")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Ichthyosis Follicularis, Alopecia, and Photophobia Syndrome in a Saudi Child: A Case Report
Authors Bin Rubaian NF, Al-Awam BS , Alluhaybi AH, Alsaati AA
Received 9 September 2023
Accepted for publication 23 November 2023
Published 7 December 2023 Volume 2023:16 Pages 3527—3533
DOI https://doi.org/10.2147/CCID.S439288
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Rungsima Wanitphakdeedecha
Nouf F Bin Rubaian,1 Bashayer S Al-Awam,2 Abdulelah Hassan Alluhaybi,1 Ahmed Abdulaziz Alsaati3
1Dermatology Department, King Fahad Hospital of the University, Imam Abdulrahman bin Faisal University, Al-Khobar, Saudi Arabia; 2Department of Pediatrics, College of Medicine, King Fahad Hospital of the University, Imam Abdulrahman bin Faisal University, Dammam, Saudi Arabia; 3Dermatology Department, College of Medicine, King Faisal University, Al-Hofuf, Saudi Arabia
Correspondence: Ahmed Abdulaziz Alsaati, Dermatology Department, College of Medicine, King Faisal University, Al-Hofuf, Saudi Arabia, Tel +966555828542, Email [email protected]
Abstract: Ichthyosis follicularis, atrichia, and photophobia (IFAP) syndrome is a rare autosomal recessive, X-linked, genetic disorder that involves a triad of follicular ichthyosis, atrichia of the scalp, and photophobia. We report a case of an 8-year-old boy with alopecia of the scalp, eyebrows, and eyelashes, which occurred in his first year of age. His birth was uneventful, and his developmental milestones were normal. The alopecia was non-scarring and was accompanied by mild generalized xerosis, photophobia, and recurrent angular cheilitis. Moreover, numerous non-inflammatory, follicular, keratotic tiny papules were noticed. His deciduous teeth had retention with gum hyperplasia, and his feet showed symmetrical plantar keratoderma and nail dystrophy of the right big toe. The genetic testing confirmed an X-linked recessive inheritance of IFAP syndrome without BRESHECK syndrome due to the mutation in the MBTPS2 (300294) gene located on chromosome Xp22.12. The patient was given symptomatic treatment with urea cream for plantar keratoderma and was advised to apply constant moisturizers to avoid generalized xerosis. Dermatological and ophthalmological follow-ups were recommended. This is the first case reported from Saudi Arabia. This case report throws light on the characteristics of IFAP syndrome and denotes the points of differentiation from similar conditions.
Keywords: X-linked, genetic disorder, dermatology, follicular plugging
Introduction
Ichthyosis follicularis, atrichia, and photophobia (IFAP) syndrome is a rare autosomal recessive, X-linked, genetic disorder that involves a triad of follicular ichthyosis, atrichia of the scalp, and photophobia. Other clinical features that might be found in some of the patients include brain anomalies, severe mental retardation, ectodermal dysplasia, skeletal deformities (especially the vertebrae), Hirschsprung disease, ear and eye anomalies, cleft palate or cryptorchidism, and kidney dysplasia/hyperplasia. These manifestations represent the criteria of BRESHECK syndrome.1 The IFAP syndrome has two subtypes. The IFAP syndrome-1 (IFAP1) with or without BRESHECK syndrome is the consequence of mutation in membrane-bound transcription factor peptides site 2 (MBTPS2) gene on chromosome Xp22. Its counterpart, IFAP syndrome-2, occurs due to heterozygous mutation in the sterol regulatory element-binding protein 1 (SREBF1) gene on chromosome 17p11. The MBTPS2 gene is a membrane-embedded zinc metalloprotease, which is required for cleavage of sterol regulatory element-binding protein (SREBPs) and endoplasmic reticulum (ER) stress response. Therefore, the mutation leads to functional deficiency of MBTPS2 that disrupts sterol or endoplasmic reticulum (ER) homeostasis, leading to impairment of the differentiation of epidermal structures. This disruption gives rise to the expression of the triad of IFAP.2–4 Patients with IFAP syndrome may be misdiagnosed as their dermatological manifestations overlap with a wide range of other dermatological diseases. Demonstration and discussion of these cases help clinicians, particularly dermatologists keep in mind their presentation, so they would be able to suspect and identify such patients, thus preventing any unnecessary delays in management. We report a case of an 8-year-old Saudi boy who was diagnosed with IFAP-1 syndrome.
Case Report
We report a case of an 8-year-old boy who was brought by his parents to the dermatology clinic at King Fahd Hospital of the University in Al-Khobar City. The parents were concerned about the loss of the child’s hair from the scalp, eyebrows, and eyelashes. Hair loss occurred in his first year of age, even though the father did not notice any previous hair loss on his child upon his birth. History taking showed that the child was delivered at full term by an uneventful Cesarean section. However, he was admitted to the neonatal intensive care unit for 14 days due to a urinary tract infection and hematuria that were treated accordingly. The child had normal developmental milestones. The loss of hair was observed during his first year of life and was accompanied by mild skin dryness, sensitivity to light, and deformed, thickened, and discolored nail of the right big toe. The father noted recurrent, cracked, and painful sores at the corners of the mouth with constant closing of the child’s both eyes. The boy had atopic manifestation of eczematous plaques that spontaneously resolved after one year of age. The history of consanguinity between the parents was negative and the patients had five elder brothers who did not share similar suffering or had any of the features of their younger brother. Furthermore, no similar history was found in any relatives within the boy’s family.
Clinical dermatological examination has confirmed a non-scarring type of alopecia of the scalp, eyelashes, and eyebrows. Moreover, numerous non-inflammatory follicular keratotic tiny papules were noticed [Figure 1]. The patient was not able to open his eyes in a room with good lighting, indicating the presence of photophobia. Furthermore, his dry lips showed angular cheilitis and fissures [Figure 2]. Additionally, retention of deciduous teeth was observed along with gum hyperplasia upon oral cavity examination [Figure 3]. The patient had general xerosis, which was observed on his neck, trunk, as well as both upper and lower extremities. His feet showed symmetrical focal thickened skin with fissures on the plantar surface [Figure 4] and nail dystrophy of the right big toe [Figure 5]. General examination did not reveal any other significant findings.
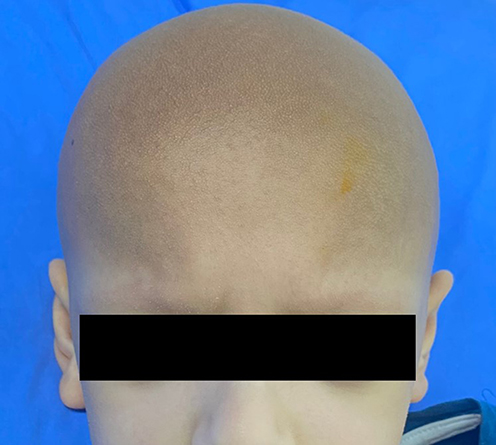 |
Figure 1 Numerous non-inflammatory follicular keratotic tiny papules on the patient’s scalp, eyebrows, and eyelashes. |
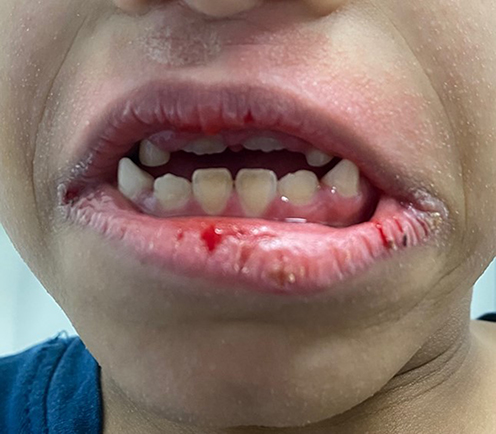 |
Figure 2 Dry lips with fissures and angular cheilitis. |
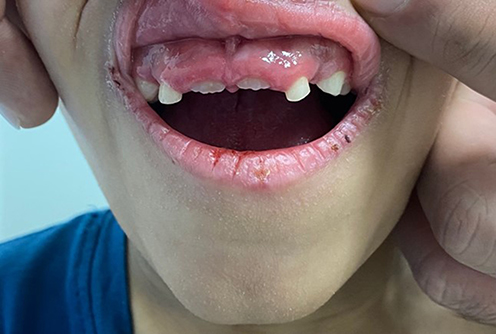 |
Figure 3 Retention of primary teeth with gum hyperplasia. |
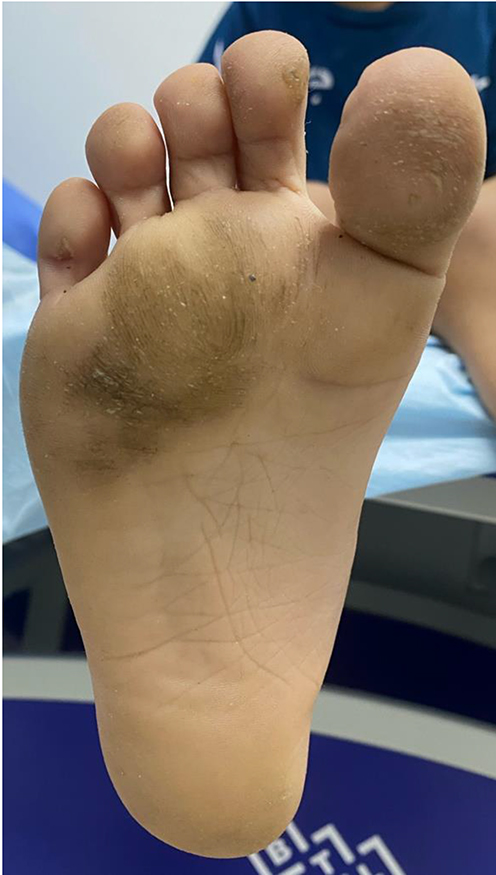 |
Figure 4 Symmetrical focal thickness and fissures of the skin of both feet revealing plantar keratoderma. |
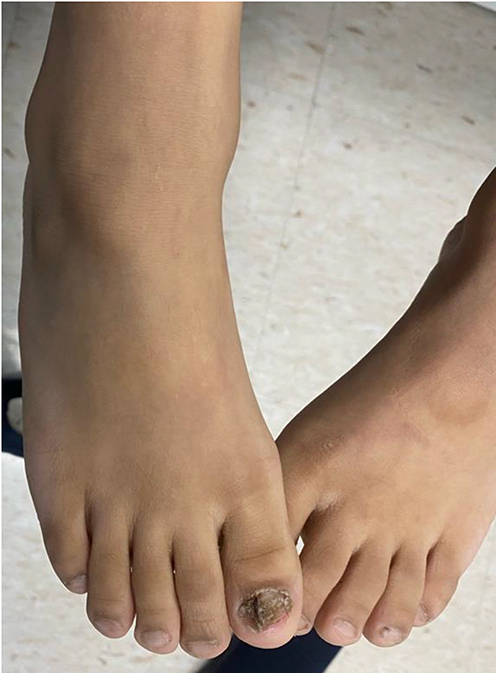 |
Figure 5 Nail dystrophy of the patient’s right big toe with sparing of the contralateral big toe. |
Based on the aforementioned features, we considered the probability of ectodermal dysplasia (ED); however, further testing was needed to confirm and discover the subtype of ED. Therefore, following the parents’ consent, genetic testing was done for the patient, confirming an X-linked recessive inheritance of ichthyosis follicularis, atrichia, and photophobia (IFAP) syndrome. A missense mutation (NM 0158843: C.1499G>A; p. (Gly 500Asp)) in the MBTPS2 (300294) gene located on chromosome Xp22.12 was identified. It was classified as likely pathogenic (Class 2) according to the recommendations of CENTOGENE and ACMG. The result showed an X-linked recessive inheritance of IFAP syndrome without BRESHECK syndrome (absent brain anomalies, severe mental retardation, skeletal deformities, Hirschsprung disease, ear and eye anomalies, cleft palate, cryptorchidism, and kidney dysplasia/hyperplasia), and it was consistent with a genetic diagnosis of MBTPS2-related X-linked Ectodermal Dysplasia with no further clinically relevant variants related to the described phenotypes. The hemizygous pathogenic variant was identified. Neither relevant variants in the genes for which secondary findings were reported in the patient nor did it identify relevant pathogenic or likely pathogenic variant. With the clinical features and genetic testing, the diagnosis of IFAP syndrome was confirmed. The patient and his parents were advised regarding the necessity of skin moisturizing, and they were given urea cream as a symptomatic treatment for the plantar keratoderma. The patient was scheduled for dermatological and ophthalmological follow-up.
Discussion
IFAP syndrome is a rare genetic disorder that is composed of the triad of follicular ichthyosis, atrichia of the scalp, and photophobia. The syndrome was first described in 1909 by Macleod and Collins, in which IFAP was evidenced by the finding of hair follicle plugging with horny spines and was associated with general dry skin. They had reported that the condition started during the first year of life or at the end of the second year, even if the birth history was uneventful and healthy. There was no evidence of inflammation preceding the growth of those spiny papules.5 More than 61 cases of IFAP syndrome have been reported worldwide since the first case reported by Macleod and Collins, and to the best of our knowledge, no case reports of IFAP syndrome have been documented in Saudi Arabia.
Further clinical features of IFAP syndrome include developmental delay, hypotonia, short stature, microcephaly, frontal bossing, dystrophin nails, seizures, and intellectual disability. Some of these features occur from birth and others develop in the first years of life.6 As a rare X-linked genetic condition, the syndrome predominantly targets males; nevertheless, the triad of IFAP is mostly seen in all of the affected patients. The follicular papules are commonly distributed over the scalp, trunk, and upper and lower extremities, whereas the alopecia involves the scalp, eyebrows, and eyelashes. Photophobia is due to corneal defects like erosions, ulcers, scars, and neovascularization.7 Magnetic resonance imaging of such patients can reveal enlargement of the cisterna magna similar to the case of Nakayama et al in which irregular deficiency in the medial occipital lobe, indicating schizencephaly, was found with irregular distortion on the anterior horn of the lateral ventricle, expanding in the lateral superior direction.8
In the current case, the patient developed multiple non-inflammatory follicular keratotic papules on his scalp and had a generalized dryness that was more pronounced on his neck, trunk, and extremities. These findings correspond with the description given by Macleod and Collins in their statement of the absence of evident inflammation preceding the spiny papules. Furthermore, the current case developed alopecia of the scalp, eyebrows, and eyelashes along with photophobia in the first year of age, which is also in line with the age at onset reported by Macleod and Collins.5
Variants in the MBTPS2 gene have been reported to cause a broad phenotypic spectrum of X-linked genodermatoses, such as keratosis follicularis spinulosa decalvans (KFSD) and Olmsted syndrome. The KFSD is a hereditary disorder featuring a cicatricial-type of alopecia with follicular papules, while Olmsted syndrome is a rare congenital disorder characterized by mutilating palmoplantar keratoderma, perioral/perianal keratotic plaques, leukokeratosis of oral mucosa, onychodystrophy, and follicular keratosis. Even though these conditions share similar allelic mutations as that of IFAP-1 syndrome, yet they differ in some of their characteristics.9–12
Although it constitutes a part of the BRESHECK syndrome, ED may occur as an independent disorder with many subtypes. One of these subtypes, dermotrichic syndrome, shares the same expression of X-linked monogenic disorder as that of IFAP syndrome. Both disorders share similar features regarding the atrichia onset from birth, generalized ichthyosiform lesions, short stature, mental retardation, seizures, and X-linked recessive inheritance. Nevertheless, both syndromes are considered separate entities even when they share similar features. Thus, they need to be differentiated from each other. The main differences include the occurrence of hypohidrosis without hyperthermia, as well as intestinal and skeletal congenital anomalies in dermotrichic syndrome, while recurrent respiratory infections, ocular, and respiratory disorders along with atopic manifestations occur with the IFAP syndrome.13 Dermatologists should be vigilant to identify these cases and differentiate them from other conditions causing skin xerosis or hair loss. History can be deceiving, as in this case report, where the past and family histories did not include any risk factors that may point to the presence of a genetic disorder, such as consanguinity or the presence of manifestations in another family member. However, the development of the triad of IFAP after a year gap of uneventful symptoms raises the probability towards the syndrome.
As there is no permanent cure for IFAP syndrome, genetic counseling is of great importance. The therapeutic goal of the syndrome is to crucially maintain the protection of the skin barrier, reduce microbial colonization, and support immune regulation to ensure an optimum quality of life for those patients. Constant moisturization is needed for the dry skin, while keratolytic urea emollients can be used for follicular hyperkeratosis to temporarily relieve cutaneous lesions. While photophobia and seizures may resolve spontaneously, anti-seizure medications may occasionally be necessary, and ocular lubrication is an integral therapeutic option.14 Therefore, the presenting case was given symptomatic treatment with urea cream for plantar keratoderma along with moisturizers for the generalized xerosis. Follow-up visits were scheduled to ensure optimum management of cutaneous manifestations.
The present case report was limited by the absence of follow-up data as the patient was recently diagnosed and was scheduled to start his follow-up. In addition, data were lacking considering his previous presentations at other healthcare facilities and the prescribed treatment before being correctly diagnosed at our institution.
Conclusion
IFAP syndrome can present as a part of BRESHECK syndrome or may lack any of BRESHECK features as the present case. Dermatologists should bear in mind the probability of IFAP diagnosis even in the absence of other features of BRESHECK syndrome and even if the history lacks risk factors of a genetic disorder. This case study has been reported to further increase the awareness of healthcare providers and physicians about this rare syndrome to prevent misdiagnosis and avoid delaying treatment, as well as preventing patients from receiving ineffective treatments for a mistaken diagnosis of another dermatological condition.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of King Fahad Hospital of the University, Imam Abdulrahman bin Faisal University, Al-Khobar, Saudi Arabia. An informed, written consent was obtained from the patient’s guardians.
Consent for Publication
Consent for publication of this case report and the patient’s images was obtained from the patient’s guardians.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Naiki M, Mizuno S, Yamada K, et al. MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet A. 2012;158a:97–102. doi:10.1002/ajmg.a.34373
2. Kamo M, Ohyama M, Kosaki K, et al. Ichthyosis follicularis, alopecia, and photophobia syndrome: a case report and a pathological insight into pilosebaceous anomaly. Am J Dermatopathol. 2011;33(4):403–406. doi:10.1097/DAD.0b013e3181e8b562
3. Oeffner F, Fischer G, Happle R, et al. IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet. 2009;84(4):459–467. doi:10.1016/j.ajhg.2009.03.014
4. Wang H, Humbatova A, Liu Y, et al. Mutations in SREBF1, encoding sterol regulatory element binding transcription factor 1, cause autosomal-dominant IFAP syndrome. Am J Hum Genet. 2020;107(1):34–45. doi:10.1016/j.ajhg.2020.05.006
5. Macleod JM, Collins ET. Two cases of advanced “Keratosis follicularis”, associated with baldness. Proc R Soc Med. 1908;1:27–31.
6. Mégarbané H, Mégarbané A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome. Orphanet J Rare Dis. 2011;6(1):29. doi:10.1186/1750-1172-6-29
7. Höpker LM, Ribeiro CG, Oliveira LM, et al. Ichthyosis follicularis, alopecia and photophobia syndrome (IFAP): report of the first case with ocular and cutaneous manifestations in Brazil with a favorable response to treatment. Arq Bras Oftalmol. 2011;74(1):55–57. doi:10.1590/S0004-27492011000100013
8. Nakayama J, Iwasaki N, Shin K, et al. A Japanese case of ichthyosis follicularis with atrichia and photophobia syndrome with an MBTPS2 mutation. J Hum Genet. 2011;56(3):250–252. doi:10.1038/jhg.2010.163
9. Malvankar DD, Sacchidanand S. Keratosis follicularis spinulosa decalvans: a report of three cases. Int J Trichology. 2015;7:125–128. doi:10.4103/0974-7753.167461
10. Tao J, Huang CZ, Yu NW, et al. Olmsted syndrome: a case report and review of literature. Int J Dermatol. 2008;47:432–437. doi:10.1111/j.1365-4632.2008.03595.x
11. Wang HJ, Tang ZL, Lin ZM, et al. Recurrent splice-site mutation in MBTPS2 underlying IFAP syndrome with Olmsted syndrome-like features in a Chinese patient. Clin Exp Dermatol. 2014;39:158–161. doi:10.1111/ced.12248
12. Bolognia JL, Schaffer JV, Cerroni L. Dermatology: 2-Volume Set.
13. Martino F, D’Eufemia P, Pergola MS, et al. Child with manifestations of dermotrichic syndrome and ichthyosis follicularis-alopecia-photophobia (IFAP) syndrome. Am J Med Genet. 1992;44(2):233–236. doi:10.1002/ajmg.1320440222
14. Elharrouni AA, Baybay H, Douhi Z, et al. Ichthyosis Follicularis with Alopecia and Photophobia Syndrome (IFAP): a case report and review of the literature. J Dermatol Sci Res Rev Rep. 2020;1:1–2.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.