Back to Archived Journals » Hypoxia » Volume 2
Hypoxia-inducible factor-mediated induction of WISP-2 contributes to attenuated progression of breast cancer
Authors Fuady J, Bordoli M, Abreu-Rodríguez I, Kristiansen G, Hoogewijs D, Stiehl D, Wenger R ![]()
Received 22 December 2013
Accepted for publication 4 February 2014
Published 31 March 2014 Volume 2014:2 Pages 23—33
DOI https://doi.org/10.2147/HP.S54404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jerry H Fuady,1,* Mattia R Bordoli,1,* Irene Abreu-Rodríguez,1,* Glen Kristiansen,2 David Hoogewijs,1,** Daniel P Stiehl,1,** Roland H Wenger1,**
1Institute of Physiology and Zurich Center for Human Physiology, University of Zurich, Zurich, Switzerland; 2University Hospital Bonn, Institute of Pathology, Bonn, Germany
*,**These authors contributed equally to this work
Abstract: Hypoxia and the hypoxia-inducible factor (HIF) signaling pathway trigger the expression of several genes involved in cancer progression and resistance to therapy. Transcriptionally active HIF-1 and HIF-2 regulate overlapping sets of target genes, and only few HIF-2 specific target genes are known so far. Here we investigated oxygen-regulated expression of Wnt-1 induced signaling protein 2 (WISP-2), which has been reported to attenuate the progression of breast cancer. WISP-2 was hypoxically induced in low-invasive luminal-like breast cancer cell lines at both the messenger RNA and protein levels, mainly in a HIF-2α-dependent manner. HIF-2-driven regulation of the WISP2 promoter in breast cancer cells is almost entirely mediated by two phylogenetically and only partially conserved functional hypoxia response elements located in a microsatellite region upstream of the transcriptional start site. High WISP-2 tumor levels were associated with increased HIF-2α, decreased tumor macrophage density, and a better prognosis. Silencing WISP-2 increased anchorage-independent colony formation and recovery from scratches in confluent cell layers of normally low-invasive MCF-7 cancer cells. Interestingly, these changes in cancer cell aggressiveness could be phenocopied by HIF-2α silencing, suggesting that direct HIF-2-mediated transcriptional induction of WISP-2 gene expression might at least partially explain the association of high HIF-2α tumor levels with prolonged overall survival of patients with breast cancer.
Keywords: invasion, metastasis, motility, oxygen, tumor, transcriptional regulation
Introduction
Temporally and spatially variable tissue hypoxia is characteristic of solid tumors.1 Hypoxia-inducible factors (HIFs) allow cancer cells to adapt to microenvironmental tissue hypoxia, affecting all aspects of tumor progression, including metabolism, proliferation, inflammation, angiogenesis, and metastasis.2–5 Importantly, HIFs are also involved in resistance to cancer therapy, and overall survival correlates with HIFα levels in a cancer type-specific manner.6–8 Transcriptionally active HIFs are heterodimers usually composed of a constitutively expressed β subunit and either a HIF-1α or a HIF-2α subunit, the stability and activity of which is regulated by oxygen-dependent protein hydroxylation.9,10 Despite their high structural similarity and identical DNA sequence recognition, several studies have identified specific roles for HIF-1 and HIF-2 in tumorigenesis.11–13 We and others previously showed in a variety of cancer cell lines that HIF-1α protein levels decreased under prolonged hypoxia while HIF-2α levels increased, suggesting HIFα isoform-specific kinetics of target gene expression.8,14,15 In contrast with the many known HIF-1 and HIF-1/HIF-2 target genes,8,14,16 only a few genes have been reported to be regulated exclusively by HIF-2, including erythropoietin, ephrin A1, VE-cadherin, protein tyrosine phosphatase receptor-type Z polypeptide 1, amphiregulin, and Wnt-1 induced signaling protein 2 (WISP-2).8,14,17–21
WISP-2 is a secreted protein member of the connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family, and is also known as CCN5.22,23 WISP-2 has been detected in adult skeletal muscle, colon, and ovary, and in the fetal lung, as well as in the stroma of breast tumors derived from Wnt-1 transgenic animals.24 WISP-2 expression has been shown, in most studies, to correlate inversely with the aggressiveness of breast, pancreatic, and colon cancer, suggesting tumor suppressor-like activity.25,26 WISP-2 shows transiently elevated levels during progression of breast cancer; while it is almost undetectable in normal human mammary epithelial cells, it is highly expressed in estrogen receptor-positive noninvasive breast cancer cell lines (including MCF-7, BT-474, ZR-75-1, and T-47D), and is again undetectable in highly invasive estrogen receptor-negative cells (including MDA-MB-231, MDA-MB-468, BT-20, and DU-4475).27–30 Loss of WISP-2 in MCF-7 cells induced estrogen-independent growth and promoted epithelial-to-mesenchymal transition, consistent with a more invasive phenotype, whereas forced WISP-2 expression in MDA-MB-231 cells reduced proliferation and invasiveness.30 WISP-2 expression is induced by estrogens as well as by epidermal growth factor and insulin-like growth factor in estrogen receptor-positive cells, and WISP-2 is necessary for estrogen-induced and insulin-like growth factor-induced proliferation.31–35
A functional estrogen response element has been identified in the WISP2 promoter which is required for inducibility of the WISP2 gene by estrogen.29 Estrogen receptor-α recruits the histone acetyl transferase cAMP response element (CRE) binding (CREB) protein as well as the cyclin-dependent kinase inhibitor p21WAF1/CIP1 to the WISP2 promoter, suggesting cooperative control of WISP2 gene expression.29,36 Hypoxia has been identified as another stimulus of WISP2 gene expression, and is mediated specifically by HIF-2α in cooperation with the ETS oncogene family member ETS-like gene 1 (ELK-1) in MCF-7 cells.14,37 We previously demonstrated that the WISP2 promoter is induced specifically by HIF-2α in MCF-7 cells.8 Here, we identified the hypoxia response elements (HREs) and characterized a microsatellite region responsible for HIF-2α-specific induction of the WISP2 promoter. Furthermore, we assessed the impact of hypoxia and HIF-2α on WISP-2-mediated cell proliferation, clonogenic growth, and motility.
Materials and methods
Cell culture and transfection
Human breast cancer cell lines (MCF-7, T-47D, ZR-75-1, BT-474, MDA-MB-468, and MDA-MB-231) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich, St Louis, MO, USA). Hypoxic experiments were performed at 1% oxygen and 5% CO2 in a gas-controlled glove box (Invivo2 400, Ruskinn Technology Limited, Bridgend, UK) as described previously.38 Viral particles were generated in HEK293T cells following cotransfection of the transfer vector (3 μg) together with the packaging plasmids, ie, pLP1 (4.2 μg), pLP2 (2 μg), and pVSV-G (2.8 μg) (Invitrogen, Carlsbad, CA, USA) using polyethylenimine (Polysciences, Warrington, PA, USA). MCF-7 cells were infected with viral particles and cell pools were selected with puromycin as described before.38 Two independent WISP-2 short hairpin RNA (shRNA) vectors (TRCN0000033355 and TRCN0000033357) were obtained from Sigma-Aldrich. shRNA-resistant HIF-2α was used to rescue MCF-7 cells with stable HIF-2α knockdown, as described before.8
Messenger RNA (mRNA) and protein detection
Total cellular RNA was extracted as previously described.39 Total RNA (2 μg) was reverse transcribed using AffinityScript reverse transcriptase (Agilent, Santa Clara, CA, USA) and complementary DNA (cDNA) levels were estimated by quantitative polymerase chain reaction (PCR) using a SYBR® Green quantitative PCR reagent kit (Sigma-Aldrich) in a MX3000P light cycler (Agilent). Transcript levels were calculated by comparison with a calibrated standard and expressed as ratios relative to ribosomal protein L28 mRNA levels. Immunoblots were performed as previously described.40 Antibodies against the following proteins were used: WISP-2 (Abcam, Cambridge, UK), HIF-1α (BD Transduction Laboratories, Allschwil, Switzerland), HIF-2α (Novus Biologicals, Littleton, CO, USA), Sp1 (Santa Cruz Biotechnology, Dallas, TX, USA), and β-actin (Sigma-Aldrich). Breast cancer tissue microarray analysis has been described previously.8
Plasmid construction and reporter gene assays
The promoter truncations −1919/+16, −808/+16, −520/+16, and −422/+16 have been reported elsewhere29 and were kindly provided by M Sabbah (Paris, France); −422/−75, −83/+16, and MS deletions were generated by standard cloning techniques. The WRE1, CRE, ELK-1, and WRE2 sites in the −422/+16 construct were mutated by site-directed mutagenesis. The −252/+16 truncation was generated by PCR cloning. The −422 (CA)n promoter region was amplified from T-47D genomic DNA, transformed into bacteria, and isolated for sequencing to determine the CA repeat length. The −112/+16 and −112/+16 HRE2/3 double-mutant constructs were generated by elongation of an internal MluI site using double-stranded oligonucleotides. HRE2 and HRE3 in the −422/+16 construct were mutated by site-directed mutagenesis (5′-CGTG-3′ to 5′-CAAA-3′). Dual luciferase reporter gene assays were performed as described previously.41 The promoterless luciferase vector pGL3basic was included as negative control in all experiments.
Genomic DNA extraction and sequencing
Cells were washed with phosphate-buffered saline, resuspended in high salt lysis buffer (200 mM NaCl, 50 mM Tris/HCl pH 8.0, 50 mM ethylenediaminetetraacetic acid [EDTA] pH 8.0, 1% sodium dodecyl sulfate) and treated overnight with proteinase K (0.5 mg/mL). After addition of saturated NaCl, DNA was precipitated with isopropanol, washed with 70% ethanol, and resuspended in Tris/EDTA buffer. WISP-2 genomic DNA was amplified by PCR using forward primer 5′-TACGGGTACCACGGACAGGCACCCCCTTGGTGG-3′ and reverse primer 5′-CAGATGTGCAGAGCCAGCAGCTT-3′. PCR products were gel-purified and sequenced (Microsynth AG, Balgach, Switzerland).
Cellular proliferation, colony formation and motility assays
To determine cell proliferation and viability, 1010 cells per well were seeded into six-well plates, allowed to adhere overnight, exposed to normoxia or hypoxia for 0 to 72 hours, detached by trypsin/EDTA, and counted using a Vi-cell™ XR cell viability analyzer (Beckman-Coulter, Brea, CA, USA). For low cell density colony-forming assays, 2 × 103 cells per well were plated into six-well plates, allowed to adhere overnight, and exposed to normoxia or hypoxia for 10 days, with the medium replaced every 3 days. The colonies were fixed with methanol, stained with 0.5% crystal violet, and counted. For anchorage-independent colony formation assays, 104 cells were resuspended in 2 mL of 0.4% low melting agarose (Sigma-Aldrich) in Dulbecco’s Modified Eagle’s Medium, poured on top of a 2% low melting agarose layer in six-well plates, and allowed to settle overnight. Following exposure to normoxia or hypoxia for 14 days, the soft agar was washed with phosphate-buffered saline, the colonies were stained with 0.005% crystal violet in methanol for one hour at room temperature, and counted. For the scratch assay, cells were allowed to grow to 100% confluency in 12-well plates. Following crosswise scratching with a 200 μL pipette tip, the cells were exposed to normoxia or hypoxia in fetal calf serum-free Dulbecco’s Modified Eagle’s Medium for 24 hours. The cell-free area was measured and converted to percent recovery.
Statistical analysis
If not indicated otherwise, unpaired Student’s t-tests were applied. Differences between two values at the P<0.05 level were considered to be statistically significant.
Results
HIFα isoform responsible for hypoxic WISP-2 induction is specific for distinct breast cancer cell lines
To determine the HIFα isoform directing WISP-2 expression in breast cancer, four luminal-like (MCF-7, BT-474, T-47D, and ZR-75-1), one basal A-like (MDA-MB-468), and one basal B-like (MDA-MB-231) human breast cancer cell line were analyzed. Similar to data published elsewhere,8,30 constitutive WISP-2 mRNA levels were high in MCF-7 cells, moderate in T-47D and BT-474 cells, low in ZR-75-1 cells, and almost undetectable in MDA-MB-231 and MDA-MB-468 cells (Figure 1A). Although WISP-2 immunoblotting is notoriously difficult,42 probably because the WISP-2 protein is partially secreted we managed to detect WISP-2 protein in MCF-7 cells where it was found to be upregulated following hypoxic stimulation (Figure 1B), as previously published for the WISP-2 mRNA levels.8
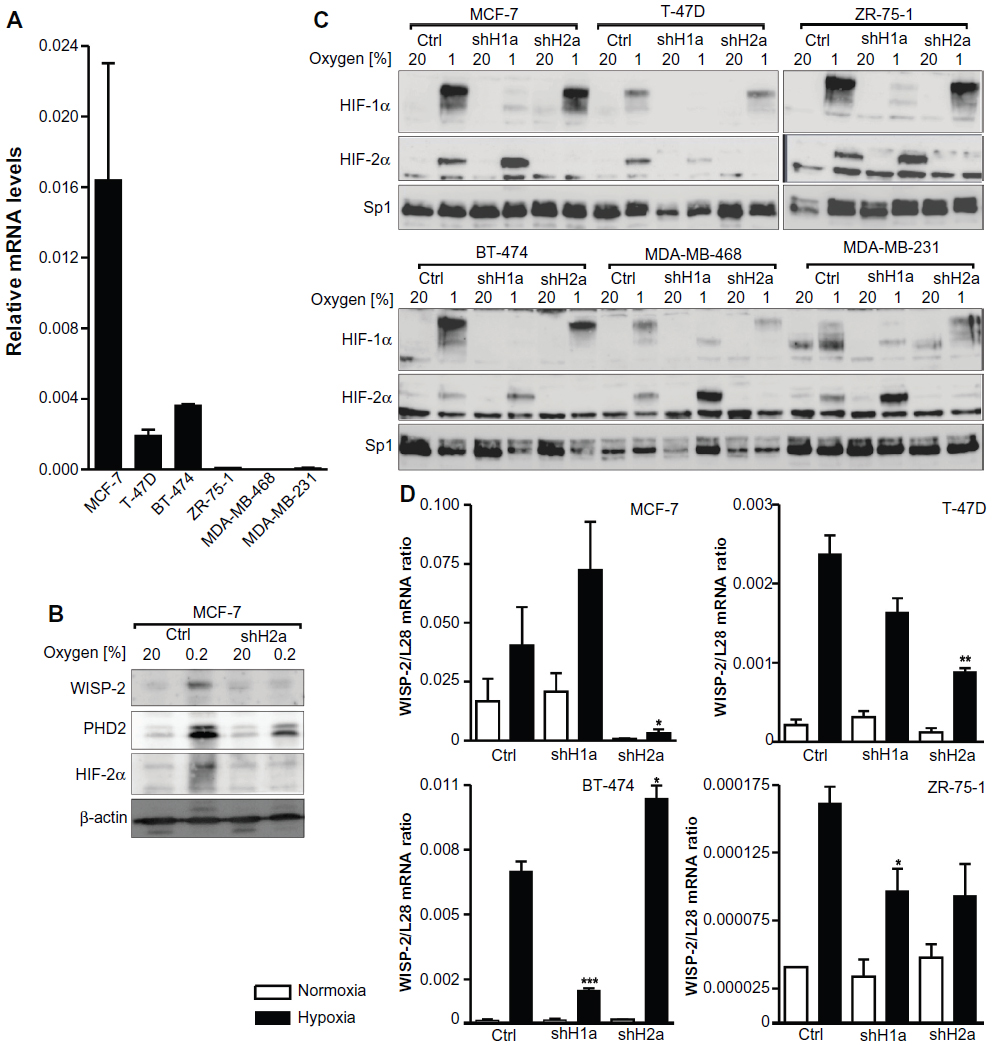 | Figure 1 HIFα isoform-specific regulation of WISP-2 transcription. (A) Basal WISP-2 mRNA levels were determined by reverse transcription quantitative polymerase chain reaction in the six breast cancer cell lines indicated using a calibrated internal standard. Shown are mean values ± the standard error of the mean of three independent experiments. (B) MCF-7 cells were cultured in normoxic conditions or exposed to hypoxia for 24 hours, and analyzed by immunoblotting using antibodies derived against WISP-2, the HIF target prolyl-4-hydroxylase domain 2 (PHD2), HIF-2α, or the constitutively expressed control protein, β-actin. (C) Cells were stably transfected with either control shRNA or shRNAs targeting HIF-1α (shH1a) or HIF-2α (shH2a) and analyzed by immunoblotting of nuclear extracts using antibodies derived against HIF-1α, HIF-2α, or the constitutively expressed control transcription factor, Sp1. (D) Hypoxic WISP-2 mRNA induction was determined in the indicated cell lines which were stably transfected with either control or shRNA constructs as in (C). WISP-2 mRNA levels were quantified by reverse transcription quantitative polymerase chain reaction and normalized to the mRNA levels of the ribosomal protein, L28. Shown are mean values ± the standard errors of the mean of three independent experiments. (D) For statistical evaluation of the hypoxically exposed cells, the effects of HIF-1α or HIF-2α silencing were compared with the control shRNA transfected cells. *P<0.05; **P<0.01; ***P<0.001. |
Immunoblot analyses revealed that all six cell lines expressed both HIF-1α and HIF-2α in a hypoxia-inducible manner (Figure 1C). The variable relative molecular weight of HIF-1α between approximately 98 kDa and 120 kDa, as observed in the different cell lines, is due to varying degrees of phosphorylation as reported previously.43 HIF-1α and HIF-2α were stably knocked down by viral transduction of shRNA constructs. Exogenous shRNA expression efficiently reduced HIFα levels (Figure 1C). In the absence of HIF-1α, increased HIF-2α protein levels could be observed in most cell lines, a currently unexplained phenomenon that we and others have described previously.8,44
As in MCF-7 cells,8 robust hypoxic WISP-2 mRNA induction was also found in T-47D, BT-474, and ZR-75-1 cells (Figure 1D). No hypoxic WISP-2 induction could be detected in MDA-MB-231 and MDA-MB-468 cells (data not shown). Knockdown of the HIFα isoforms demonstrated that WISP-2 mRNA is predominantly regulated by HIF-2α in MCF-7 and T-47D cells (Figure 1D). In MCF-7 cells, HIF-2α shRNA also prevented the hypoxic induction of WISP-2 but not of prolyl-4-hydroxylase domain 2 protein levels, the latter being an HIF-1α target (Figure 1B). However, in BT-474 cells, hypoxic WISP-2 mRNA induction was HIF-1α-dependent but not HIF-2α-dependent, and in ZR-75-1 cells hypoxic WISP-2 mRNA induction was attenuated in the absence of both HIF-1α and HIF-2α (Figure 1D). These data demonstrate that WISP-2 is regulated by hypoxia in low-invasive luminal-like breast cancer cell lines and that cancer-specific HIFα isoforms confer hypoxic WISP-2 induction.
Potential enhancer elements involved in HIF-inducible WISP2 promoter activity
To further investigate the HIFα isoform-specific effect on WISP-2 transcription, we tested various WISP2 promoter truncations driving firefly luciferase reporter gene expression in MCF-7 cells. All constructs ranging from −1929/+16 to −422/+16 relative to the transcriptional start site of the human WISP2 gene displayed similar promoter activities. In contrast with the robust hypoxic WISP-2 mRNA induction (Figure 1C), only weak responses to hypoxia could be observed with these reporter gene constructs (Figure 2A). Whereas moderate increases in promoter activity, mainly under hypoxic conditions, were observed after HIF-1α overexpression, strong increases in normoxic as well as hypoxic promoter activities followed HIF-2α overexpression, suggesting that HREs likely are present on these constructs but are not sufficient to confer full hypoxic inducibility. Additional truncations of the −422/+16 construct resulted in complete loss of promoter activity in the absence of the region close to the TATA-like box (construct −422/−75), demonstrating that the first 75 base pairs are essential for basal promoter activity. Normal basal activity but unresponsiveness to HIFα overexpression was observed for construct −83/+16 (Figure 2B).
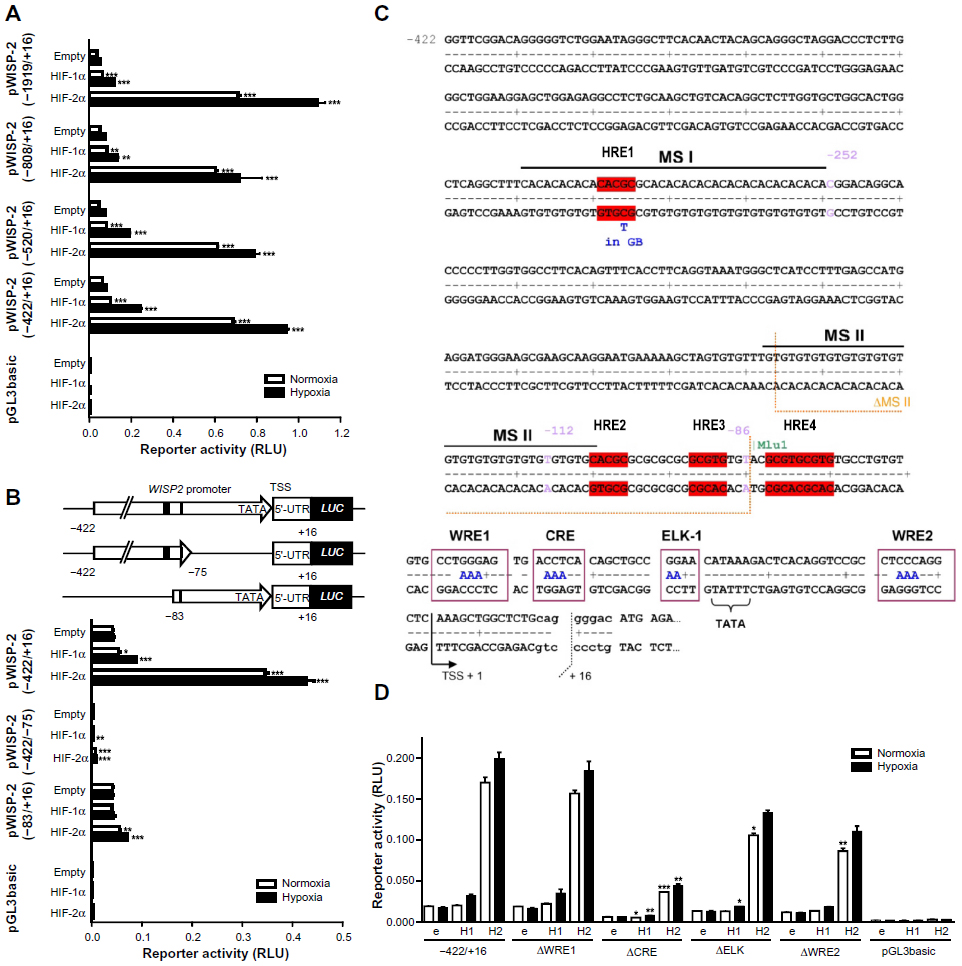 | Figure 2 Characterization of the WISP2 promoter. Dual luciferase reporter gene experiments in MCF-7 cells transfected with either HIF-1α or HIF-2α overexpression vectors together with the indicated firefly luciferase vectors and a renilla luciferase control vector, after 24 hours under normoxic or hypoxic (1% O2) conditions. (A) Promoter truncations from positions −1919 to −422 upstream of the transcriptional start site. (B) Potential hypoxia response elements are depicted by black marks (upper panel). HIF responsiveness was lost in the minimal promoter (lower panel). (C) Four potential hypoxia response elements in the WISP2 5′ flanking region are indicated in red. (D) MCF-7 shH2a cells were cotransfected with either the wild-type (−422) promoter construct or constructs bearing the indicated mutations, together with either an empty (e) vector, or a HIF-1α (H1) or HIF-2α (H2) overexpression vector. Shown are mean values ± the standard errors of the mean of three (four for D) independent experiments performed in triplicate. For statistical evaluation, the effects of HIF-1α and HIF-2α overexpression were compared with the respective empty vector controls (A and B), or were first normalized to the empty vector control values and then compared with the respective normalized values of the wild-type −422/+16 promoter construct (D). *P<0.05; **P<0.01; ***P<0.001. |
Three single putative HREs (HRE1, HRE2, and HRE3) and a double HRE (HRE4) were identified in the WISP2 promoter region (Figure 2C). Of note, the three single HREs were all located within microsatellite (MS) regions. HRE1 was found in a CA repeat region denominated MS I, HRE2 was located at the transition between a GT and a GC repeat, collectively named MS II, and HRE3 was identified within the GC repeat of MS II (Figure 2C). Furthermore, a phylogenetic footprint analysis of the promoter region revealed a strong sequence conservation around the TATA-like box, comprising a potential CRE, a binding site for ELK-1, and two WNT response elements (WREs, Figure 2C). However, when tested in MCF-7 shH2a cells with suppressed endogenous HIF-2α levels, WRE1 mutation neither affected basal promoter activity nor HIFα responsiveness. Mutation of the CRE site strongly decreased promoter activity but partially retained HIF-2α-mediated induction. Similarly, the ELK and WRE2 mutations reduced overall promoter activity and slightly affected HIF-2α responsiveness (Figure 2D). In conclusion, these conserved elements are mainly required for basal promoter activity but do not seem to confer HIF-2α-dependent induction of the WISP2 promoter, although we cannot exclude a partial cooperation between these cis-acting elements and the HREs.
Two HREs within an MS repeat regulate HIF-dependent WISP-2 transcription
To identify the HRE(s) responsible for HIF-mediated WISP-2 regulation, MS I (containing HRE1) was removed by a −252/+16 promoter truncation (ΔMS I), and MS II (containing HRE2 and HRE3) was removed by a promoter deletion (ΔMS II) as indicated in Figure 2C. Overexpression of both HIFα isoforms in stable HIF-2α knockdown MCF-7 cells was used to drive reporter gene expression. HIFα-dependent induction of the ΔMS I construct in stable HIF-2α knockdown MCF-7 cells was indistinguishable from the −411/+16 construct, indicating that MS I does not contain a functional HRE. In contrast, HIFα-dependent induction of reporter gene activity was strongly reduced when MS II was deleted (Figure 3A).
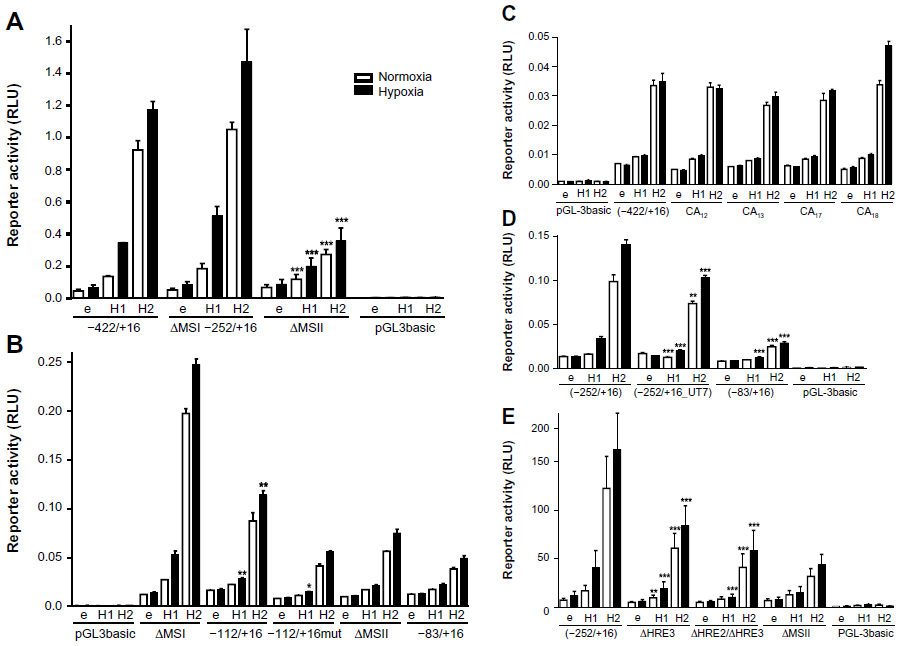 | Figure 3 Identification of the hypoxia response elements responsible for HIF-mediated induction of WISP-2 transcription. Dual luciferase reporter gene experiments in MCF-7 shH2a cells transfected with either an empty vector (e), or a HIF-1α (H1) or HIF-2α (H2) overexpression vector together with the indicated firefly luciferase constructs and a renilla luciferase control vector, after 24 hours under normoxic or hypoxic (1% O2) conditions. (A) WISP2 promoter constructs lacking either MS I (ΔMSI) or MS II (ΔMSII) were compared with the −422/+16 construct. (B) WISP2 promoter constructs containing wild-type (−112/+16) or mutant HRE2 and HRE3 (−112/+16mut) were compared with the ΔMSI, ΔMSII or minimal promoter (−83/+16) constructs. (C) WISP2 promoter constructs containing the indicated MS repeat length polymorphisms identified in T-47D cells were compared with the −422/+16 fragment derived from MCF-7 cells. (D) WISP2 promoter constructs containing the HRE2-deficient sequence identified in UT-7 cells (−252/+16_UT7) were compared with the corresponding fragment (−252/+16) and the minimal promoter (−83/+16) derived from MCF-7 cells. Shown are the mean values ± standard deviations of representative experiments performed in quadruplicate. (E) WISP2 promoter constructs containing the −252/+16 fragment, either wild-type (−252/+16), HRE2 mutant (ΔHRE2) or HRE2 and HRE3 double mutant (ΔHRE2/ΔHRE3), were compared with the ΔMSII construct. Shown are the mean values ± standard deviations of three independent experiments performed in quadruplicate. For statistical evaluation, the effects of HIF-1α and HIF-2α overexpression were first normalized to the empty vector control values and then compared with the respective normalized values of the wild-type −422/+16 (A and C) or −252/+16 (D and E) promoter constructs. (B) Similarly, the activities of −112/+16 and −112/+16mut were compared with the −83/+16 minimal promoter. *P<0.05; **P<0.01; ***P<0.001.
|
Deletion of the MS II resulted in basal reporter gene expression comparable with minimal WISP2 promoter (−83/+16) activity (Figure 3B). These experiments were repeated in wild-type MCF-7 cells with a similar outcome (data not shown). Furthermore, the −83/+16 minimal promoter construct was extended with oligonucleotides containing either wild-type (−112/+16) or mutated (−112/+16mut) HRE2 and HRE3, as indicated in Figure 2C. While no differences in activity were observed between the −112/+16mut and the −83/+16 constructs, a partial but significant reconstitution of HIF-2α-mediated WISP2 promoter induction was observed with the −112/+16 construct under hypoxic conditions (Figure 3B). In conclusion, these results demonstrate that HRE2 and/or HRE3 within MS II mainly confer HIF responsiveness to the WISP2 promoter.
Because MS instability is a hallmark of cancer,45 we tested whether differences in the length of the CA repeats in MS II might influence HIFα-inducible promoter activity. Therefore, genomic DNA from T-47D cells was amplified and cloned. CA repeat length variants, CA12, CA13, CA17, and CA18, were obtained and compared with the parental −422/+16 construct derived from MCF-7 cells (Figure 3C). However, no change in reporter gene activity could be observed, suggesting that MS instability does not affect HIFα-mediated WISP2 promoter activity.
Unexpectedly, sequencing of the WISP2 upstream regulatory region revealed the presence of HRE2 in all breast cancer cell lines analyzed (Figure 1), as well as in Hep3B and HepG2 hepatoma, SK-N-MC neuroepithelioma, and HeLa cervical carcinoma, but not in three published sequences (GRCh37.p2, HuRef, and Hs_Celera). Only UT-7 megakaryoblastic leukemia cells lacked HRE2 and showed the same sequence as the database entries (data not shown). Therefore, the −252/+16 region was amplified from UT-7 cells (−252/+16_UT7) and compared with the promoter constructs derived from MCF-7 cells. Only a small reduction in response to HIFα overexpression was observed with the −252/+16_UT7 construct (Figure 3D). Also, mutation of HRE3 (ΔHRE3) resulted in a modest reduction in reporter gene activity, and a double mutation of HRE2 and HRE3 (ΔHRE2/ΔHRE3) further abrogated reporter gene activity comparable with that observed after deletion of MS II (Figure 3E). Taken together, these results indicate that the two HREs within MS II mediate at least partially HIFα-dependent induction of the WISP2 promoter.
WISP-2 negatively correlates with tumor infiltration by macrophages
We previously reported that high WISP-2 levels correlate positively with high HIF-2α levels and prolonged overall survival in patients with breast cancer.8 Because tumor macrophage infiltration, associated with the hypoxic tumor microenvironment and high HIFα levels, not only represents an important step during progression of breast cancer but also an important prognostic marker,46,47 we analyzed whether WISP-2 expression levels are related to tumor-associated macrophage counts. Therefore, over 300 breast cancer samples were immunostained and scored for the macrophage markers CD68 and CD163 and the pan-leukocyte marker CD45 (Figure 4A). Interestingly, a negative correlation between WISP-2 and tumor-associated macrophage counts was found (Figure 4B). As expected, overall survival of these patients was negatively associated with macrophage infiltration (Figure 4C). These results suggest that HIF-2α-mediated WISP-2 expression is a marker for (and maybe is even causally involved in) breast cancer progression stage, with low cancer cell proliferation/invasiveness as well as low macrophage infiltration, and both contributing to an improved prognosis.
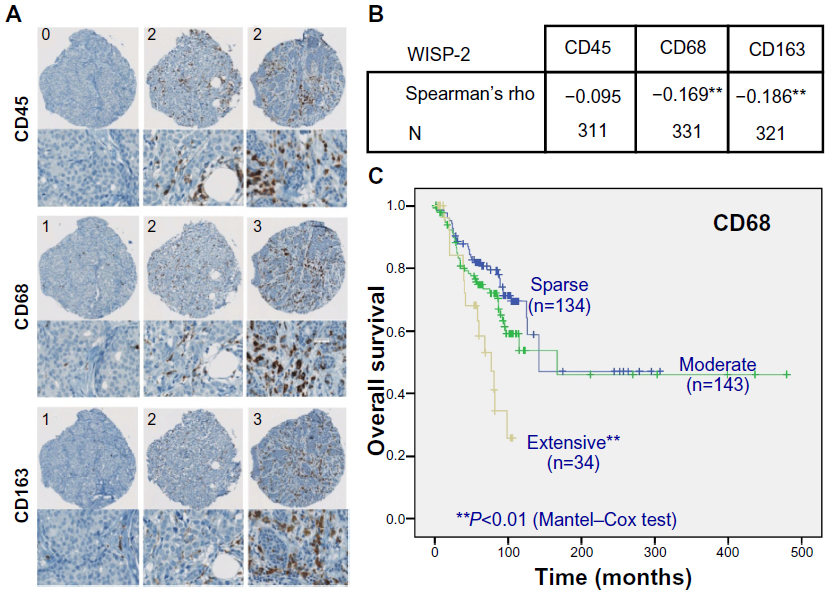 | Figure 4 WISP-2 correlation with macrophage infiltration in human breast cancer tissues. (A) Examples of spots of breast cancer tissue microarrays immunostained for CD45, CD68, and CD163 macrophage markers. Numbers in upper left corners indicate categorization of staining intensity (0/1, sparse; 2, moderate; 3, extensive). (B) Negative correlation of WISP-2 expression with macrophage markers CD68 and CD163 in human breast cancer samples 331 and 321, respectively. (C) Negative prognostic correlation between overall survival of breast cancer patients and high infiltration levels with CD68-positive tumor-associated macrophages. **P<0.01. |
HIF modulates the WISP-2 suppressed motility of MCF-7 cells
Silencing of WISP-2 in noninvasive MCF-7 cells has been reported to enhance motility and modulate the expression of genes involved in cancer invasiveness.30,48,49 To investigate the functional consequences of HIF-mediated WISP-2 regulation, we stably transfected MCF-7 cells with WISP-2 shRNA, resulting in a robust knockdown of WISP-2 mRNA using two independent shRNA constructs (shW#1 and shW#2) under normoxic as well as hypoxic conditions (Figure 5A). Proliferation of both WISP-2 knockdown cells, but not of previously established HIF-2α knockdown cells,8 was significantly impaired under both normoxic and hypoxic conditions (Figure 5B). As determined by automated trypan blue exclusion and video microscopy analysis, no difference in gross cell morphology or viability could be observed (data not shown). Low cell density colony formation was somewhat attenuated by knockdown of both HIF-2α and WISP-2 (Figure 5C). In contrast, anchorage-independent colony formation was increased, especially in normoxic cells (Figure 5D). Similarly, recovery from scratches in confluent cell layers was significantly increased by knockdown of both HIF-2α and WISP-2 (Figure 5E). Probably owing to remaining weak WISP-2 induction (see Figure 5A), under hypoxic conditions a slightly impaired anchorage-independent colony formation (Figure 5D) as well as scratch recovery of MCF-7 cells was observed (Figure 5E), although the values were still higher than in the hypoxic control cells. In summary, these results are consistent with a role of HIF-2 in WISP-2-mediated suppression of MCF-7 anchorage-independent growth and cell motility, which are two hallmarks of cancer progression.
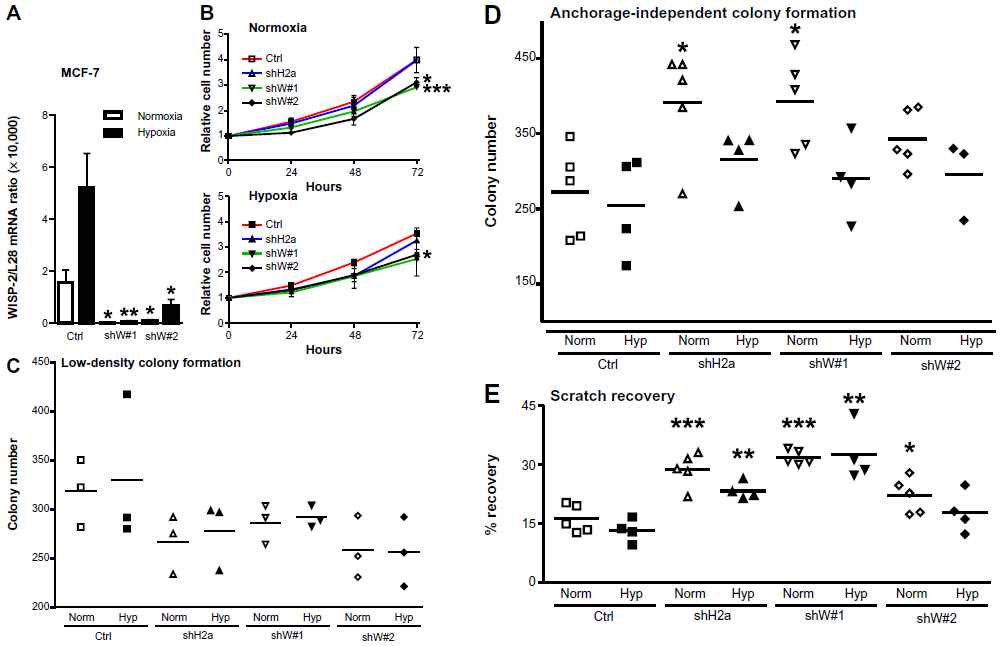 | Figure 5 Role of HIF-2α and WISP-2 in MCF-7 proliferation and motility. (A) MCF-7 cells were stably transfected by lentiviral infection with control or two independent WISP-2 shRNA constructs (shW#1 and shW#2). Following normoxic or hypoxic (24 hours, 0.2% O2) exposure, WISP-2 mRNA (messenger RNA) levels were determined by reverse transcription quantitative polymerase chain reaction and normalized to the mRNA levels of the ribosomal protein L28. Shown are the mean values ± standard deviations of five independent experiments. (B) Proliferation of MCF-7 cells kept under normoxic or hypoxic conditions for up to 3 days was determined by cell counting, and cell numbers were normalized to the number of cells initially seeded. Shown are the mean values ± standard deviations of three independent experiments performed in triplicate. Differences after 3 days relative to the nontransfected controls were analyzed by Student’s t-test. (C) Low-density colony formation of MCF-7 cells cultured under normoxic or hypoxic conditions for 10 days. Shown are the colony numbers of three measurements in triplicate and the corresponding mean values. (D) Anchorage-independent colony formation of MCF-7 cells cultured in soft agar under normoxic or hypoxic conditions for 14 days. Shown are the colony numbers of 4–5 triplicate measurements and the corresponding mean values. (E) Recovery of scratched confluent MCF-7 cell layers after 24 hours of normoxic or hypoxic (0.2% O2) exposure. Shown are the results of 4–5 independent experiments. For statistical evaluation, all values were compared with the respective control shRNA transfected cells. *P<0.05; **P<0.01; ***P<0.001. |
Discussion
In this study, we could corroborate the previously reported hypoxic induction of WISP-28 which was HIF-2-dependent in most but not all low-invasive breast cancer cell lines tested, suggesting that currently unknown cell type-specific cofactors determine the HIFα isoform responsible for hypoxic WISP-2 induction, rather than intrinsic gene selectivity of the HIFα isoforms themselves. Previous studies have suggested a cooperation of HIF-2α, but not HIF-1α, with ETS factors resulting in target selectivity,14,50 but whether a different composition of ETS factors explains the HIF-1α-specific WISP-2 expression in BT-474 cells remains to be explored. Although we observed lower basal levels of WISP2 promoter activity following mutation of the conserved CRE, ELK-1, and WRE sites, HIF-2α-mediated WISP-2 regulation remained unaffected, suggesting that none of these factors confers HIFα isoform selectivity.
Two functional HREs were identified which are essential for HIF-2α-specific induction of WISP2 promoter activity in MCF-7 cells, but even promoter constructs up to 1,919 base pairs upstream of the transcriptional start site did not fully recapitulate the hypoxic induction factors determined at the mRNA level. Of note, in a genome-wide chromatin immunoprecipitation study, both HIFα isoforms were reported to bind to a region within the first intron of WISP-2,51 suggesting that these elements might contribute to endogenous regulation of WISP-2. However, at least in MCF-7 cells, we could not detect any further increase in hypoxic WISP-2 transcription by this region in combination with the upstream regulatory elements (data not shown). The two functional HREs are located within the highly polymorphic MS II region of the WISP2 promoter. Such unstable polymorphic dinucleotide repeats are known to play an important role in tumor progression. MS instability seems to be mostly due to the loss of a functional mismatch repair machinery, and is implicated in prognosis and therapy response because it alters the expression levels of the affected genes.45,52 However, we could not find any role for MS II length in regulating reporter constructs driven by the WISP2 promoter.
By immunohistochemical analysis of breast cancer samples, we recently demonstrated that HIF-2α and WISP-2 levels correlate with a more differentiated tumor cell type and consistently with a better prognosis.8 These results are in line with the findings presented herein showing that WISP-2 negatively correlates with tumor macrophage invasion, which provides an additional marker for a better tumor prognosis. Silencing of WISP-2 elevated two parameters of cancer cell aggressiveness, ie, anchorage-independent colony formation and recovery from scratches in confluent cell layers. These effects could not simply be explained by changes in proliferation rates and anchorage-dependent colony formation, which were actually slightly decreased rather than increased. Interestingly, increased anchorage-independent colony formation and scratch recovery following WISP-2 knockdown could be phenocopied by HIF-2α silencing. While we cannot exclude that additional signaling pathways are recruited by the hypoxic tumor microenvironment, our data strongly suggest that HIF-2-mediated WISP-2 induction contributes to a less aggressive tumor type.
Conclusion
Taken together, our data suggest that the previously reported association between high HIF-2α levels and an increased overall survival rate in patients with breast cancer could be explained at least partially by HIF-2α-mediated direct induction of WISP-2, maintaining a less aggressive breast cancer phenotype.
Acknowledgments
We thank M Sabbah for providing the plasmids and P Spielmann for technical assistance. This work was supported by Swiss National Science Foundation grants 31003A_129962 (to RHW and DPS) and 31003A_146203 (to RHW), and by the European Union (FP7 grant agreement 246539 to IAR).
Disclosure
The authors report no conflicts of interest in this work.
References
Lehmann S, Stiehl DP, Honer M, et al. Longitudinal and multimodal in vivo imaging of tumor hypoxia and its downstream molecular events. Proc Natl Acad Sci U S A. 2009;106:14004–14009. | |
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. | |
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. | |
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. | |
Ratcliffe PJ. Oxygen sensing and hypoxia signaling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591:2027–2042. | |
Unruh A, Ressel A, Mohamed HG, et al. The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. | |
Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14:191–201. | |
Stiehl DP, Bordoli MR, Abreu-Rodríguez I, et al. Non-canonical HIF-2α function drives autonomous breast cancer cell growth via an AREG-EGFR/ErbB4 autocrine loop. Oncogene. 2012;31:2283–2297. | |
Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. | |
Holmquist-Mengelbier L, Fredlund E, Lofstedt T, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423. | |
Patel SA, Simon MC. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death Differ. 2008;15:628–634. | |
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22. | |
Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66:5641–5647. | |
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1α, HIF-2α, and other pathways. J Biol Chem. 2006;281:15215–15226. | |
Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. | |
Warnecke C, Zaborowska Z, Kurreck J, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004;18:1462–1464. | |
Le Bras A, Lionneton F, Mattot V, et al. HIF-2α specifically activates the VE-cadherin promoter independently of hypoxia and in synergy with Ets-1 through two essential ETS-binding sites. Oncogene. 2007;26:7480–7489. | |
Yamashita T, Ohneda K, Nagano M, et al. Hypoxia-inducible transcription factor-2α in endothelial cells regulates tumor neovascularization through activation of ephrin A1. J Biol Chem. 2008;283:18926–18936. | |
Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. Characterization of the activation of protein tyrosine phosphatase, receptor-type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor-2α. PLoS One. 2010;5:e9641. | |
Bordoli MR, Stiehl DP, Borsig L, et al. Prolyl-4-hydroxylase PHD2- and hypoxia-inducible factor 2-dependent regulation of amphiregulin contributes to breast tumorigenesis. Oncogene. 2011;30:548–560. | |
Brigstock DR. The CCN family: a new stimulus package. J Endocrinol. 2003;178:169–175. | |
Katsube K, Sakamoto K, Tamamura Y, Yamaguchi A. Role of CCN, a vertebrate specific gene family, in development. Dev Growth Differ. 2009;51:55–67. | |
Pennica D, Swanson TA, Welsh JW, et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in Wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci U S A. 1998;95:14717–14722. | |
Russo JW, Castellot JJ. CCN5: biology and pathophysiology. J Cell Commun Signal. 2010;4:119–130. | |
Banerjee SK, Banerjee S. CCN5/WISP-2: a micromanager of breast cancer progression. J Cell Commun Signal. 2012;6:63–71. | |
Saxena N, Banerjee S, Sengupta K, Zoubine MN, Banerjee SK. Differential expression of WISP-1 and WISP-2 genes in normal and transformed human breast cell lines. Mol Cell Biochem. 2001;228:99–104. | |
Zoubine MN, Banerjee S, Saxena NK, Campbell DR, Banerjee SK. WISP-2: a serum-inducible gene differentially expressed in human normal breast epithelial cells and in MCF-7 breast tumor cells. Biochem Biophys Res Commun. 2001;282:421–425. | |
Fritah A, Redeuilh G, Sabbah M. Molecular cloning and characterization of the human WISP-2/CCN5 gene promoter reveal its upregulation by oestrogens. J Endocrinol. 2006;191:613–624. | |
Fritah A, Saucier C, De Wever O, et al. Role of WISP-2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol Cell Biol. 2008;28:1114–1123. | |
Inadera H, Hashimoto S, Dong HY, et al. WISP-2 as a novel estrogen-responsive gene in human breast cancer cells. Biochem Biophys Res Commun. 2000;275:108–114. | |
Banerjee S, Saxena N, Sengupta K, Tawfik O, Mayo MS, Banerjee SK. WISP-2 gene in human breast cancer: estrogen and progesterone inducible expression and regulation of tumor cell proliferation. Neoplasia. 2003;5:63–73. | |
Banerjee S, Sengupta K, Saxena NK, Dhar K, Banerjee SK. Epidermal growth factor induces WISP-2/CCN5 expression in estrogen receptor-a-positive breast tumor cells through multiple molecular cross-talks. Mol Cancer Res. 2005;3:151–162. | |
Dhar G, Mehta S, Banerjee S, et al. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: a potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 2007;254:63–70. | |
Dhar K, Banerjee S, Dhar G, Sengupta K, Banerjee SK. Insulin-like growth factor-1 (IGF-1) induces WISP-2/CCN5 via multiple molecular cross-talks and is essential for mitogenic switch by IGF-1 axis in estrogen receptor-positive breast tumor cells. Cancer Res. 2007;67:1520–1526. | |
Fritah A, Saucier C, Mester J, Redeuilh G, Sabbah M. p21WAF1/CIP1 selectively controls the transcriptional activity of estrogen receptor a. Mol Cell Biol. 2005;25:2419–2430. | |
Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1a and HIF-2a. Mol Biol Cell. 2007;18:4528–4542. | |
Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J Biol Chem. 2006;281:23482–23491. | |
Barth S, Nesper J, Hasgall PA, et al. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27:3758–3768. | |
Balamurugan K, Luu VD, Kaufmann MR, et al. Onconeuronal cerebellar degeneration-related antigen, Cdr2, is strongly expressed in papillary renal cell carcinoma and leads to attenuated hypoxic response. Oncogene. 2009;28:3274–3285. | |
Wollenick K, Hu J, Kristiansen G, et al. Synthetic transactivation screening reveals ETV4 as broad coactivator of hypoxia-inducible factor signaling. Nucleic Acids Res. 2012;40:1928–1943. | |
Hammarstedt A, Hedjazifar S, Jenndahl L, et al. WISP2 regulates preadipocyte commitment and PPARγ activation by BMP4. Proc Natl Acad Sci U S A. 2013;110:2563–2568. | |
Katschinski DM, Le L, Heinrich D, et al. Heat induction of the unphosphorylated form of hypoxia-inducible factor-1a is dependent on heat shock protein-90 activity. J Biol Chem. 2002;277:9262–9267. | |
Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. | |
Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2012;7:153–162. | |
Shay JE, Celeste Simon M. Hypoxia-inducible factors: crosstalk between inflammation and metabolism. Semin Cell Dev Biol. 2012;23:389–394. | |
Tang X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013;332:3–10. | |
Banerjee S, Dhar G, Haque I, et al. CCN5/WISP-2 expression in breast adenocarcinoma is associated with less frequent progression of the disease and suppresses the invasive phenotypes of tumor cells. Cancer Res. 2008;68:7606–7612. | |
Dhar G, Banerjee S, Dhar K, et al. Gain of oncogenic function of p53 mutants induces invasive phenotypes in human breast cancer cells by silencing CCN5/WISP-2. Cancer Res. 2008;68:4580–4587. | |
Elvert G, Kappel A, Heidenreich R, et al. Cooperative interaction of hypoxia-inducible factor-2a (HIF-2a) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J Biol Chem. 2003;278:7520–7530. | |
Mole DR, Blancher C, Copley RR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1a and HIF-2a DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. | |
Karran P. Microsatellite instability and DNA mismatch repair in human cancer. Semin Cancer Biol. 1996;7:15–24. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.