Back to Journals » OncoTargets and Therapy » Volume 11
Icotinib hydrochloride enhances chemo- and radiosensitivity by inhibiting EGFR signaling and attenuating RAD51 expression and function in Hela S3 cells
Authors Wang X ![]() , Gu Y, Liu H, Shi L, Sun X
, Gu Y, Liu H, Shi L, Sun X ![]()
Received 26 September 2017
Accepted for publication 18 January 2018
Published 5 March 2018 Volume 2018:11 Pages 1245—1258
DOI https://doi.org/10.2147/OTT.S152613
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Xuanxuan Wang, Yanjun Gu, Hai Liu, Liming Shi, Xiaonan Sun
Department of Radiation Oncology, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Background: Radiotherapy and cisplatin-based chemotherapy are currently considered as standard treatments employed for advanced cervical cancer (CC). However, patients with local recurrence or distant metastasis continue to have poor outcomes. EGFR overexpression correlated with chemo/radioresistance, and disease failure has been well proved in the previous studies. Hence, the aim of this study was to explore the therapeutic efficacy and underlying mechanism of the sensitization to radiation or cisplatin of icotinib hydrochloride (IH), a high-selective EGFR tyrosine kinase inhibitor (TKI), in the Hela S3 human CC cell line.
Methods: Cell proliferation was measured with cell counting kit-8 (CCK-8) assay. Flow cytometry analysis was performed to examine cell cycle distribution and apoptosis. The phosphorylation of EGFR and its downstream signaling molecules were measured by Western blot analysis. γ-H2AX foci and RAD51 foci in the cellular nucleus were visualized using immunofluoresence staining. Expression levels of RAD51 in the whole cells and subceullar fractions were detected to demonstrate the impact of IH on DNA repair.
Results: IH can significantly inhibit cell proliferation, redistribute cell cycle, enhance apoptosis and impair DNA damage response of Hela S3 cells following radiation or cisplatin treatment through suppressing the activation of the EGFR signaling pathway and attenuating the expression and function of homologous recombination (HR) protein RAD51.
Conclusion: This study suggests that IH is a potential sensitizer in radiotherapy and cisplatin-based chemotherapy for CC and RAD51 may serve as a prognosis biomarker for this combination treatment.
Keywords: icotinib hydrochloride, cervical cancer, EGFR, radiotherapy, chemotherapy
Introduction
Cervical cancer (CC) remains one of the most frequent female malignancies worldwide, especially in the developing countries.1 Although patients in early stage could be cured by surgery, the prognosis for the patients in advanced stage who are commonly treated with radiotherapy or cisplatin-based chemoradiotherapy is still poor.2,3 Both ionizing radiation (IR) and cisplatin work by inducing DNA damage;4,5 double-stranded DNA breaks (DSBs) are among the most toxic form.6 Following induction of DSBs, several DNA damage response (DDR) pathways will be activated. The two major DSB repair pathways are non-homologous end joining (NHEJ) and homologous recombination (HR).4,6 NHEJ is generally considered as an error-prone repair pathway that is predominantly responsible for DSB repair during G0, G1 and early S phases, whereas HR is an error-free pathway that occurs only in the late S and G2 phases of the cell cycle.7–9 RAD51 is a protein that plays a central role in the homologous recombination repair (HRR) pathway.10 The elevated expression of RAD51 has been reported in different cancer types including CC and is associated with poor prognosis of patients,11–13 which may result from the increase in genomic instability and radiation/chemotherapy resistance.14,15
EGFR is a cell surface tyrosine kinase receptor belonging to the ErbB family, and the signaling downstream pathways play vital roles in regulating cellular survival, proliferation, metastasis and angiogenesis.16,17 A handful of studies have demonstrated its role in modulating DDR, and agents inhibiting EGFR signaling have been proven as effective radiation and chemotherapy sensitizers.18–21 However, results of EGFR-targeted agents plus chemo- and/or radiation combination therapy are confusing when applied in CC.22–24 Therefore, further studies of these combinations and seeking other predictive biomarkers as integral components are warranted.
Icotinib hydrochloride (IH), as a new first-generation oral EGFR tyrosine kinase inhibitor (TKI), was developed and patented by Zhejiang Beta Pharma Co., Ltd. (Hangzhou, China). It has shown positive antitumor activities both in vivo and in vitro25,26 and was approved by the State Food and Drug Administration (SFDA) of China in 2011 for advanced non-small-cell lung cancer (NSCLC) patients.27 A randomized, double-blind Phase III study showed that IH was not inferior compared to gefitinib in terms of efficacy, while it had a lower proportion of drug-related adverse events.28 However, it is still not clear whether combining IH with chemotherapy or radiotherapy can be effective in advanced CC. This study not only evaluated the therapeutic efficacy of this combination in vitro but also clarified the potential underlying mechanism that IH could suppress EGFR signaling activation and attenuate RAD51 expression and nuclear foci formation induced by irradiation or cisplatin, therefore suggesting a potential clinical application for CC.
Materials and methods
Cell lines and culture
The human CC cell lines Hela S3 and SiHa were purchased from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China), and the cells were maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) or DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 100 mg/mL penicillin/streptomycin (Solarbio, Beijing, China) at 37°C in a humidified atmosphere with 5% CO2. IH was kindly provided by Zhejiang Beta Pharma Co., Ltd. and dissolved in 100% dimethyl sulfoxide (DMSO) (Solarbio) to a final concentration of 50 μM and stored at −20°C.
Cell Counting Kit-8 (CCK-8) assay
Cells were seeded in 96-well cell culture plates at a density of 5×103 cells/well and incubated in 100 μL of RPMI 1640 medium overnight. On the following day, different concentrations of IH or DMSO were added 2 hours before irradiation (0, 2, 4 and 6 Gy) with 6 MV X-rays generated by a linear accelerator (Siemens, Erlangen, Germany) or cisplatin (0, 1, 2 and 4 μg/mL) (Hospira, Mulgrave, Australia) treatment. For cell viability at the indicated time point, 10 μL of CCK-8 reagent (Dojindo Laboratories, Kumamoto, Japan) was added into each well, and the cells were incubated at 37°C for 2.5 hours. Optical density (OD) values were measured according to the absorbance at 450 nm using a microplate reader (Thermo Fisher Scientific).
Flow cytometry analysis
For cell cycle analysis, 24 hours after exposure to IH (5 μM), radiation (6 Gy), cisplatin (1.5 μg/mL), IH–radiation or IH–cisplatin combination treatment, the cells were harvested and washed twice with ice-cold PBS. After being fixed in 70% cold ethanol for 4 hours, the cells were stained with propidium iodide in the presence of RNase A at 37°C for 30 minutes. The stained cells were then subjected to analysis with a flow cytometer (BD Biosciences, San Jose, CA, USA), and the results of the cell cycle distribution were analyzed by ModFit LT software.
Flow cytometry analysis for apoptosis was performed using the Annexin V-APC/7-AAD apoptosis kit (MultiSciences, Shanghai, China) according to the manufacturer’s protocol. Data were analyzed with FlowJo vX 0.7 software.
Western blotting
Cells were harvested and lysed in RIPA buffer (Beyotime, Haimen, China) for total protein extraction, and cellular cytoplasmic and nuclear fractions were obtained by using the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime) according to the manufacturer’s instructions. After being quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and boiled for 10 minutes in loading buffer containing SDS, proteins were separated via SDS PAGE and subjected to Western blotting. The membrane was detected by using the following primary antibodies: antiphospho-EGFR (Y1068), anti-EGFR, antiphospho-AKT (S473), anti-AKT, antiphospho-ERK1/2 (Y204) and anti-ERK1/2, which were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA); anti-RAD51, anti-β-tubulin, anti-LaminB1 and anti-β-actin which were purchased from Abcam (Cambridge, MA, USA). ImageJ software was used to perform the grayscale analysis.
Colony forming assay
Cells were seeded in 25 mm culture dishes and allowed to adhere overnight. Then, the cells were incubated with 5 μM IH for 2 hours followed by exposure to 2 Gy irradiation or 0.25 μg/mL cisplatin. The cells were further cultured for 10–14 days, and then stained with 0.5% crystal violet in methanol.
Immunofluorescence staining
Cells were seeded on coverslips and treated with 5 μM IH for 2 hours followed by exposure to IR (6 Gy) or cisplatin (1.5 μg/mL). After 24 hours, the cells were fixed using 4% paraformaldehyde for 15 minutes at room temperature and subsequently permeabilized with 0.3% Triton solution for 20 minutes. After being blocked with 5% BSA, the coverslips were stained with anti-γ-H2AX (Ser139) or anti-RAD51 antibodies (Abcam) at a dilution of 1:500 in phosphate buffered saline with Tween 20 (PBST) buffer overnight at 4°C. The cells were then washed three times with PBST, followed by incubation with Alexa Fluor® dye-conjugated secondary antibody (Abcam) for 2 hours at room temperature. Cell nuclei were counterstained with DAPI and visualized using an Axiovert 200M inverted microscope (Zeiss, Oberkochen, Germany).
RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNAs from cell lines were extracted using the RNAiso plus Reagent (Takara, Dalian, China) and reverse transcribed using the PrimeScript® RT reagent kit containing gDNA eRaser (Takara). Then, qRT-PCR was performed using the SYBR® PremixDimer Eraser kit (Takara). The primer sequences used for specific gene amplification were as follows:
RAD51 primer: Forward – CAGTGATGTCCTGGATAATGTAGC
Reverse – TTACCACTGCTACACCAAACTCAT
EGFR primer: Forward – AGGCACGAGTAACAAGCTCAC
Reverse – ATGAGGACATAACCAGCCACC
GAPDH primer: Forward – TGCACCACCAACTGCTTAG
Reverse – AGAGGCAGGGATGATGTTC.
Statistical analysis
Statistical analysis was performed with SPSS software. All results are expressed as mean ± SD, and P<0.05 indicates significance.
Results
IH enhances cisplatin- or radiation-induced proliferation inhibition in Hela S3 cells
In order to identify the proliferation inhibitory effect of different treatments on human CC cells, Hela S3 cells were chosen, grouped and incubated with IH at final concentrations of 0, 2.5, 5, 10 and 20 μmol/L for 2 hours, and then treated with 0, 2, 4 and 6 Gy radiation or 0, 1, 2 and 4 μg/mL cisplatin, respectively. After further culturing for 24 hours, the CCK-8 assay was carried out to detect the cell viability. The results demonstrated that IH inhibited the viability of Hela S3 cells in a concentration-dependent manner, and this suppression effect on proliferation was more obvious when combined with irradiation or cisplatin (Figure 1A and B). The viability of Hela S3 cells separately in the following 72 hours after monotherapy or combination therapy was examined. Compared with the control group, the radiation alone group did not show notable inhibitory effect on cell viability until 72 hours after treatment, while in the combined group, the cell growth rates were significantly decreased from the second day and became more obvious at the third day after treatment (Figure 1C). Besides, 2 μg/mL of cisplatin strongly suppressed cell proliferation in the following 48 hours after treatment, and when combined with IH, the Hela S3 cells totally stopped growing and even cell death appeared in the following 72 hours after treatment (Figure 1D). The proliferation inhibitory effect of IH on the basis of differences in viability of another type of human CC cell line, SiHa, in the following 72 hours, after different treatments, was further confirmed. The IH–IR or IH–cisplatin combination treatment, compared with IR or cisplatin alone, significantly suppressed the proliferation of SiHa cells (Figure S1A). Together, our results suggested that combining IH with chemotherapy or radiotherapy had more significant antiproliferative effect.
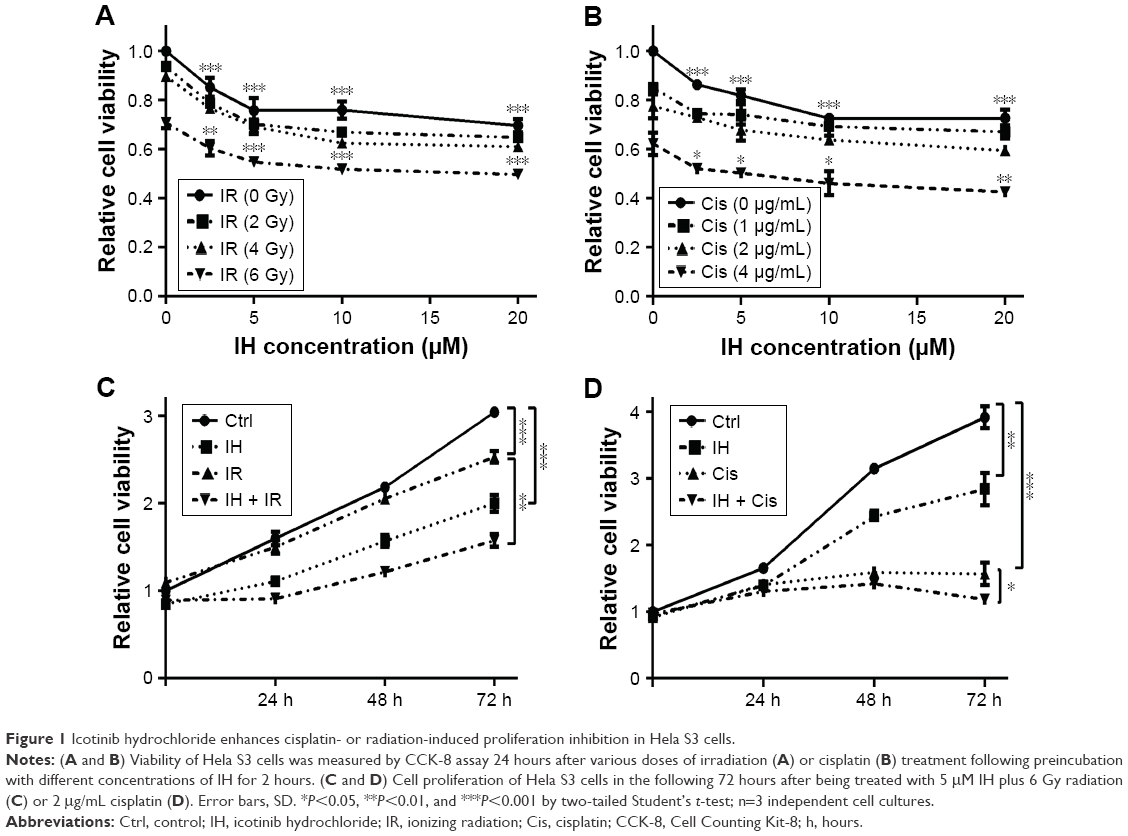 | Figure 1 Icotinib hydrochloride enhances cisplatin- or radiation-induced proliferation inhibition in Hela S3 cells. |
Effect of IH on irradiation- or cisplatin-induced cell cycle arrest
The influence of IH on cell cycle distribution in vitro was measured via flow cytometry (Figure 2). The Hela S3 cells treated with IH alone showed an increase in population in the G0/G1 phase compared with the control group (P<0.01). Radiation alone induced G2/M phase arrest in the Hela S3 cells, and when combined with IH, the G2/M phase arrest was significantly prolonged (29.91%±0.17% vs 23.31%±1.48%, P<0.01) (Figure 2A and C). Additionally, the combined treatment of IH and cisplatin significantly increased the percentage of Hela S3 cells in the G0/G1 phase compared with cisplatin alone (23.01%±0.95% vs 15.67%±1.09%, P<0.01) (Figure 2B and D). Although no significant differences were observed, IH pretreatment also prolonged IR-induced G2/M phase arrest and cisplatin-induced S phase arrest in SiHa cells (Figure S1B). In aggregate, the results of this study suggested that IH treatment could lead to cell cycle redistribution and therefore inhibit cell proliferation and improve the efficacy of radiotherapy and chemotherapy.
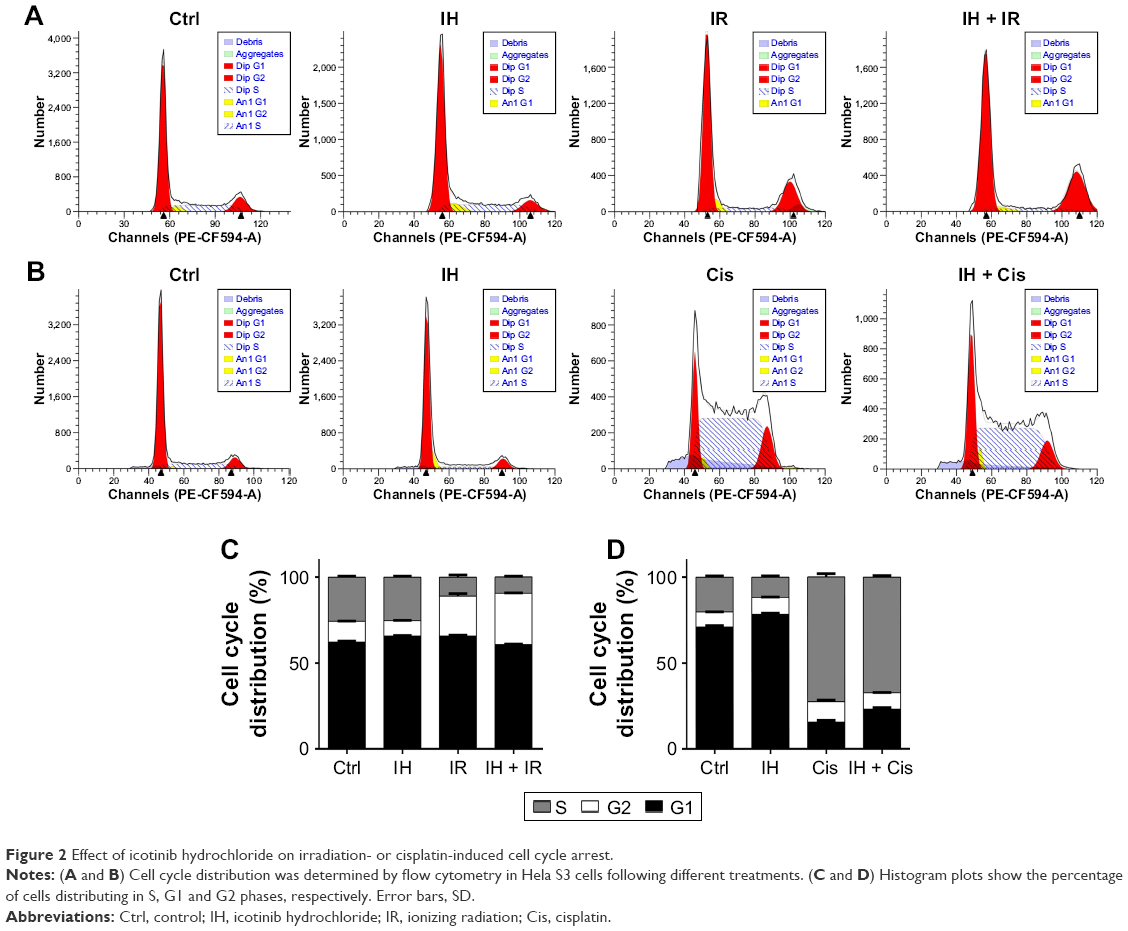 | Figure 2 Effect of icotinib hydrochloride on irradiation- or cisplatin-induced cell cycle arrest. |
Effect of IH on cell apoptosis following irradiation or cisplatin treatment
Whether IH increased IR- or cisplatin-induced apoptosis in Hela S3 cells was evaluated next. The apoptotic rate of the IH plus radiation combination group was 16.34%±2.15%, while it was 9.44%±0.17% in the radiation monotherapy group (P<0.05, Figure 3A and C). For cells in the cisplatin monotherapy group, the apoptotic rate was 23.86%±0.36%, while it increased to 38.70%±0.94% with IH pretreatment (P<0.01, Figure 3B and D). Significant differences were observed between the combined group and the monotherapy group (P<0.05, Figure 3). Similar results were obtained in SiHa cells as the combination group showed higher apoptotic rates than irradiation alone or cisplatin monotherapy groups (26.45%±1.09% vs 22.08%±0.83% and 18.96%±0.95% vs 11.64%±0.52%, respectively) (P<0.05, Figure S1C). Collectively, IH–radiation or IH–cisplatin combination treatment significantly augmented the apoptosis of CC cells compared with monotherapy.
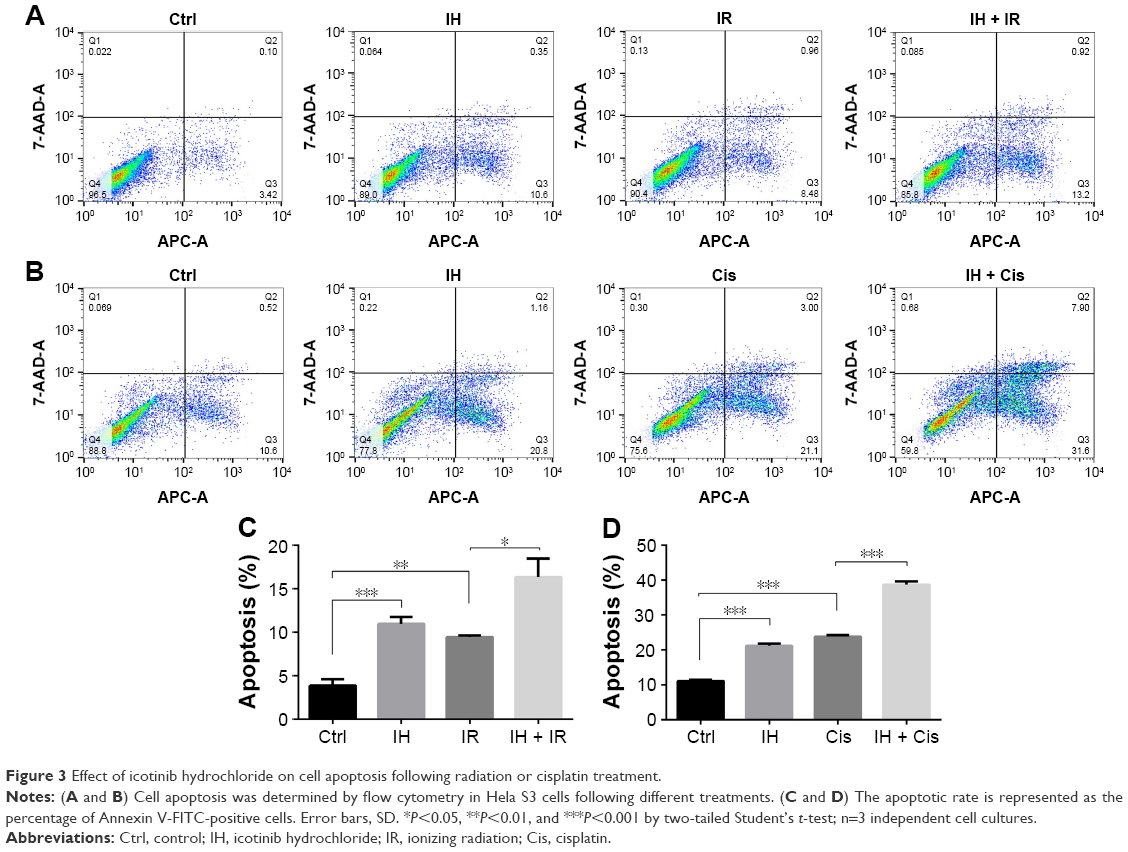 | Figure 3 Effect of icotinib hydrochloride on cell apoptosis following radiation or cisplatin treatment. |
IH inhibits the EGFR signaling pathway in Hela S3 cells
To investigate in depth the biological function of IH, the phosphorylation levels of EGFR and its downstream signaling molecules, including AKT and ERK, in the Hela S3 cells after pretreatment with IH followed by irradiation or cisplatin exposure, were examined (Figure 4). The data showed that both radiation and cisplatin treatments significantly induced high phosphorylation levels of EGFR at the Y1068 residue in Hela S3 cells, as compared with the remaining three groups. IH pretreatment not only inhibited radiotherapy- or chemotherapy-activated phosphorylation of EGFR (Y1068) but also strongly diminished the phosphorylation of the downstream molecules AKT (S473) and ERK (Y204). These results demonstrated the potent inhibitory effect of IH on the EGFR signaling pathway.
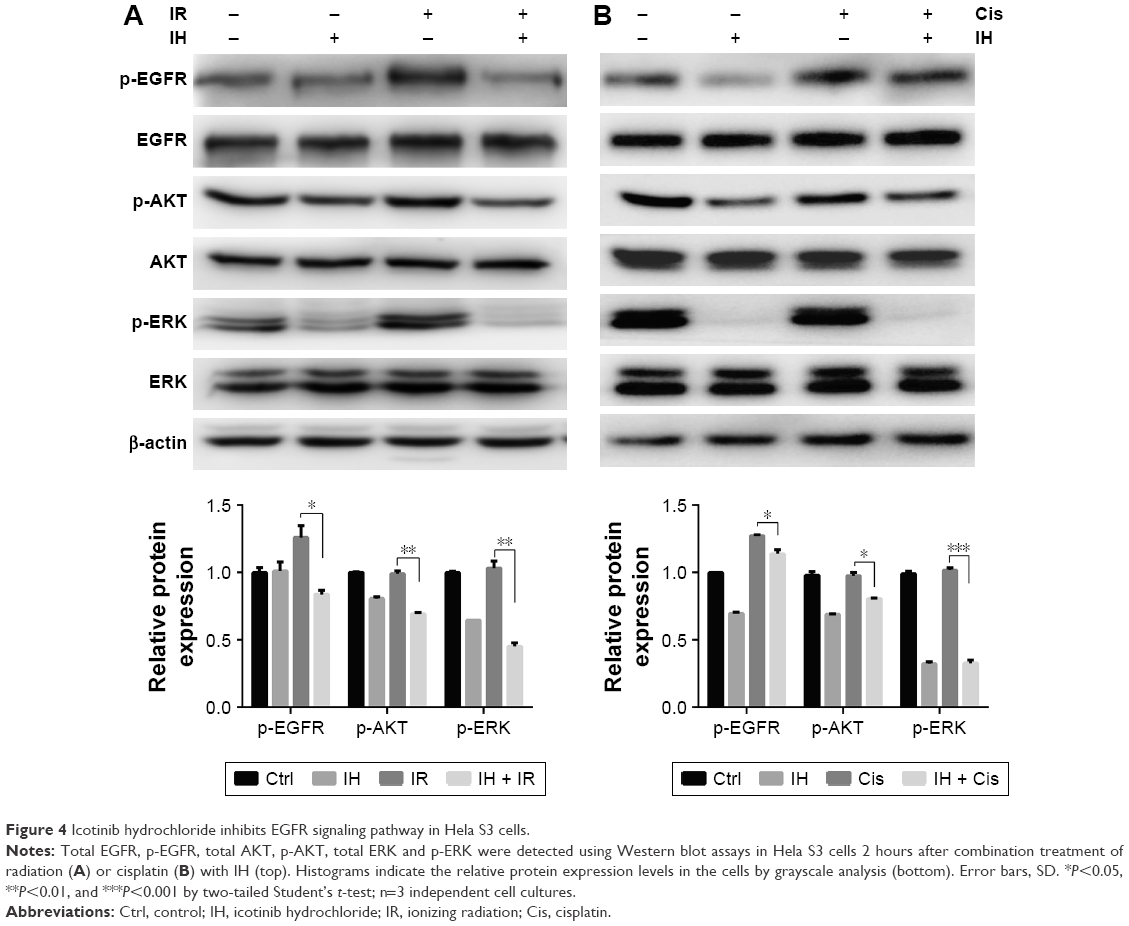 | Figure 4 Icotinib hydrochloride inhibits EGFR signaling pathway in Hela S3 cells. |
Radiation and chemotherapy sensitization of IH by delaying DNA damage repair progress
To further clarify the sensitization of IH to chemotherapy and radiotherapy, colony formation assays were performed, which reflected the long-term biological effects of treatment. The present results confirmed the sensitization effect of IH to chemo/radiotherapy as the combined group grew a lesser number and smaller size of clones at 2 weeks after treatment compared with the monotherapy groups (Figures 5A and S1D). DNA damage repair efficiency after irradiation or cisplatin exposure was measured using γ-H2AX foci formation assays (Figures 5B and C, S1E and F). The γ-H2AX foci number reflected DNA damage initiation and resolution in irradiation- or cisplatin-treated cells. Pretreatment with IH significantly increased the number of γ-H2AX foci positive cells (cells with five or more γ-H2AX foci) 24 hours after exposure to irradiation or cisplatin (Figures 5D and S1G), suggesting that the combined treatment delayed DNA damage repair and improved the efficacy of chemo- and radiotherapy in CC cells.
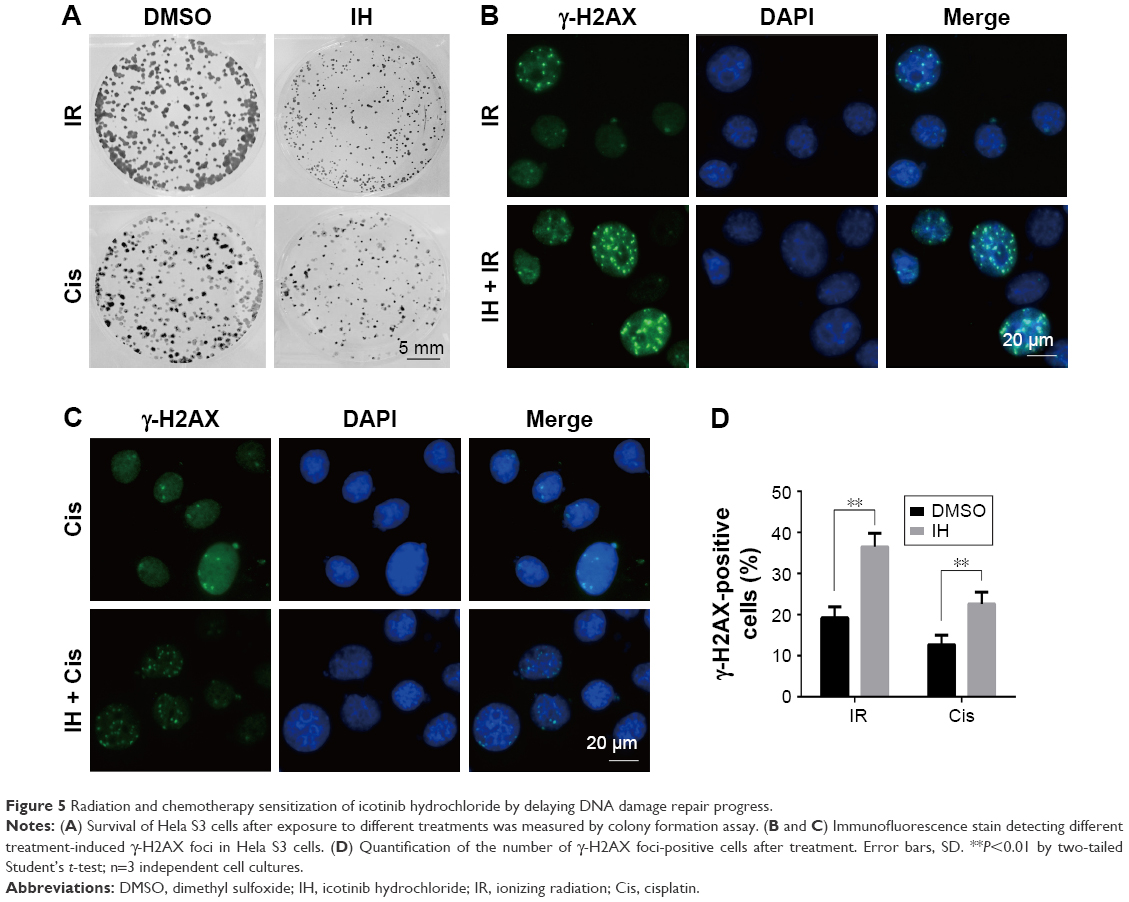 | Figure 5 Radiation and chemotherapy sensitization of icotinib hydrochloride by delaying DNA damage repair progress. |
IH attenuates chemotherapy- or radiotherapy-induced HR protein RAD51 upregulation and nuclear foci formation
RAD51 recombinase is a key protein involved in DSB repair through HR. In this study, the data showed that, in response to DNA damage, the expressions of RAD51 were increased dramatically at both the transcriptional and translational levels in Hela S3 cells, whereas pretreatment with IH suppressed its upregulation significantly (Figure 6A–D). According to previous studies, RAD51 was known to accumulate into subnuclear structures to form foci at the sites of ssDNA that were undergoing repair in response to DNA damage,29 and therefore, RAD51 foci were commonly used as a specific measure of local HRR activity. Results of immunofluorescence staining revealed that the number of cells with more than five RAD51 foci remarkably decreased after IH pretreatment (Figure 6E–G). The same results were obtained when detecting the RAD51 protein levels in nucleus and cytoplasm separately using Western blot assays (Figure 6H). To further confirm this finding, the effects of IH on RAD51 protein expression and function in SiHa cells were analyzed. The present results indicated that IH could also significantly inhibit IR- or cisplatin-induced RAD51 upregulation (Figure S1K and L) as well as nuclear foci formation (Figure S1H–J) in SiHa cells. Taken together, the results of this study showed that the expression and function of RAD51 in CC cells were significantly diminished by pretreatment with IH.
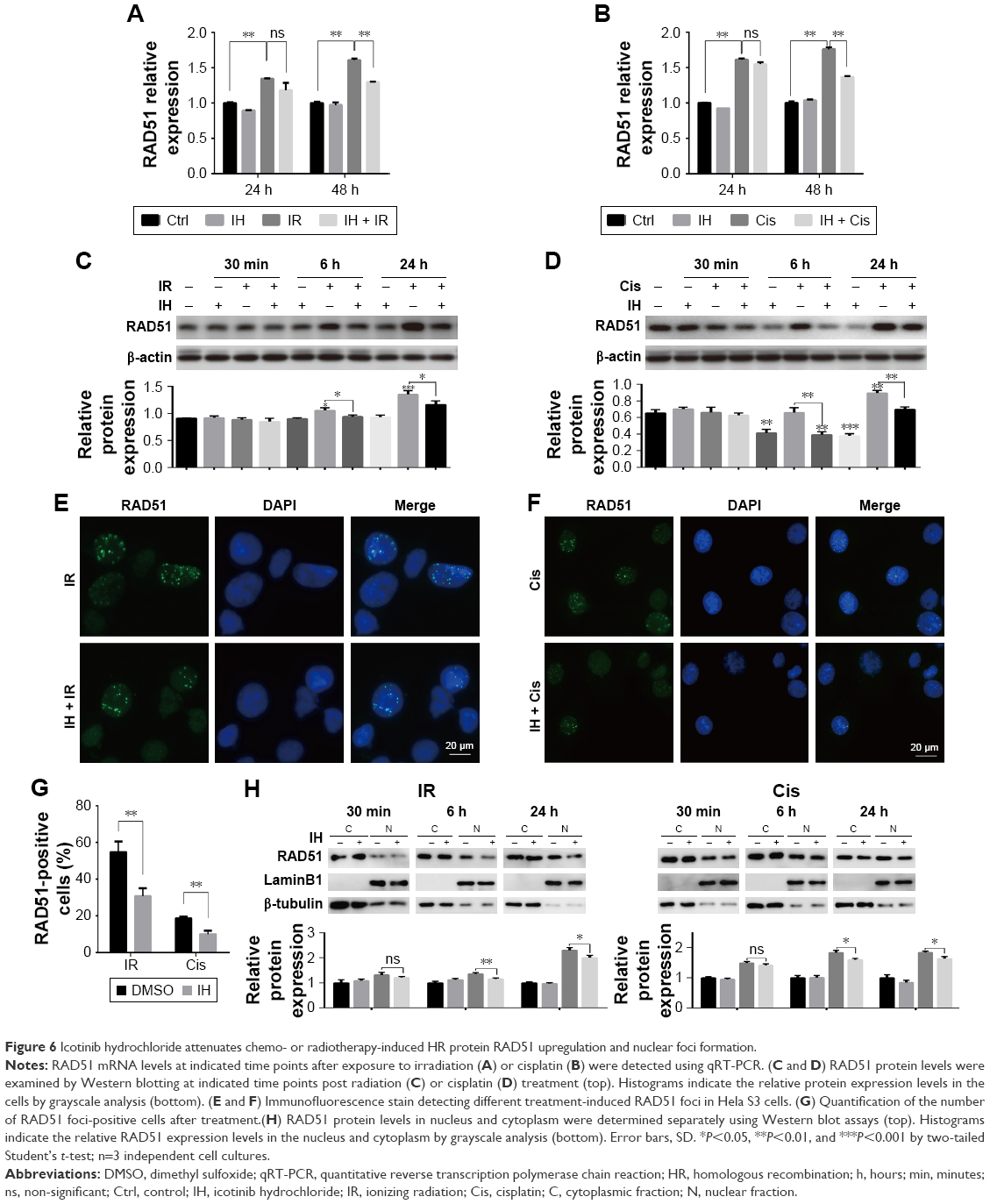 | Figure 6 Icotinib hydrochloride attenuates chemo- or radiotherapy-induced HR protein RAD51 upregulation and nuclear foci formation. |
Discussion
Although EGFR is frequently overexpressed in CC,30–32 and many research data so far, in vitro, have been extremely inspiring,33,34 the results are confusing when applying EGFR inhibitors into clinical trials.22,23,35,36 For example, no clinical responses were detected in a Phase II trial, which evaluated the efficacy and tolerability of cetuximab in persistent or recurrent carcinoma of the cervix.24 However, a multicenter, Phase II trial by another research group about gefitinib treating patients with recurring locoregionally advanced or metastatic CC observed that 20% of the patients had stable disease.22 Besides, another Phase II trial demonstrated that a combination of erlotinib and chemoradiotherapy was well tolerated and had promising activity in locally advanced CC, as 94.4% of the patients achieved a complete response (CR), the 2-year and 3-year overall survival (OS) rates were 91.7 and 80.6% and the progression-free survival (PFS) rates were 80 and 73.8%, respectively.23
It is well-known that EGFR is an important growth factor which binds to its ligand, phosphorylates the tyrosine kinase receptor and stimulates its downstream signaling pathways and therefore participates in the regulation of a number of physiological and pathological processes including cell proliferation.17,37 In this study, a significant proliferation inhibitory effect of IH was observed, especially when it was combined with radiation or cisplatin (Figures 1, 5A and S1A and D). However, compared with Hela S3 cells, the impact of IH on the viability of SiHa cells was relatively minor (Figure S1A). Then, the protein and mRNA levels of EGFR between these two cell lines were analyzed, and the results revealed that SiHa cells expressed significantly lower levels of EGFR than Hela S3 cells (Figure S2). Therefore, it was hypothesized that the efficiency of proliferation inhibition of IH may in part be determined by the EGFR gene expression levels of CC cell lines.
DSBs are highly toxic lesions and lethal injury to the cell.4,38 While IR is one of the most common ways to induce DSBs,8,38 progressive accumulation of DNA lesions following cisplatin treatment could also result in the formation of DSBs.39 Once DNA damage happens, the DDR system will be activated which is followed by either delaying the cell cycle progression to examine and repair the damage or inducing apoptosis to eliminate the cells.40 Using flow cytometry, the impact of IH on cell cycle distribution and apoptosis was studied. We found that, for both Hela S3 and SiHa CC cells, IH treatment alone increased the number of cells in the G0/G1 phase, and when it was combined with radiotherapy, the proportion of cells in G2/M arrest was augmented (Figure 2A and C, Figure S1B). However, opposite results were obtained in these two cell lines treated with IH plus cisplatin as IH pretreatment increased cisplatin-induced S phase arrest in SiHa cells but decreased the proportion of Hela S3 cells in the S phase after cisplatin exposure (Figure 2B and D, Figure S1B). Although further studies are needed to clarify the underlying mechanism, the results of this study demonstrated that IH could cause CC cell cycle redistribution and facilitate the apoptosis induced by radiation or cisplatin (Figures 2, 3, S1B and S1C).
In the presence of DSBs, histone H2AX rapidly posphorylated by ataxia telangiectasia mutated (ATM) on serine 139 and aggregates at the DSB, recruiting other DDR proteins and forming detectable foci.41 Previous studies have demonstrated that γ-H2AX foci reflect the temporal and spatial distribution of DSB formation and will subsequently be abolished with the elimination of DNA damage.42,43 Therefore, visualization and quantification of γ-H2AX via immunofluorescence staining are commonly used ways to evaluate the cell damage repair efficiency. The results of this study showed that IH pretreatment significantly increased the intracellular γ-H2AX foci of Hela S3 and SiHa cells compared with the radiation group and the cisplatin alone group (Figures 5B–D and S1E–G), which suggested that IH could enhance chemo- and radiotherapy sensitivity of CC cells by increasing DSBs and/or blocking DNA repair progression.
The activation of EGFR induced by irradiation or chemotherapeutic agents is associated with cancer cells escaping from death through modulating DDR.19,44,45 Previous studies have demonstrated that EGFR nuclear translocation and interaction with DNA-PKcs resulted in stimulation of DNA-PK kinase activity following exposure to cisplatin or radiation.19,45 Inhibition of EGFR by cetuximab or gefitinib could therefore inhibit the repair of radiation-induced DSBs through impairing the EGFR–DNA–PKcs interaction.46,47 But we failed to observe the inhibitory effect of IH on EGFR nuclear accumulation in Hela S3 cells following irradiation (Figure S3A). In our previous study, we discovered that nimotuzumab, a humanized anti-EGFR monoclonal antibody, could decrease the phosphorylation of DNA repair-related proteins DNA-PKcs and ATM and regulate the RAD51 expression in human esophageal carcinoma cell line KYSE-150R.48 In this study, however, the phosphorylation of DNA-PKcs and ATM induced by exposure to radiation was not diminished (Figure S3B), but the expression of RAD51 was significantly downregulated both in mRNA and protein levels by IH pretreatment in Hela S3 cells (Figure 6A–D). Intriguingly, it was observed that RAD51 in the protein levels was reduced as early as 6 hours after exposure to radiation or cisplatin, while in the mRNA levels it decreased significantly in the following 48 hours after treatment. These results suggested that IH inhibited RAD51 expression not only through modulating RAD51 gene transcription but also promoting RAD51 protein degradation. Notably, Chen et al49 discovered previously that RAD51 expression was regulated by ERK activation. The expression of constitutively active MKK1/2 vectors (MKK1/2-CA) significantly rescued the reduced RAD51 levels on gefitinib treatment, and the blocking of ERK1/2 activation could lower RAD51 protein levels in emodin-treated H1650 and A549 cells. Given the strong inhibitory effect of IH on the EGFR pathway especially on the phosphorylation of ERK (Figure 4), we therefore hypothesized that there might exist an EGFR-ERK-RAD51 axis and that IH regulates RAD51 levels indirectly by inhibiting EGFR activity. In addition, the immunofluorescence staining results (Figures 6E–G and S1H–J) indicated that the recruitment of RAD51 to DNA damage foci induced by exposure to irradiation or cisplatin was also hampered when combined with IH. These results were confirmed by separating and detecting subcellular protein expressions using Western blot assays (Figures 6H and S1l) as nuclear RAD51 was significantly decreased in the IH combination groups compared with radiation or cisplatin monotherapy groups.
Conclusion
This study demonstrated that IH, an oral potent EGFR-TKI, could enhance the sensitivity of Hela S3 cells to chemo- and radiotherapy by efficiently inhibiting the EGFR downstream signaling pathway, delaying DNA damage repair and suppressing RAD51 upregulation and nuclear foci formation.
Our work suggested that IH is a potent clinical application option in radiotherapy and cisplatin-based chemotherapy for CC, and RAD51 may serve as a candidate biomarker for identifying patients who are most likely to benefit from this combination treatment.
Acknowledgment
This study was supported in part by grants from the Chinese National Natural Science Foundation (No 81441086 and 81672976), Natural Science Foundation of Zhejiang Province (No LY14H160016), and Major Science and Technology Program of Zhejiang Province (No 2013C03044-6).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Vaccarella S, Lortet-Tieulent J, Plummer M, Franceschi S, Bray F. Worldwide trends in cervical cancer incidence: impact of screening against changes in disease risk factors. Eur J Cancer. 2013;49(15):3262–3273. | ||
Long HJ 3rd, Bundy BN, Grendys EC Jr, et al; Gynecologic Oncology Group Study. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2005;23(21):4626–4633. | ||
Featherstone C, Jackson SP. DNA double-strand break repair. Curr Biol. 1999;9(20):R759–R761. | ||
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. | ||
Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair. 2004;3(8–9):781–796. | ||
Cromie GA, Connelly JC, Leach DR. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell. 2001;8(6):1163–1174. | ||
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–143. | ||
Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–5715. | ||
San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. | ||
Maacke H, Opitz S, Jost K, et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;88(6):907–913. | ||
Li Y, Wang WY, Xiao JH, et al. Overexpression of Rad51 predicts poor prognosis in colorectal cancer: our experience with 54 patients. PLoS One. 2017;12(1):e0167868. | ||
Chen Q, Cai D, Li M, Wu X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol Rep. 2017;38(2):767–774. | ||
Mason JM, Logan HL, Budke B, et al. The RAD51-stimulatory compound RS-1 can exploit the RAD51 overexpression that exists in cancer cells and tumors. Cancer Res. 2014;74(13):3546–3555. | ||
Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair. 2008;7(5):686–693. | ||
Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. | ||
Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 suppl):21–26. | ||
Kriegs M, Kasten-Pisula U, Rieckmann T, et al. The epidermal growth factor receptor modulates DNA double-strand break repair by regulating non-homologous end-joining. DNA Repair. 2010;9(8):889–897. | ||
Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011;71(3):1103–1114. | ||
Morgan MA, Parsels LA, Kollar LE, Normolle DP, Maybaum J, Lawrence TS. The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin Cancer Res. 2008;14(16):5142–5149. | ||
Welsh JW, Komaki R, Amini A, et al. Phase II trial of erlotinib plus concurrent whole-brain radiation therapy for patients with brain metastases from non-small-cell lung cancer. J Clin Oncol. 2013;31(7):895–902. | ||
Goncalves A, Fabbro M, Lhomme C, et al. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol. 2008;108(1):42–46. | ||
Nogueira-Rodrigues A, Moralez G, Grazziotin R, et al. Phase 2 trial of erlotinib combined with cisplatin and radiotherapy in patients with locally advanced cervical cancer. Cancer. 2014;120(8):1187–1193. | ||
Santin AD, Sill MW, McMeekin DS, et al. Phase II trial of cetuximab in the treatment of persistent or recurrent squamous or non-squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;122(3):495–500. | ||
Tan F, Shen X, Wang D, et al. Icotinib (BPI-2009H), a novel EGFR tyrosine kinase inhibitor, displays potent efficacy in preclinical studies. Lung Cancer. 2012;76(2):177–182. | ||
Zhao Q, Shentu J, Xu N, et al. Phase I study of icotinib hydrochloride (BPI-2009H), an oral EGFR tyrosine kinase inhibitor, in patients with advanced NSCLC and other solid tumors. Lung Cancer. 2011;73(2):195–202. | ||
Camidge DR. Icotinib: kick-starting the Chinese anticancer drug industry. Lancet Oncol. 2013;14(10):913–914. | ||
Shi Y, Zhang L, Liu X, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14(10):953–961. | ||
Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750. | ||
Kersemaekers AM, Fleuren GJ, Kenter GG, et al. Oncogene alterations in carcinomas of the uterine cervix: overexpression of the epidermal growth factor receptor is associated with poor prognosis. Clin Cancer Res. 1999;5(3):577–586. | ||
Li Q, Tang Y, Cheng X, Ji J, Zhang J, Zhou X. EGFR protein expression and gene amplification in squamous intraepithelial lesions and squamous cell carcinomas of the cervix. Int J Clin Exp Pathol. 2014;7(2):733–741. | ||
Noordhuis MG, Eijsink JJ, Ten Hoor KA, et al. Expression of epidermal growth factor receptor (EGFR) and activated EGFR predict poor response to (chemo)radiation and survival in cervical cancer. Clin Cancer Res. 2009;15(23):7389–7397. | ||
Baguley BC, Marshall ES, Holdaway KM, Rewcastle GW, Denny WA. Inhibition of growth of primary human tumour cell cultures by a 4-anilinoquinazoline inhibitor of the epidermal growth factor receptor family of tyrosine kinases. Eur J Cancer. 1998;34(7):1086–1090. | ||
Dai Q, Ling YH, Lia M, et al. Enhanced sensitivity to the HER1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib hydrochloride in chemotherapy-resistant tumor cell lines. Clin Cancer Res. 2005;11(4):1572–1578. | ||
de la Rochefordiere A, Kamal M, Floquet A, et al. PIK3CA pathway mutations predictive of poor response following standard radiochemotherapy ± cetuximab in cervical cancer patients. Clin Cancer Res. 2015;21(11):2530–2537. | ||
Nogueira-Rodrigues A, do Carmo CC, Viegas C, et al. Phase I trial of erlotinib combined with cisplatin and radiotherapy for patients with locally advanced cervical squamous cell cancer. Clin Cancer Res. 2008;14(19):6324–6329. | ||
Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203–212. | ||
Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. | ||
Nowosielska A, Marinus MG. Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA Repair. 2005;4(7):773–781. | ||
Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:16. | ||
Rass E, Chandramouly G, Zha S, Alt FW, Xie A. Ataxia telangiectasia mutated (ATM) is dispensable for endonuclease I-SceI-induced homologous recombination in mouse embryonic stem cells. J Biol Chem. 2013;288(10):7086–7095. | ||
Mah LJ, Orlowski C, Ververis K, et al. Evaluation of the efficacy of radiation-modifying compounds using gammaH2AX as a molecular marker of DNA double-strand breaks. Genome Integr. 2011;2(1):3. | ||
Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24(4):679–686. | ||
Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol. 2007;83(11–12):781–791. | ||
Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res. 2009;1(3):249–258. | ||
Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005;76(2):157–161. | ||
Friedmann B, Caplin M, Hartley JA, Hochhauser D. Modulation of DNA repair in vitro after treatment with chemotherapeutic agents by the epidermal growth factor receptor inhibitor gefitinib (ZD1839). Clin Cancer Res. 2004;10(19):6476–6486. | ||
Liu H, Yang W, Gao H, et al. Nimotuzumab abrogates acquired radioresistance of KYSE-150R esophageal cancer cells by inhibiting EGFR signaling and cellular DNA repair. Onco Targets Ther. 2015;8:509–518. | ||
Chen RS, Jhan JY, Su YJ, et al. Emodin enhances gefitinib-induced cytotoxicity via Rad51 downregulation and ERK1/2 inactivation. Exp Cell Res. 2009;315(15):2658–2672. |
Supplementary materials
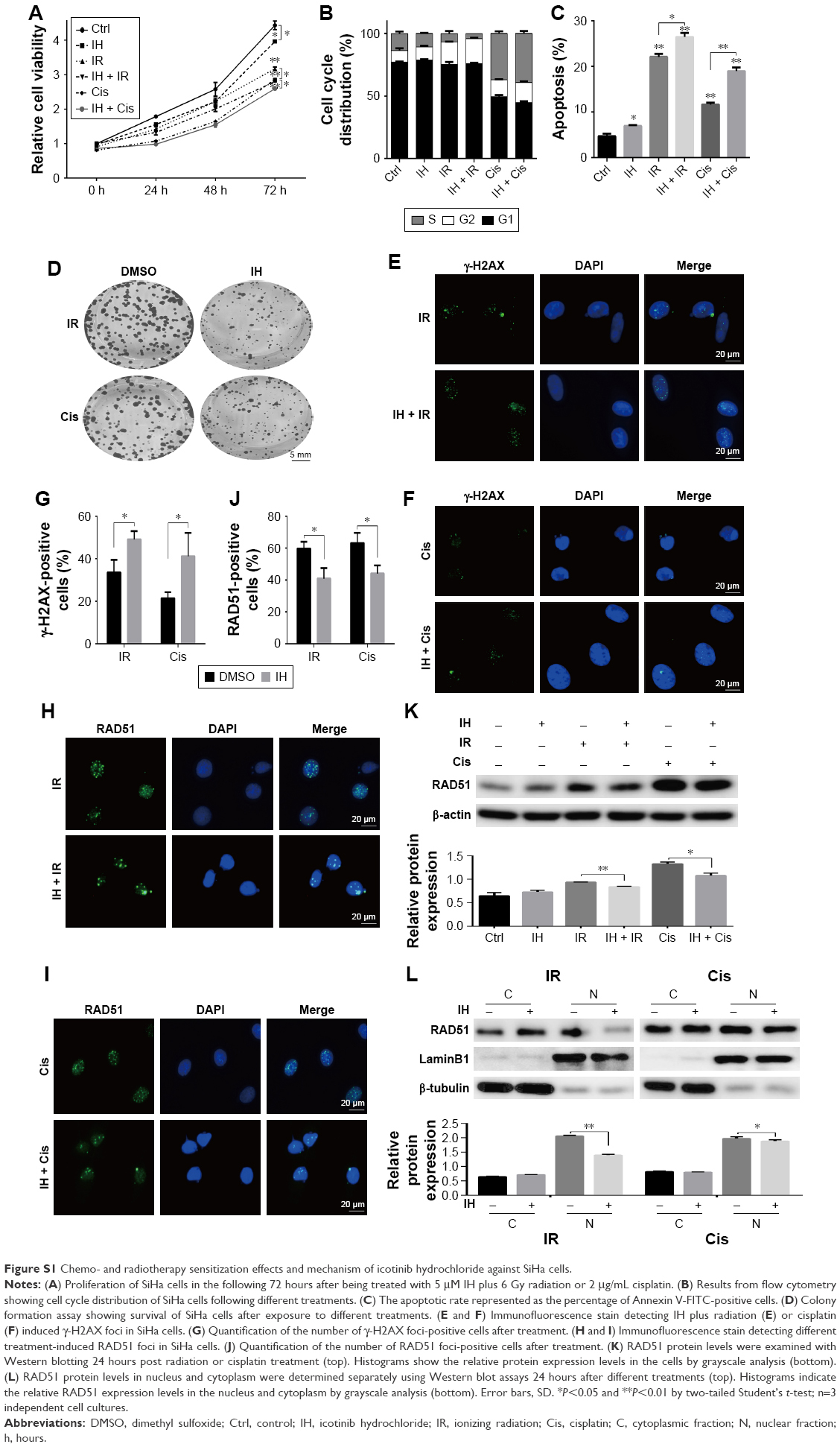 | Figure S1 Chemo- and radiotherapy sensitization effects and mechanism of icotinib hydrochloride against SiHa cells. |
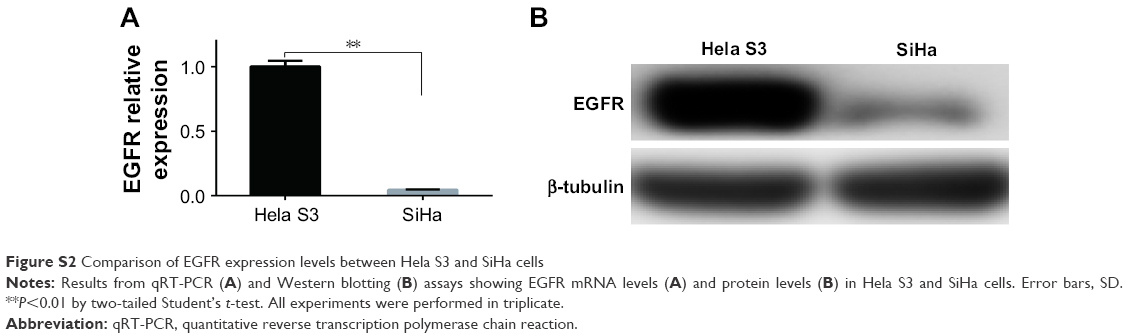 | Figure S2 Comparison of EGFR expression levels between Hela S3 and SiHa cells |
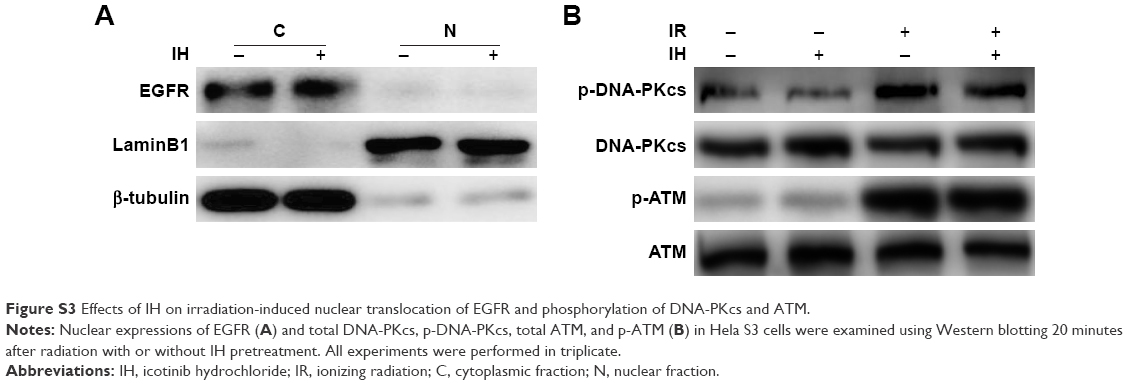 | Figure S3 Effects of IH on irradiation-induced nuclear translocation of EGFR and phosphorylation of DNA-PKcs and ATM. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.