation) and may act as the miRNA sponge regulating the hsa_circRNA_0008672/miR-1265/MAPK1 axis.
Conclusion: Our findings demonstrate critical roles of the circRNAs in COPD molecular etiology. The data support a plausible mechanism that circRNAs may be involved in the development of COPD by affecting the immune balance. Moreover, the hsa_circRNA_0008672/miR-1265/MAPK1 axis may contribute to the pathogenesis of COPD, warranting further investigation.
Keywords: circular RNA, competing endogenous RNAs, chronic obstructive pulmonary disease, expression profile, co-expression network
Introduction
Characterized by persistent respiratory symptoms and limitations in airflow, chronic obstructive pulmonary disease (COPD) is a heterogeneous disease associated with significant exposure to noxious particles or gases and is influenced by host factors including abnormal lung development.1 COPD has become a global public health issue due to its association with high rates of morbidity, disability, and mortality. According to the China Pulmonary Health [CPH] study, the prevalence of COPD among Chinese adults over the age of 40 is 13.7%, representing a 55% rise compared to ten years prior.2 A Global Burden of Disease (GBD) study reported that the disease resulted in the death of an estimated 3.2 million people worldwide in 2017.3 COPD is associated with various environmental and genetic factors. The pathogenesis of COPD involves chronic inflammation, oxidative stress, and protease/anti-protease imbalance.4,5 Furthermore, an increasing body of evidence has implicated immune response imbalances in the pathogenesis of COPD.6–8
Circular RNAs (circRNAs) are non-coding RNAs that play key roles in regulating gene expression. The structure of circRNAs consists of a covalently closed loop formed by reverse splicing and the consequent removal of the 5ʹ cap and 3ʹ poly-A tail, making them resistant to exonuclease RNase R degradation and hence, relatively stable in the plasma.9 CircRNAs are widely expressed in eukaryotic cells and exhibit cell- and tissue-specificity.10 When first observed in the 1970s, circRNAs were considered transcriptional “noise” with no biological function.11 With the development of high-throughput sequencing technology and bioinformatics, an increasing amount of data has shown that circRNAs play an important role in regulating gene expression by binding to specific microRNAs (miRNAs) or RNA-binding proteins, regulating transcription and interfering with splicing, and even in the translation of peptides.12–14
Research has further revealed that competing endogenous RNA (ceRNA), also known as an miRNA sponge, helps to mediate the functions of circRNAs.15,16 CircRNAs bind to miRNAs via miRNA response elements (MREs) to carry out their function.17 For example, circTP63 is up-regulated in patients with lung squamous cell carcinoma and can function as a ceRNA regulating the expression of human Forkhead Box M1 (FOXM1) by competitively binding to miR-873-3p.18 Similarly, hsa_circ_0016070 promotes the proliferation of pulmonary artery smooth muscle cells (PASMC) and is involved in vascular remodeling in COPD associated pulmonary arterial hypertension (PAH) via sponging of miR-942 and in turn up-regulating cyclin D1 (CCND1).19 Moreover, the circRNA expression profile has been analyzed in the context of a variety of respiratory diseases, including lung cancer,20,21 pulmonary fibrosis,22 pulmonary tuberculosis23 and acute lung injury.24 To date, the expression profile and ceRNA network of circRNAs in COPD patients have been largely unexplored.
In the current study, we analyze PBMCs from COPD patients and normal controls to investigate, for the first time to our knowledge, the roles of circRNAs in the molecular etiology of COPD.
Patients and Methods
Study Population
The COPD group consisted of patients in the China-Japan Friendship Hospital from July 2017 to December 2018. The inclusion criteria were 1) age ≥ 45 and ≤ 75 years, 2) the ratio of forced expiratory volume in 1st second (FEV1) to forced vital capacity (FVC) less than 70% after 20 min of albuterol administration, and 3) stable disease upon enrollment. Stable COPD was defined as the absence of exacerbations, and a lack of hospitalizations, oral corticosteroids, and antibiotics in the past three months. The age- and sex-matched normal controls who received a physical examination during the same time period in the China-Japan Friendship Hospital were enrolled if they met the following criteria: 1) no history of chronic cough, expectoration, wheezing, or other symptoms, 2) FEV1/FVC score ≥ 0.7 on a spirometry test after inhalation of albuterol, and 3) no COPD or other chronic respiratory diseases. The exclusion criteria for both COPD patients and normal controls included the following: 1) Acute heart and/or cerebrovascular disease, liver and kidney insufficiency, diabetes mellitus or rheumatic disease; 2) past or present diagnosis of a malignant tumor; 3) pregnancy; and 4) acute infectious disease. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the China-Japan Friendship Hospital (2017–19), and written informed consent of the study including genetic analysis was obtained from each participant.
PBMC Isolation and RNA Extraction
We collected 6 mL of peripheral venous blood from each participant, and peripheral blood mononuclear cells (PBMCs) were isolated within 2 h by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare) and were stored at −80°C in Trizol (Life technologies, CA, US). Total RNA was extracted from PBMCs using Trizol reagent and purified using a Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA was then quantified using a spectrophotometer (NanoDrop 1000, Thermo Fisher Scientific, Waltham, MA, United States) at 260 nm absorbance, and its integrity was assessed with agarose gel electrophoresis. All experimental devices were RNase-free.
Microarray Profiling and Data Analysis
The expression profiles were assessed by microarray according to the recommended protocols provided by Cnkingbio Biotechnology Corporation (Beijing, China), which included RNA labeling, hybridization, scanning, and data analysis. Briefly, the purified RNA was amplified and transcribed into fluorescent cDNA, and the labeled cDNA was then hybridized at 45°C for 16 h with GeneChip (Affymetrix Clariom D Human array). The GeneChips were washed and stained in the Affymetrix Fluidics Station 450. All arrays were scanned with an Affymetrix GeneChip Command Console, which was installed in the GeneChip Scanner 3000 7G. The data were analyzed using the Robust Multichip Analysis (RMA) algorithm with the default Affymetrix analysis settings and global scaling as a normalization method. Values are presented as log2RMA signal intensities. Differentially expressed circRNAs and mRNAs were then further analyzed with hierarchical clustering.25 All samples used for microarray analysis were included in the same batch, so there is no batch effect.
GO and KEGG Enrichment Analyses
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) molecular pathway enrichment analyses were performed to analyze the primary function of the differentially expressed mRNAs. The GO analysis consisted of the biological process (BP), cellular component (CC), and molecular function (MF).
CircRNA Target Pathway Network
The circRNA target pathway network was built using Cytoscape26 (version 3.6.0, https://js.cytoscape.org/) according to the relationships between significant pathways and genes as well as the relationships between circRNAs and pathways. Using the graph theory methods, we evaluated the regulatory status of circRNAs and pathways. The evaluation criteria included the degrees of circRNAs and pathways in the network. The degree of each circRNA refers to the number of pathways regulated by the circRNA, and the degree of each pathway refers to the number of circRNAs that regulated the pathway.
Constructed the ceRNA Network (CircRNA-miRNA-mRNA Network)
Firstly, TargetScan27 (http://www.targetscan.org/vert_71/) and miRWalk28 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) databases were used to predict the target relationships between circRNA-miRNA and miRNA-mRNA. The Pearson correlation coefficient between miRNA-mRNA and miRNA-circRNA was then calculated. For a given circRNA-mRNA pair, both the mRNA and circRNA were targeted with a common miRNA and co-expression negatively correlated with the miRNA. The visualization of the network was built with Cytoscape.
Validation with RT-qPCR
We used quantitative real-time PCR (RT-qPCR) to validate the differentially expressed circRNAs and mRNAs that were identified in microarray analysis on all samples. Specific primers for each gene are listed in Supplementary Figure 1. The GAPDH gene was used as an internal control. For each RT-qPCR reaction, we performed technical triplicates using the SYBR Green reagent (Bio-Rad, Hercules, CA, USA) approach, and took the sample mean to ensure data stability. The comparative CT (2−ΔΔCT) method was used to obtain the fold change of circRNA and mRNA expression levels.
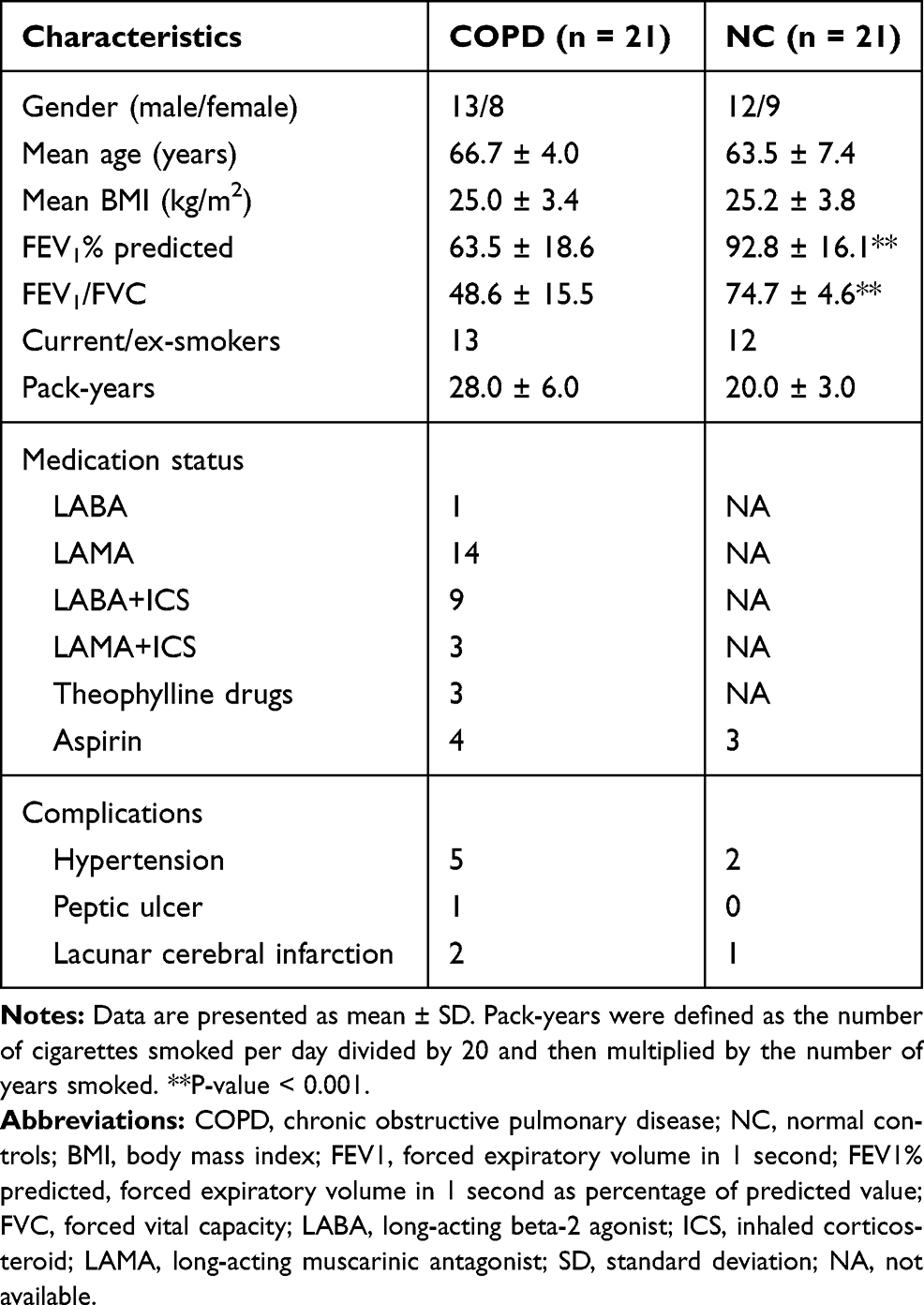 |
Table 1 Basic Clinical Information of Participants |
Statistical Analysis
Statistical analyses were conducted with the Statistical Package for Social Sciences software package (SPSS, version 22.0) and GraphPad Prism 7.0 (GraphPad Software, CA, USA). Student’s t-tests were used to identify between-group differences in the expression of circRNAs and mRNAs. A |fold change| (FC) > 1.5 and P-value < 0.05 were considered significant in gene expression. For GO and KEGG analysis, statistical analyses were performed using two-tailed Fisher’s exact tests and a P-value < 0.05 was considered to be statistically significant. For RT-qPCR, experimental data are represented as mean ± SEM, Student’s t-tests or Mann–Whitney test were used to determine statistical significance, and a P-value < 0.05 was considered statistically significant.
Results
Subjects’ Clinical Characteristics
A total of 21 COPD patients and 21 normal controls were included in our study, and the clinical characteristics of the subjects are shown in Table 1. There were no significant differences in age, sex, BMI, or smoking history (pack-years) between COPD patients and the normal control group. The lung function of patients with COPD was decreased, and both FEV1/FVC and FEV1 predicted (FEV1%) were significantly lower in COPD patients compared to those in the normal control group (P-value < 0.001). Two patients with COPD had an acute exacerbation of their disease in the previous year (not in the past 3 months), while the remaining patients had no history of acute exacerbation in the previous year.
Overview the CircRNA and mRNA Expression Profiles
A total of 12,960 circRNAs were assessed in PBMC samples, among which 2132 circRNAs (22 circRNAs up-regulated and 2110 circRNAs down-regulated) were identified as significant with cutoff criteria of |FC| > 1.5 and P-value < 0.05 (Figure 1A). In addition, 20,666 dysregulated mRNAs were detected, including 50 up-regulated and 2684 down-regulated mRNAs, between the two groups (Figure 1B). Hierarchical clustering was used to distinguish patients with COPD from normal controls based on their expressions of circRNAs and mRNAs (Supplementary Figure 1A and B). Although a small degree of heterogeneity was observed between the groups, the expression of these selected circRNAs or mRNAs informed the division of the samples into COPD and normal control groups. The top ten most significant circRNAs and mRNAs ranked by FC are summarized in Table 2 and Table 3, respectively.
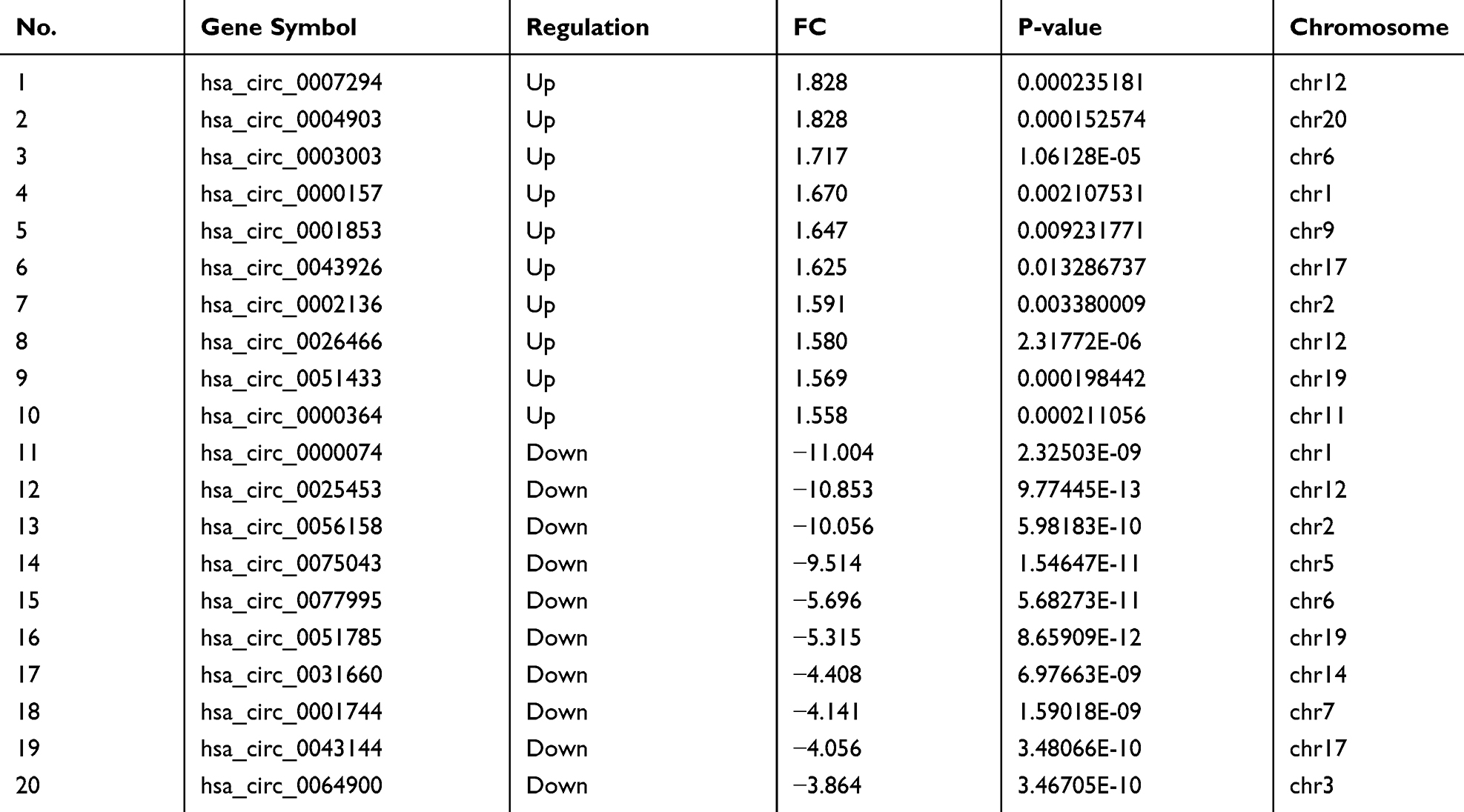 |
Table 2 Ten Most Up-Regulated and Down-Regulated Circular RNAs in COPD Patients Compared to Normal Controls |
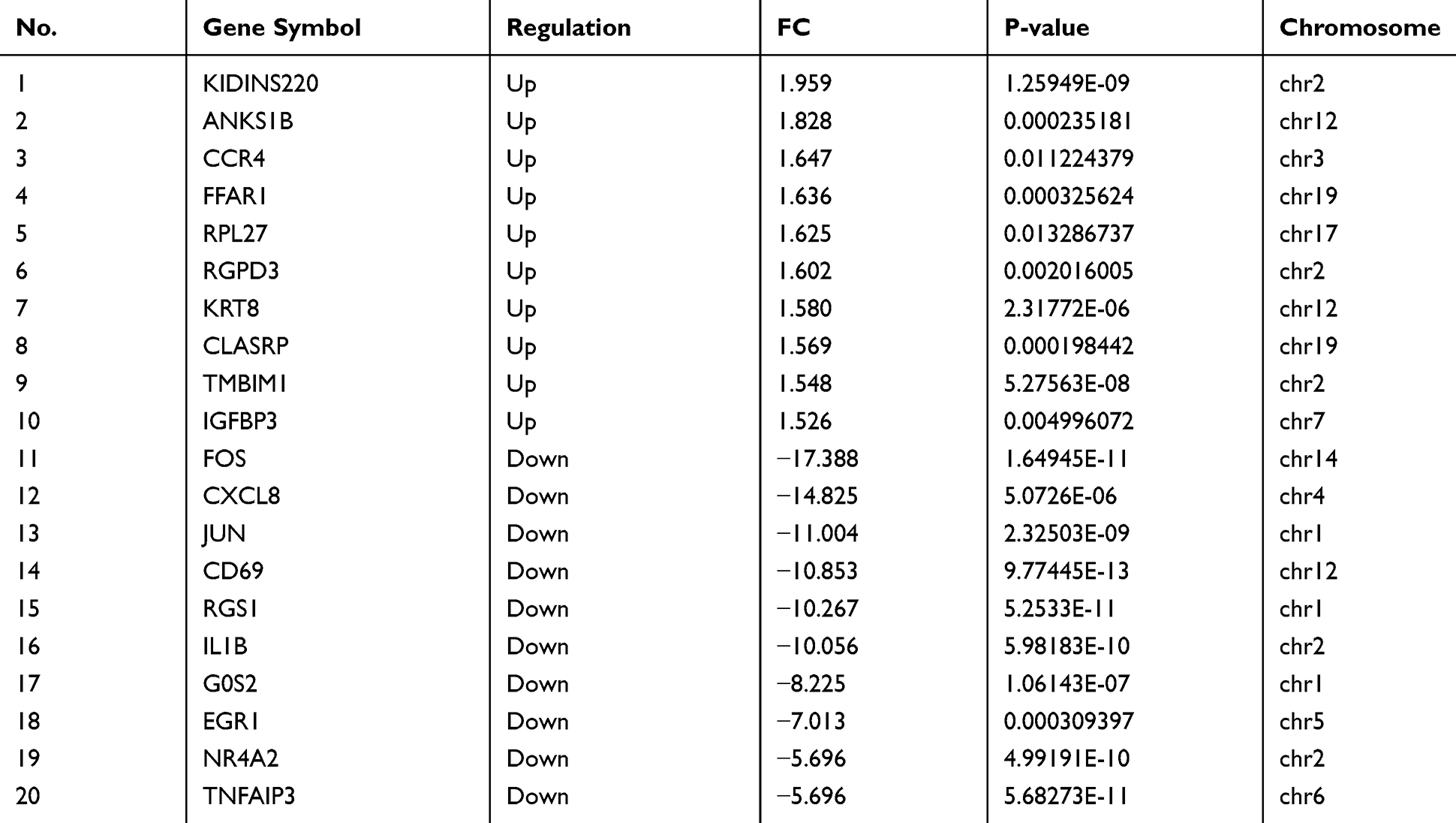 |
Table 3 Ten Most Up-Regulated and Down-Regulated mRNAs in COPD Patients Compared to Normal Controls |
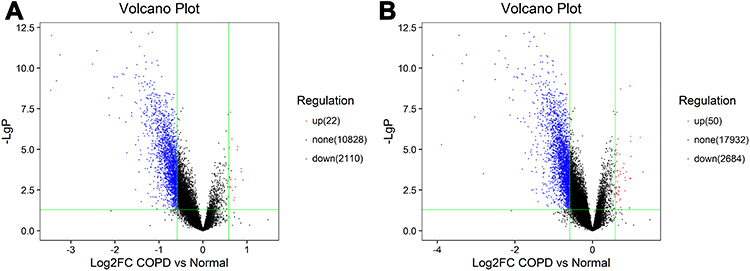 |
Figure 1 CircRNA and mRNA expression profile in COPD patients. Volcano plots were used to distinguish differentially expressed circRNAs (A) and mRNAs (B). Red and blue indicate up-regulation and down-regulation, respectively. |
Functional Analysis of the Differentially Expressed Genes
For the up-regulated mRNAs, we found the most significant GO terms for BP primarily involved immune reactions, cell death and apoptosis, inflammatory factor activation, and signaling transduction, all of which play important roles in the development of COPD. The GO terms of MF included protein kinase regulator activity and chemokine receptor activity translation factor activity. In the CC, the three most significant terms were insulin-like growth factor binding protein complex, insulin-like growth factor ternary complex, and insulin-like growth factor ternary complex eukaryotic translation initiation factor 4F complex. (Figure 2A).
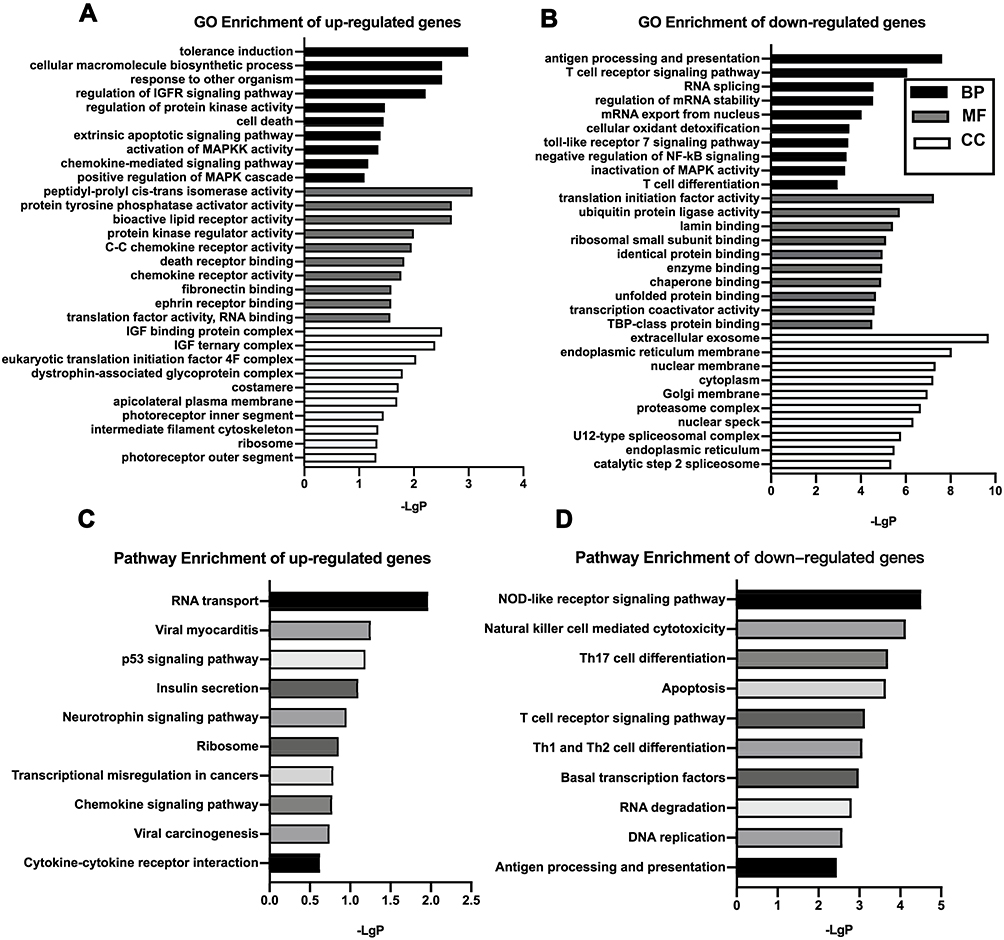 |
Figure 2 GO and KEGG pathway analysis. GO analysis of significantly up-regulated (A) and down-regulated (B) mRNAs clustered in the CC, MF, and BP. KEGG analysis of differentially up-regulated (C) and down-regulated mRNAs (D). The x‐axis shows ‐lgP, and the y-axis shows GO terms or KEGG pathways.Abbreviations: GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, biological process; CC, cellular component; MF, molecular function. |
For the down-regulated mRNAs, the most significant GO terms for BP were antigen processing and presentation, the T-cell receptor signaling pathway, T-cell activation, RNA splicing, and mRNA stability, as well as the negative regulation of inflammatory signaling pathways, which are primarily associated with immune and inflammatory responses. The MF was primarily related to the activation of transcription and translation processes and the binding of different kinds of proteins (lamin binding, ribosomal small subunit binding, enzyme binding, etc). In the CC, the most significant terms were extracellular exosome and cellular component organization (endoplasmic reticulum membrane, nuclear membrane, cytoplasm, etc) (Figure 2B).
The top three pathways in the KEGG pathway enrichment analysis for the up-regulated genes were RNA transport, viral myocarditis, and p53 signaling pathway. The top three pathways in the KEGG pathway enrichment analysis for the down-regulated genes were NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity, and Th17 cell differentiation, all of which are involved in immunity and inflammation (Figure 2C and D).
CircRNA Target Pathway Network
Many of circRNAs exert unknown functions. To predict the possible functions of the differentially expressed circRNAs, the circRNA target pathway networks were constructed. We chose the top three KEGG pathways (NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity and Th17 cell differentiation) as well as their related dysregulated circRNAs to construct the network (Supplementary Figure 2). The circRNA target pathway network found that 65 circRNAs were involved in the above three pathways: 30 circRNAs target the natural killer cell mediated cytotoxicity pathway, 20 target Th17 cell differentiation, and 35 target the NOD-like receptor signaling pathway. Hsa_circRNA_0008672 was the only circRNA targeted in all three pathways.
Construction of the ceRNA Network
We constructed the ceRNA network based on the microarray data to examine the regulatory relationships between circRNA, miRNA, and mRNA (Supplementary Figure 3). As shown in the figure, the ceRNA network includes 254 dysregulated circRNAs, 254 dysregulated mRNAs, and 16 predicted miRNAs. In addition, we constructed the functional ceRNA network regulatory modules based on the circRNAs involved in the NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity, and Th17 cell differentiation pathway (Supplementary Figure 4). Nine circRNAs, seven miRNAs, and nine mRNAs were contained within this network, which will help us further understand the immune-related mechanisms in COPD.
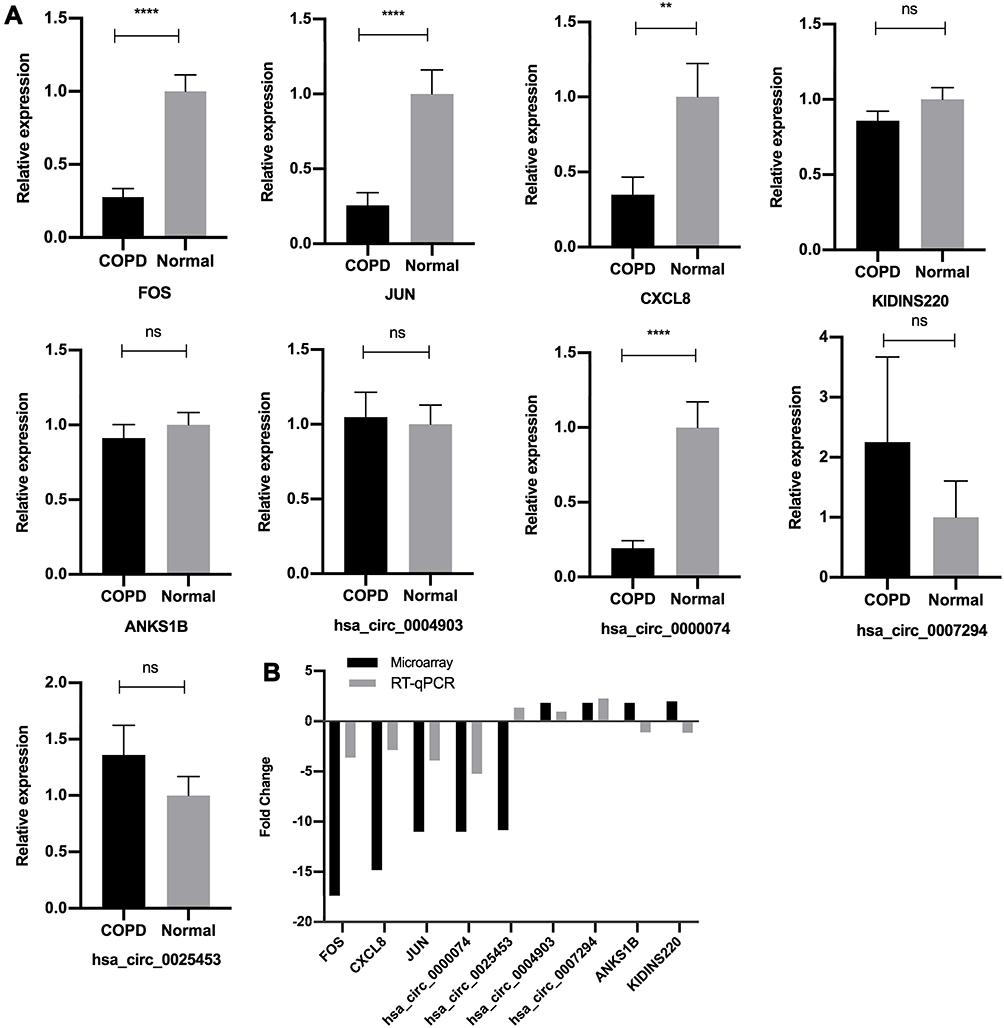 |
Figure 3 Validation of microarray data by RT‐qPCR. (A) Relative expression levels of the circRNAs and mRNAs between the COPD group and normal control group, assessed by RT-qPCR. The heights of the columns in the chart represent the relative expression (2−ΔΔCT). Data are mean ± SEM. All data were normalized to GAPDH gene expression. (B) Comparison of mean fold changes between microarray data and RT-qPCR results. SEM, standard error of the mean. ****P-value < 0.0001. **P-value < 0.001. ns, no significance. |
Validation of the Differentially Expressed CircRNAs and mRNAs
To validate the reliability of the microarray data, we selected five circRNAs and five mRNAs from the top up-regulated and down-regulated transcripts for RT-qPCR. The results are shown in Figure 3A and B. The RT-qPCR assays were successful for all the selected RNAs except hsa_circ_00056158. Six of the remaining nine RNAs showed consistent direction as shown by the microarray data. Four RNAs, Hsa_circ_0000074, FOS, JUN, and CXCL8 were validated with statistical significance (P-value < 0.05). The expression tendency of hsa_circ_0004903 and hsa_circ_0007294 were akin to the microarray data; however, the relative expression showed no significant difference between the two groups (P-value > 0.05). The remaining three RNAs (hsa_circ_0025453, ANKS1B, KIDINS220) were not consistent with the microarray data.
Discussion
COPD is a complex disease that causes damage not only to the airway and lung parenchyma, but also to other organs such as the heart and muscles. Currently, there is no effective treatment strategy once it is diagnosed. Previous studies found that epigenetic mechanisms significantly contribute to COPD pathophysiology and may present potential therapeutic targets.29,30 Because circRNAs are an epigenetic modifier, their dysregulation has been implicated in the pathogenesis of several diseases and they have attracted much attention as biomarkers or therapeutic targets.31 However, the expression profiles and regulatory network of circRNAs in COPD remained unknown. To the best of our knowledge, the present study is the first to identify the expression profiles of circRNAs in the PBMCs of patients with COPD. We identified 2132 circRNAs and 2734 mRNAs that were dysregulated in patients with COPD compared to normal controls. These results were validated by RT-qPCR, which revealed that six out of nine agreed with the microarray data. The association of some of these validated genes with COPD has been previously reported. For example, CXCL8 is a member of the chemokine family as well as the main chemokine for neutrophils, and plays a pivotal role in the inflammation process. It was shown that the abnormal regulation of CXCL8 and its receptors has been shown to play an important role in COPD.32 The association between the dysregulated circRNAs and COPD has not been previously reported, which is consistent with the fact that little is currently known about the function of circRNAs in COPD. Alternatively, some of the dysregulated circRNAs have been known to be associated with COPD. For example, hsa_circRNA_0003060 was found to be significantly down-regulated in the primary human small-airway epithelial cell (HSAECs) model of COPD triggered by cigarette smoke extract, which is consistent with our findings.33 In addition, other major mRNAs have been known to be associated with COPD, such as IGFBP3 and CD69. The expression of IGFBP3 was increased in lung tissue after cigarette smoke extract exposure and has been observed to play key roles in airway inflammation and remodeling.34 Furthermore, CD69 has been reported to have a protective role in acute pulmonary inflammation and subsequent emphysema development.35,36 In line with these studies, our microarray data revealed that CD69 was down-regulated with a FC of 10.853 in patients with COPD. These findings indicate that the dysregulation of the circRNAs and mRNAs identified in our study are involved in the development of COPD.
We also performed functional enrichment analyses to identify the potential roles of the dysregulated circRNAs in COPD development. The GO analysis for the mRNAs targeted by dysregulated circRNAs revealed that tolerance induction, cell death and apoptosis, mitogen-activated protein kinase (MAPK) activation, and signaling transduction are involved in the biological and cellular processes that relate to the pathogenesis of COPD. It is well documented that activation of the MAPK cascade is the center of multiple signaling pathways, therefore regulating a variety of important pathophysiological processes including inflammatory responses. Many studies have demonstrated that the MAPK signaling pathway also plays a key role in the chronic inflammatory immune response of COPD.37–39 Furthermore, apoptosis is a key pathological process of COPD, and the relationship between apoptosis of alveolar epithelial cells and the development of emphysema has been well documented.40,41 The GO analysis provided us with potential biological functions of the dysregulated circRNAs associated with COPD.
The KEGG pathway analysis indicated that these dysregulated circRNAs might contribute to the development of COPD by affecting the NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity, Th17 cell differentiation, the T-cell receptor signaling pathway, Th1 and Th2 cell differentiation, and the Toll-like receptor-signaling pathway. All of these pathways are involved in immune imbalances, and previous studies have indicated that the pathogenesis of COPD is associated with both innate and adaptive immune imbalances.42 Further, evidence suggests that human natural killer cell cytotoxicity is inhibited in severe COPD, making the lung less resistant to respiratory pathogens and leading to secondary inflammation. This in turn accelerates the progression of COPD.43 Providing further support for our findings, previous studies have found that decreased TLR4 signal transduction is associated with development of emphysema,6,44 which suggests that circRNA may play a role in COPD by influencing the immune system.
To further understand which circRNAs are involved in the relevant immune response pathways, we constructed a circRNA target pathway network. To this end, we selected three pathways: the NOD-like receptor signaling pathway, natural killer cell mediated cytotoxicity, and Th17 cell differentiation. The network allowed us to identify the circRNAs that may be involved in these three pathways. Hsa_circRNA_0008672 is at the core of these three pathways and we therefore speculate that it may play a central role. Hence, our networks provide new directions for future investigation into the functions of circRNAs in COPD.
We also constructed a ceRNA network to further explore the miRNA sponge role of circRNA in regulating gene expression. In the visible ceRNA network, we can conclude that circRNAs may regulate mRNAs by sponging one or several miRNAs. For example, the activation of MAPK1 leads to a cascade of inflammatory and immune responses and thus plays a key role in the pathogenesis of COPD, while hsa_circRNA_0008672 can bind to miR-1265 to affect the expression of MAPK1. These RNA interactions indicate that the tripartite regulation among circRNA, miRNA, and mRNA was comprehensive, providing a novel perspective for the pathogenesis of COPD.
Our study was subject to certain limitations. First, the RT-qPCR validation was performed in the same COPD population, and hence these results must be further validated in a larger COPD cohort. Second, our study of the circRNA functions was only based on bioinformatics analysis technology. CircRNA overexpression or knockdown experiments should be performed to determine the precise functions of dysregulated circRNAs in COPD. Finally, PBMCs contain a variety of cells, including monocytes, lymphocytes, NK cells, etc. It may therefore be informative to define circRNA expression in different types of PBMCs, which is the direction of our future research.
Conclusion
This study elucidates the comprehensive expression profiles of circRNAs as well as their possible functions and contribution to a regulatory network in COPD patients. Our study showed that circRNAs might be involved in the development of COPD by promoting immune imbalance. Moreover, the hsa_circRNA_0008672/miR-1265/MAPK1 axis may be involved in the pathogenesis of COPD and should be further investigated.
Acknowledgments
We would like to thank all of the study participants from China-Japan Friendship Hospital. We thank Drs. Yong Li for performing the Lung function test with patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease 2020 report; 2020. Available from: https://goldcopd.org/gold-reports/.
2. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717.
3. Roth GA, Abate D, Abate KH, et al. Global, regional and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392(10159):1736–1788.
4. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940.
5. Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013.
6. Zhang X, Shan P, Jiang G, Cohn L, Lee PJ. Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest. 2006;116(11):3050–3059.
7. Donovan C, Starkey MR, Kim RY, et al. Roles for T/B lymphocytes and ILC2s in experimental chronic obstructive pulmonary disease. J Leukoc Biol. 2019;105(1):143–150.
8. van Eeden SF, Hogg JC. Immune-modulation in chronic obstructive pulmonary disease: current concepts and future strategies. Respiration. 2019;1–16.
9. Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol. 2016;14(8):1035–1045.
10. Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19(2):141–157.
11. Wang J, Zhu M, Pan J, Chen C, Xia S, Song Y. Circular RNAs: a rising star in respiratory diseases. Respir Res. 2019;20(1):3.
12. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338.
13. Zhang Z, Yang T, Xiao J. Circular RNAs: promising biomarkers for human diseases. E Bio Medicine. 2018;34:267–274.
14. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428–442.
15. Kulcheski FR, Christoff AP, Margis R. Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J Biotechnol. 2016;238:42–51.
16. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388.
17. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352.
18. Cheng Z, Yu C, Cui S, et al. circTP63 functions as a ceRNA to promote lung squamous cell carcinoma progression by upregulating FOXM1. Nat Commun. 2019;10(1):3200.
19. Zhou S, Jiang H, Li M, et al. Circular RNA hsa_circ_0016070 is associated with Pulmonary Arterial Hypertension by promoting PASMC proliferation. Mol Ther Nucleic Acids. 2019;18:275–284.
20. Ma Y, Zhang X, Wang YZ, Tian H, Xu S. Research progress of circular RNAs in lung cancer. Cancer Biol Ther. 2019;20(2):123–129.
21. de Fraipont F, Gazzeri S, Cho WC, Eymin B. Circular RNAs and RNA splice variants as biomarkers for prognosis and therapeutic response in the liquid biopsies of lung cancer patients. Fron Genet. 2019;10:390.
22. Yang L, Liu X, Zhang N, Chen L, Xu J, Tang W. Investigation of circular RNAs and related genes in pulmonary fibrosis based on bioinformatics analysis. J Cell Biochem. 2019;120(7):11022–11032.
23. Qian Z, Liu H, Li M, et al. Potential diagnostic power of blood circular RNA expression in active pulmonary tuberculosis. E Bio Medicine. 2018;27:18–26.
24. Li X, Yuan Z, Chen J, et al. Microarray analysis reveals the changes of circular RNA expression and molecular mechanism in acute lung injury mouse model. J Cell Biochem. 2019;120(10):16658–16667.
25. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–14868.
26. Cline MS, Smoot M, Cerami E, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382.
27. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005.
28. Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods. 2015;12(8):697.
29. Schamberger AC, Mise N, Meiners S, Eickelberg O. Epigenetic mechanisms in COPD: implications for pathogenesis and drug discovery. Expert Opin on Drug Discov. 2014;9(6):609–628.
30. Wu DD, Song J, Bartel S, Krauss-Etschmann S, Rots MG, Hylkema MN. The potential for targeted rewriting of epigenetic marks in COPD as a new therapeutic approach. Pharmacol Ther. 2018;182:1–14.
31. Haque S, Harries LW. Circular RNAs (circRNAs) in health and disease. Genes. 2017;8(12):353.
32. Kaur M, Singh D. Neutrophil chemotaxis caused by chronic obstructive pulmonary disease alveolar macrophages: the role of CXCL8 and the receptors CXCR1/CXCR2. J Pharmacol Exp Ther. 2013;347(1):
33. Zeng N, Wang T, Chen M, et al. Cigarette smoke extract alters genome-wide profiles of circular RNAs and mRNAs in primary human small airway epithelial cells. J Cell Mol Med. 2019;23(8):5532–5541.
34. Lee H, Kim SR, Oh Y, Cho SH, Schleimer RP, Lee YC. Targeting insulin-like growth factor-I and insulin-like growth factor-binding protein-3 signaling pathways. A novel therapeutic approach for asthma. Am J Respir Cell Mol Biol. 2014;50(4):667–677.
35. Fujita T, Yoshioka K, Umezawa H, et al. Role of CD69 in the pathogenesis of elastase-induced pulmonary inflammation and emphysema. Biochem Biophys Rep. 2016;7:400–407.
36. Tsuyusaki J, Kuroda F, Kasuya Y, et al. Cigarette smoke-induced pulmonary inflammation is attenuated in CD69-deficient mice. J Recept Signal Transduct Res. 2011;31(6):434–439.
37. Chung KF. p38 Mitogen-activated protein kinase pathways in Asthma and COPD. Chest. 2011;139(6):1470–1479.
38. Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41.
39. Wu D, Yuan Y, Lin Z, et al. Cigarette smoke extract induces placental growth factor release from human bronchial epithelial cells via ROS/MAPK (ERK-1/2)/Egr-1 axis. Int J Chron Obstruct Pulmon Dis. 2016;11:3031–3042.
40. Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction, and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28(5):555–562.
41. Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7(1):53.
42. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026.
43. Shaykhiev R, Crystal RG. Innate immunity and chronic obstructive pulmonary disease: a mini-review. Gerontology. 2013;59(6):481–489.
44. An CH, Wang XM, Lam HC, et al. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2012;303(9):L748–L757.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.