Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Icosapent ethyl for the treatment of severe hypertriglyceridemia
Authors Fares H, Lavie CJ, DiNicolantonio J, O'Keefe J, Milani R
Received 8 January 2014
Accepted for publication 19 February 2014
Published 24 June 2014 Volume 2014:10 Pages 485—492
DOI https://doi.org/10.2147/TCRM.S36983
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Hassan Fares,1 Carl J Lavie,2,3 James J DiNicolantonio,4 James H O'Keefe,5 Richard V Milani2
1Department of Hospital Medicine, Ochsner Medical Center, New Orleans, LA, 2Department of Cardiovascular Diseases, John Ochsner Heart and Vascular Institute, Ochsner Clinical School, University of Queensland School of Medicine, New Orleans, LA, 3Department of Preventive Medicine, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, LA, 4Mid America Heart Institute at Saint Luke's Hospital, Kansas City, MO, 5Mid America Heart Institute, University of Missouri–Kansas City, Kansas City, MO, USA
Abstract: Hypertriglyceridemia is a highly prevalent lipid abnormality and it is associated with atherosclerosis, with a growing body of evidence linking elevated triglycerides (TGs) with cardiovascular disease. The current major omega-3 polyunsaturated fatty acids, eicosapentaenoic acid (EPA)/docosahexaenoic acid (DHA) combination, lowers serum TGs while often increasing levels of low-density lipoprotein cholesterol. Icosapent ethyl is an omega-3 polyunsaturated fatty acid with a 96% pure ethyl ester of EPA that has been recently approved for lowering TG levels in patients with very high TGs (≥500 mg/dL), and it does so without significantly affecting serum low-density lipoprotein cholesterol. The potential benefits of omega-3 fatty acid therapy for dyslipidemias will be discussed, including the potential pros and cons of EPA alone versus the more common and readily available EPA/DHA combination therapy.
Keywords: triglycerides, low-density lipoprotein, eicosapentaenoic acid, docosahexaenoic acid
Introduction
Hypertriglyceridemia (HTG) is a highly prevalent lipid abnormality that is frequently associated with other lipid and metabolic aberrations. HTG is classified into two categories: primary and secondary. Primary HTG is the result of genetic defects leading to disordered triglyceride (TG) metabolism such as familial combined hyperlipidemia and familial HTG, lipoprotein lipase deficiency, apolipoprotein C-2 deficiency, and familial dysbetalipoproteinemia.1 Secondary HTG is an acquired state, which can be attributed to lifestyle factors such as diet with high carbohydrate or high glycemic index content, alcohol consumption, uncontrolled diabetes mellitus, hypothyroidism, medications such as nonselective beta-blockers, thiazide diuretics, oral estrogens, glucocorticosteroids, and tamoxifen.2 Elevated serum levels of TGs are associated with atherosclerosis, and there is a growing body of evidence that links HTG with cardiovascular disease (CVD).3 However, the role of TGs in CVD risk assessment is still controversial. Although very high levels of serum TGs (≥500 mg/dL) is a well-established risk factor for acute pancreatitis,4 and while some meta-analyses have suggested that TGs are an independent risk factor for CVD,4 there is an emerging opinion that TGs may be better suited as a biomarker of risk for CVD rather than an independent risk factor.5 Indeed, a recent ruling by the United States Food and Drug Administration (FDA) that it will no longer approve drugs that lower TGs (in the <500 mg/dL range) without evidence that such drugs actually reduce risk for CHD events reflects this opinion.6
In patients with fasting TG levels of ≥200 mg/dL and <500 mg/dL, low-density lipoprotein cholesterol (LDL-C) becomes the primary lipid target, and statins are the mainstay of treatment for LDL-C-lowering therapy.7 If the TGs levels fail to improve with optimal statin therapy, then adjunctive therapy can be instituted. Omega-3 polyunsaturated fatty acids (PUFAs) are a type of adjunctive therapy that has been heavily investigated.7 The current guidelines to lower serum TGs, as recommended by the National Cholesterol Education Program Adult Treatment Panel III and the 2011 American Heart Association scientific statement, include weight loss by consuming a low-fat diet and engaging in physical activity, smoking cessation, limiting alcohol intake, and treatment with statins, omega-3 PUFAs, fibrates, or niacin.3,8,9
Interest in the benefits of omega-3 PUFAs arose from numerous observational studies of the rare occurrence of CVD noted among the Inuit in Greenland, whose diet consisted primarily of fish and seafood.9 Since 1944, Sinclair reported lower cholesterol and TG levels in this population, despite the fact that their diet lacked any substantive fruits, vegetables, and complex carbohydrates, but it was high in cholesterol and saturated fats.9 Since then, there has been increasing interest in the health benefits of supplemental dietary omega-3 PUFAs, particularly in their role in disease prevention. There have been numerous studies published on this topic of cardioprotection of omega-3 PUFAs. While some of the larger trials have not shown significant benefits, several observational studies have since ascertained the favorable role of omega-3 PUFAs on CVD prevention.11
The omega-3 PUFAs that are essential in human physiology are α-linolenic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). The former is termed essential because the human body is incapable of synthesizing it de novo; the latter two can be synthesized from α-linolenic acid and thus are “conditionally” essential.11 EPA and DHA, the “marine” omega-3 PUFAs, are mainly obtained by eating oily fish, such as albacore tuna, salmon, mackerel, herring, and sardines, or by consuming fish oil supplements. Interestingly, omega-3 PUFAs are not naturally synthesized in fish either, but rather they are obtained by consuming marine microorganisms, such as microalgae, which are the primary producers of EPA and DHA.12
There is growing evidence to support the beneficial effects of prescription omega-3 PUFAs on TGs. Nonprescription fish oil dietary supplements are controversial since they are not subject to the US FDA regulations, as are prescription drugs. Prescription drugs are required to have well-controlled studies to demonstrate safety, efficacy, potency, and purity. This is not the case for dietary supplements. The US FDA has approved Lovaza® (GlaxoSmithKline, Brentford, UK), which contains an omega-3 PUFA ethyl ester formulation with both EPA and DHA, at a dosage of 4 g/day for the treatment of HTG with TG levels >500 mg/dL. Icosapent ethyl (Vascepa®; Amarin Pharma Inc., Bedminster, NJ, USA) is an omega-3 PUFA with a 96% pure ethyl ester of EPA that has been recently approved to lower TG levels in patients with elevated TGs of ≥500 mg/dL.13 This paper will attempt to review omega-3 fatty PUFAs, specifically focusing on icosapent ethyl, and their effects on lipid metabolism.
Review of the pharmacology and pharmacokinetics of icosapent
Lipid handling in human circulation is primarily achieved by the use of lipoproteins, of which five different types are responsible for the transport of TGs from the liver to the peripheral tissues. Of these, the chylomicrons (CM) and very LDL (VLDL) are the most TG-rich lipoproteins.14 Table 1 gives an overview of the main modalities of TG transport. In the peripheral tissues, TGs are hydrolyzed through the action of lipoprotein lipase (LPL) in the circulation, and the remnants of the CM and VLDL are left to circulate. It is these remnants that have been implicated in atherogenesis.15 After losing most of their TGs components in the periphery, CM become CM remnants and continue to deliver the remaining TGs to the liver.15
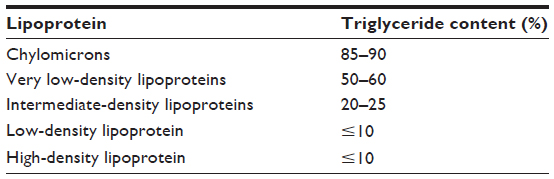 | Table 1 Packaging and transport of triglycerides |
Fasting and postprandial TG levels are reduced by omega-3 PUFAs, but the exact mechanisms are not completely understood. Some investigators have proposed that omega-3 PUFAs exert their TG-lowering effects primarily through the reduction of VLDL-based TG production, which occurs in the liver.16 Digestion of TGs in the diet results in fatty acids (FAs) that are then reformed into TGs in the enterocyte and packaged into CMs. Once in the bloodstream, nonesterified FAs, which are the main source of FAs, are released from CM remnants by degradation from LPL, and also from adipose tissue through the action of hormone-sensitive lipase.17 Omega-3 PUFAs exert their effect on hormone-sensitive lipase-mediated lipolysis by suppressing adipose tissue inflammation, hence decreasing the availability of nonesterified FAs in the circulation.17 Furthermore, EPA increases LPL-mediated extracellular lipolysis, thus enhancing VLDL clearance from the circulation.17 By decreasing the amount of free FAs delivered to the liver, less free substrates will be recruited in VLDL-TG synthesis and in their secretion from the hepatic tissue.17
There are various mechanisms that have been proposed that can account for the previously mentioned sequence of events. Results from animal studies have suggested the mechanisms by which PUFAs reduce TGs, and these include reduced FA (substrate) availability or by decreasing the activity of key enzymes that synthesize TGs, namely diacylglycerol acyltransferase or phosphatidic acid phosphodyrolase.18 Currently, it is hypothesized that EPA and/or DHA inhibit phosphatidic acid phosphodyrolase and diacylglycerol acyltransferase, thus decreasing TG synthesis. An animal study on mice that were fed for 4 months with omega-3 PUFAs showed that overall, fasting plasma levels of TGs were reduced and LPL activity was higher.19 Additionally, the study revealed that omega-3 PUFAs also facilitate the exogenous removal of TG-rich particles. These data suggest that fish sources of omega-3 PUFAs lower TGs by reducing endogenous TG synthesis by increasing LPL activity, and this increases the blood clearance of TG-rich particles.19
Some of the actions of omega-3 PUFAs are also mediated via surface or intracellular receptors that elicit gene transcription events. These include liver X receptor, hepatocyte nuclear factor 4 alpha, farnesol X receptor, and peroxisome proliferator-activated receptors (PPARs). These receptors are all controlled by sterol receptor element binding protein-1c, which is a key regulator of lipogenesis. The PPARs are transcription factors that regulate gene expression, and PPARα is an isoform that is predominantly expressed in the liver and enhances β-oxidation in response to FA in the liver and skeletal muscles.20,21 Figure 1 illustrates the different subtypes of PPARs and their roles. PPARγ is expressed in adipocytes and inflammatory cells where it mediates insulin sensitivity and the production of inflammatory mediators.22 These suggest that PPAR activation is a probable mechanism by which PUFAs are able to lower TGs, reduce inflammation, and increase insulin sensitivity.20 Therefore, reducing sterol receptor element binding protein reduction and upregulating FA oxidation in the liver and skeletal muscles through PPAR activation, and downregulation of hepatocyte nuclear factor 4 alpha will result in an increased rate of β-oxidation of FA in the liver and, hence, less TGs will be shuffled into the TG synthetic pathway.23 The net effect is less TG availability for incorporation into VLDL particles.
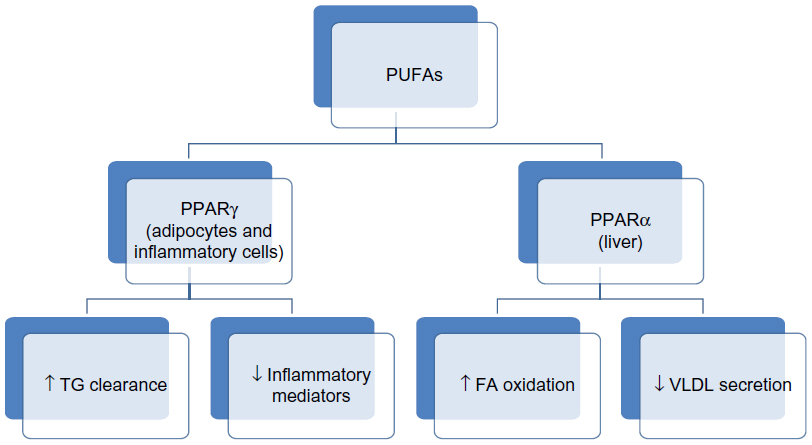 | Figure 1 Mechanism of action of PUFAs in lowering the hepatic production of TGs. |
It is important to note that the amount of omega-3s that are fed to the animal in the studies noted earlier may not be comparable, as they are generally much higher than the amounts typically consumed by humans on a per kg basis. Therefore, this is an important consideration when trying to extrapolate these data to humans. Additionally, the differences in lipoprotein metabolism between different animal species and humans also need to be taken into account.24 For these reasons, the data on animal studies need to be substantiated by additional studies on humans.
EPA requires a high-fat meal in order to be better absorbed.25–27 The peak plasma concentrations levels are reached 5 hours after oral doses are administered. The mean volume of distribution at steady state is about 88 L/kg. Only <1% of the circulating plasma levels of EPA are unesterified FA, while the remaining are incorporated in phospholipids, TGs, and cholesteryl esters.13 The liver, via beta-oxidation, metabolizes EPA and the plasma elimination half-life is approximately 89 hours, and EPA is not excreted by the renal system.7, 20
Eicosapentaenoic acid ethyl ester for treatment of hypertriglyceridemia
The MARINE study28 (Multi-Center, Placebo-Controlled, Randomized, Double-Blind, 12-week study with an open-label Extension) studied the TG-lowering effect and impact on other lipid parameters of icosapent ethyl in severe HTG patients who had TG levels ≥500 mg/dL. Other cholesterol levels including LDL, VLDL, high-density lipoprotein (HDL), non-HDL, total cholesterol (TC), and lipoprotein-associated phospholipase A3 were also assessed.29 A total of 224 patients (24.9% had TGs ≥500 mg/dL and 39.3% had TGs >750 mg/dL) were randomized to icosapent ethyl 4 g/day, icosapent ethyl 2 g/day, or placebo over a 12-week period. The results showed that 4 g/day reduced TG levels by 35.7% and 2 g/day reduced TG levels by 24.9% in patients with TGs ≥500 mg/dL. In patients with baseline TGs of >750 mg/dL, 4 g/day decreased TGs by 45.4% while 2 g/day decreased TGs by 32.9%. Overall, icosapent ethyl 4 g/day reduced TG levels by 33.1% (P<0.0001) from baseline and 2 g/day reduced TG levels by 19.7% (P=0.0051).28 More reductions in TGs were seen in patients with a baseline TG >750 mg/dL and with concurrent statin therapy, as icosapent ethyl 4 g/day with statin reduced TG levels by 65% (P=0.0001) and 2 g/day reduced TG levels by 40.7% (P=0.0276). This may indicate a possible synergy between icosapent ethyl and statins. Interestingly, icosapent ethyl did not simultaneously increase LDL-C levels. A follow-up analysis29 reported the effects of icosapent ethyl on lipoprotein particle concentration and size, showing that 4 g/day significantly reduced the concentration of the large VLDL particles by 27.9% (P=0.0211), total LDL by 16.3% (P=0.0006), small LDL by 25.6% (P<0.0001), and total HDL by 7.4% (P<0063). This information is summarized in Figure 2. The 2 g/day did not have the statistically significant reductions on these lipid parameters.
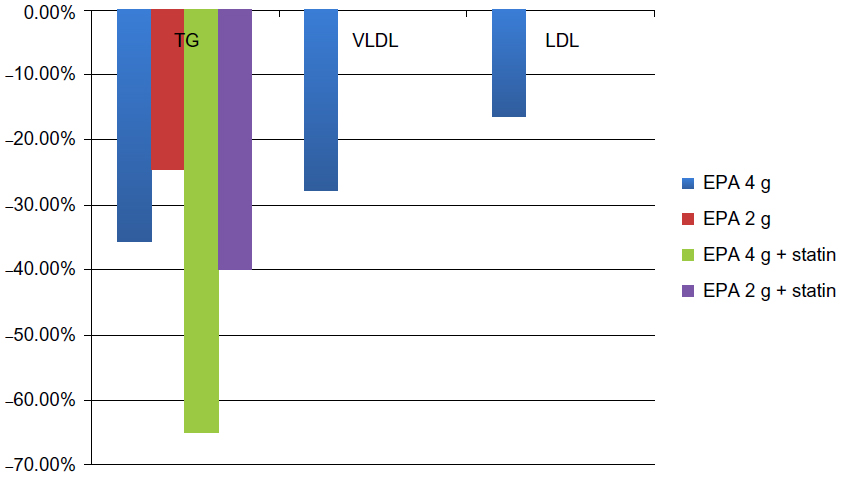 | Figure 2 Results of the MARINE trial. |
The ANCHOR study7 assessed the safety and efficacy of icosapent ethyl in patients who were at high risk of cardiovascular (CV) events, but who had adequate control of their LDL-C on statin therapy and persistently elevated TG levels of ≥200 mg/dL but <500 mg/dL. Over a 12-week period, 702 patients over age 18 years and at high risk for CVD were randomized to icosapent ethyl 4 g/day, icosapent ethyl 2 g/day, or placebo. The primary endpoint was TG level change from baseline to the end of 12 weeks. Secondary endpoints included the percent change in non-HDL and LDL-C, VLDL, apolipoprotein-B, and phospholipase A2. The ANCHOR study7 results showed that icosapent ethyl 4 g/day and 2 g/day decreased TG levels by 21.5% and 10.1%, respectively. Icosapent ethyl 4 g/day did not increase LDL-C, but rather decreased LDL levels by an additional 6.2% compared to the placebo group. Figure 3 gives an overview of these results. Icosapent ethyl also decreased non-HDL cholesterol, VLDL, TC, and HDL. Additionally, there were reductions in high-sensitivity C-reactive protein and lipoprotein-associated phospholipase A2, leading to decreased inflammation while stabilizing endothelial function, resulting in decreased CV events.
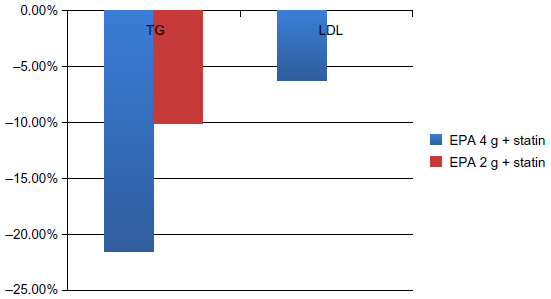 | Figure 3 Results of the ANCHOR trial. |
One limitation with regards to data interpretation in the EPA and EPA/DHA trials is that the studies used different placebos.30 EPA studies used light paraffin oil as a placebo, whereas the EPA/DHA used corn oil. The placebo had differing effects on other lipid parameters. Corn oil had no change or a very minimal decrease from the initial baseline, whereas paraffin oil increased most other lipid parameters. This is more prevalent in studies with statin-treated patients. Due to this fact, the EPA data appear to illustrate that EPA has an impressive impact in decreasing these various lipids levels.32 There were substantial changes in the placebo group that were similar to the changes observed in the active treatment arms for many lipid variables, but in an unfavorable direction, which may make the benefits of EPA (the only therapy noted in the trials mentioned earlier) seem greater. However, when comparing EPA to regular baseline levels, the data are less impressive. Therefore, caution must be taken in interpreting data when comparing different placebos that can lead to divergent effects.
While both EPA and DHA have been shown to be efficacious on TGs, studies have shown that they have very divergent effects on lipid profiles. A meta-analysis of 21 randomized controlled trials of EPA and DHA that compared serum lipids found that both EPA and DHA significantly reduced TGs when compared to placebo.31 Studies that directly compared EPA versus DHA on TGs showed a greater reduction via DHA by 6.14 mg/dL (95% confidence interval: 2.47–9.82). Another study showed that DHA had a net decrease of TGs by 6.8% compared to EPA.32 The overall pooled estimate of the studies showed that EPA had a nonsignificant reduction in LDL-C compared with placebo.31 DHA, on the other hand, significantly increased LDL by 7.23 mg/dL when compared to placebo.31 When compared against each other, DHA significantly increased LDL by 4.63 mg/dL compared to EPA.31 Another review of the studies found that DHA had a 3.3% net increase of LDL-C compared with EPA.32 EPA had a nonsignificant increase in HDL compared to placebo, whereas DHA significantly increased HDL by 4.49 mg/dL compared to placebo.31 DHA increased HDL by 3.74 mg/dL when compared with EPA.31 DHA had a net increase of 5.9% on HDL when compared with EPA.32 Figure 4 summarizes these lipid effects of EPA versus DHA.
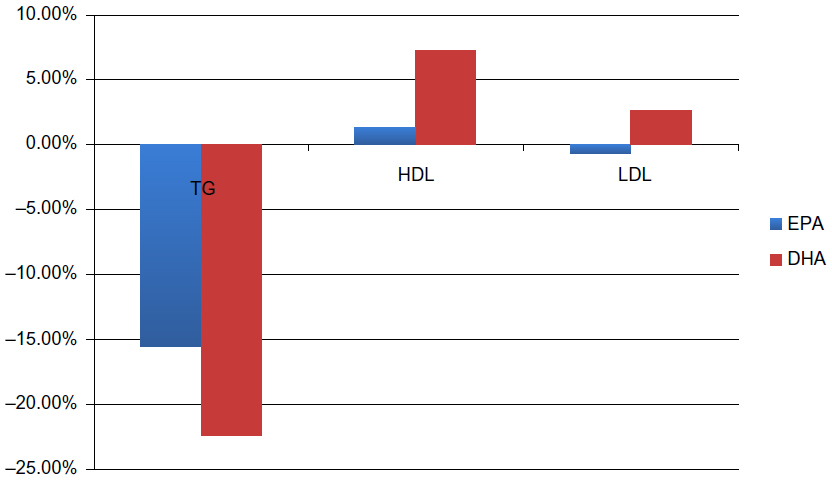 | Figure 4 Effects of EPA versus DHA on lipid parameters. |
When comparing EPA alone with the EPA/DHA combination in patients with TGs ≥500 mg/dL and <2,000 mg/dL, the EPA/DHA combination reduces TGs, non-HDL, TC, and VLDL to a greater extent than does EPA alone; EPA/DHA also increases HDL slightly, while decreasing HDL particle concentration when compared to placebo.33 EPA slightly reduced HDL compared with placebo. The greater difference is seen in the LDL-C, where the EPA/DHA combination significantly increases LDL, but it did not affect LDL particle concentration when compared with placebo.33 EPA has a slight nonsignificant decrease in LDL-C when compared with placebo. In patients on statin therapy with TGs between 200–499 mg/dL, the EPA/DHA combination showed similar results in regards to the reductions in TGs, non-HDL, TC/HDL, and to the increase in HDL, while the increase in LDL-C was non-significantly increased when compared to placebo. In the same patient population, EPA alone showed similar (and fewer) reductions in TGs, non-HDL, HDL, and slight, but significant, increases in LDL-C. The clinical impact of the relative differences in all of the atherogenic lipid particles, as well as the cardioprotective HDL, with EPA/DHA versus EPA alone has not been determined and remains controversial, and neither omega-3 formulation has demonstrated clinical event reductions in patients with HTG.
Differing effects of EPA and DHA on other conditions
A study by Nestel et al34 assessed the effects of DHA and EPA on systemic arterial compliance, which reflects arterial elasticity and is a risk factor for CV events.35 Thirty-eight patients with dyslipidemia were randomized to 3 g of EPA/day and 3 g of DHA/day or placebo. In this 7-week, double-blinded trial, the arterial function was assessed along with lipids and FAs. Components of the systemic arterial compliance include systolic and pulse pressures and total vascular resistance. In dialysis patients, oxidized LDL has been found to be more atherogenic than nonoxidixed LDL.36 EPA was shown to decrease levels of remnant lipoproteins and oxidized LDL (52% and 38%, respectively) in dialysis patients.37
Safety and tolerability
In the studies reviewed above, EPA was well tolerated and safe overall at both the 4 g/day and 2 g/day doses. The only side effect observed in the ANCHOR study7 was arthralgia, which was reported by >3% of patients. This side effect was not dose dependent and was not observed in the MARINE trial.28 EPA had no significant effect on glucose metabolism.7 This is interesting since the EPA/DHA combination showed transiently increased glucose levels.38 This may be beneficial for patients, especially since many patients with HTG also have diabetes (although the TG-lowering effect of EPA was less in those with diabetes compared with nondiabetics). In the MARINE study,28 the safety and tolerability was similar to placebo. There were no changes in serum transaminase values or creatinine levels. A common side effect of PUFAs is eructation,26 but there was no increased incidence of eructation in icosapent ethyl when compared to placebo. Caution is advised in patients with hypersensitivity to fish/shellfish. Finally, since PUFAs have been shown to prolong bleeding times (but only at very high doses above that used to treat HTG), patients on anticoagulants should be monitored closely.10,11
Conclusion
Icosapent ethyl is emerging as a promising alternative to the many available TG-lowering agents such as niacins, fibrates, and other omega-3 agents containing both EPA and DHA. Overall, icosapent ethyl is a high-purity form of EPA that reduces TG levels in patients with HTG who possess TG levels ≥500 mg/dL, as well as an adjunct therapy in patients with residual TG elevations after statin treatment. A remarkable feature of this TG-lowering effect is that it does not come at the expense of increasing LDL-C significantly. Both the MARINE28 and ANCHOR7 trials support the efficacy of EPA in lowering TG levels, and they also illustrate that EPA does not increase LDL-C as much as the EPA and DHA combinations or placebo. Ongoing studies will need to determine whether TG lowering with omega-3 confers additional CVD benefit beyond that attained via LDL-C reduction, as well as the relative clinical impact of the lipid differences between the EPA/DHA combination and EPA alone.
Disclosure
Drs O’Keefe and Lavie have both served as Speakers and Consultants to GlaxoSmithKline, and Dr Lavie serves in the same capacity for Amarin. Dr O’Keefe is the founder and has major ownership interest in CardioTabs, a company that markets omega-3s. Dr DiNicolantonio works for a company that sells omega-3 products, but he does not personally profit from these sales. The authors report no other conflicts of interest in this work.
References
Pejic RN, Lee DT. Hypertriglyceridemia. J Am Board Fam Med. 2006;19(3):310–316. | |
Sandhu S, Al-Sarraf A, Taraboanta C, Frohlich J, Francis GA. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157. | |
Christian JB, Bourgeois N, Snipes R, Lowe KA. Prevalence of severe (500 to 2,000 mg/dL) hypertriglyceridemia in United States adults. Am J Cardiol. 2011;107(6):891–897. | |
Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195–203. | |
Gandotra P, Miller M. The role of triglycerides in cardiovascular risk. Curr Cardiol Rep. 2008;10(6):505–511. | |
The EPA Drug Initiative [webpage on the Internet]. FDA puts patients at risk, now says lowering triglycerides will not protect from heart attack and stroke. 2013. Available from: http://epadruginitiative.com/press/fda-puts-patients-at-risk. Accessed February 13, 2014. | |
Ballantyne CM, Bays HE, Kastelein JJ, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol. 2012;110(7):984–992. | |
Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292–2333. | |
Oh RC, Lanier JB. Management of hypertriglyceridemia. Am Fam Physician. 2007;75(9):1365–1371. | |
Lavie CJ, Milani RV, Mehra MR, Ventura HO. Omega-3 polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardiol. 2009;54(7):585–594. | |
Lee JH, O’Keefe JH, Lavie CJ, Marchioli R, Harris WS. Omega-3 fatty acids for cardioprotection. Mayo Clin Proc. 2008;83(3):324–332. | |
Pereira SL, Leonard AE, Huang YS, Chuang LT, Mukerji P. Identification of two novel microalgal enzymes involved in the conversion of the omega3-fatty acid, eicosapentaenoic acid, into docosahexaenoic acid. Biochem J. 2004;384(Pt 2):357–366. | |
Ballantyne CM, Braeckman RA, Soni PN. Icosapent ethyl for the treatment of hypertriglyceridemia. Expert Opin Pharmacother. 2013;14(10):1409–1416. | |
Masoro EJ. Lipids and lipid metabolism. Annu Rev Physiol. 1977;39:301–321. | |
Wilhelm MG, Cooper AD. Induction of atherosclerosis by human chylomicron remnants: a hypothesis. J Atheroscler Thromb. 2003;10(3):132–139. | |
Dunbar RL, Rader DJ. Demystifying triglycerides: a practical approach for the clinician. Cleve Clin J Med. 2005;72(8):661–666, 670–672, 674–675 passim. | |
Shearer GC, Savinova OV, Harris WS. Fish oil – how does it reduce plasma triglycerides? Biochim Biophys Acta. 2012;1821(5):843–851. | |
Harris WS, Bulchandani D. Why do omega-3 fatty acids lower serum triglycerides? Curr Opin Lipidol. 2006;17(4):387–393. | |
Qi K, Fan C, Jiang J, et al. Omega-3 fatty acid containing diets decrease plasma triglyceride concentrations in mice by reducing endogenous triglyceride synthesis and enhancing the blood clearance of triglyceride-rich particles. Clin Nutr. 2008;27(3):424–430. | |
Calder PC. Mechanisms of action of (n-3) fatty acids. J Nutr. 2012; 142(3):592S–599S. | |
Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302(2):93–109. | |
Szanto A, Nagy L. The many faces of PPARgamma: anti-inflammatory by any means? Immunobiology. 2008;213(9–10):789–803. | |
Davidson MH. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am J Cardiol. 2006;98(4A):27i–33i. | |
Harris WS. n-3 fatty acids and serum lipoproteins: animal studies. Am J Clin Nutr. 1997;65(Suppl 5):1611S–1616S. | |
Lawson LD, Hughes BG. Absorption of eicosapentaenoic acid and docosahexaenoic acid from fish oil triacylglycerols or fish oil ethyl esters co-ingested with a high-fat meal. Biochem Biophys Res Commun. 1988;156(2):960–963. | |
Lawson LD, Hughes BG. Human absorption of fish oil fatty acids as triacylglycerols, free acids, or ethyl esters. Biochem Biophys Res Commun. 1988;152(1):328–335. | |
Beckermann B, Beneke M, Seitz I. [Comparative bioavailability of eicosapentaenoic acid and docasahexaenoic acid from triglycerides, free fatty acids and ethyl esters in volunteers]. Arzneimittelforschung. 1990;40(6):700–704. German. | |
Jacobson TA. A new pure ω-3 eicosapentaenoic acid ethyl ester (AMR101) for the management of hypertriglyceridemia: the MARINE trial. Expert Rev Cardiovasc Ther. 2012;10(6):687–695. | |
Bays HE, Braeckman RA, Ballantyne CM, et al. Icosapent ethyl, a pure EPA omega-3 fatty acid: effects on lipoprotein particle concentration and size in patients with very high triglyceride levels (the MARINE study). J Clin Lipidol. 2012;6(6):565–572. | |
Harris WS, Dayspring TD, Moran TJ. Omega-3 fatty acids and cardiovascular disease: new developments and applications. Postgrad Med. 2013;125(6):100–113. | |
Wei MY, Jacobson TA. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: a systematic review and meta-analysis. Curr Atheroscler Rep. 2011;13(6):474–483. | |
Jacobson TA, Glickstein SB, Rowe JD, Soni PN. Effects of eicosapentaenoic acid and docosahexaenoic acid on low-density lipoprotein cholesterol and other lipids: a review. J Clin Lipidol. 2012;6(1):5–18. | |
Maki KC, Lawless AL, Kelley KM, et al. Effects of prescription omega-3-acid ethyl esters on fasting lipid profile in subjects with primary hypercholesterolemia. J Cardiovasc Pharmacol. 2011;57(4):489–494. | |
Nestel P, Shige H, Pomeroy S, Cehun M, Abbey M, Raederstorff D. The n-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid increase systemic arterial compliance in humans. Am J Clin Nutr. 2002;76(2):326–330. | |
Glasser SP, Arnett DK, McVeigh GE, et al. Vascular compliance and cardiovascular disease: a risk factor or a marker? Am J Hypertens. 1997;10(10 Pt 1):1175–1189. | |
Drüeke TB, Nguyen Khoa T, Massy ZA, Witko-Sarsat V, Lacour B, Descamps-Latscha B. Role of oxidized low-density lipoprotein in the atherosclerosis of uremia. Kidney Int Suppl. 2001;78:S114–S119. | |
Ando M, Sanaka T, Nihei H. Eicosapentanoic acid reduces plasma levels of remnant lipoproteins and prevents in vivo peroxidation of LDL in dialysis patients. J Am Soc Nephrol. 1999;10(10):2177–2184. | |
Davidson MH, Stein EA, Bays HE, et al; COMBination of prescription Omega-3 with Simvastatin (COMBOS) Investigators. Efficacy and tolerability of adding prescription omega-3 fatty acids 4 g/d to simvastatin 40 mg/d in hypertriglyceridemic patients: an 8-week, randomized, double-blind, placebo-controlled study. Clin Ther. 2007;29(7):1354–1367. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.