Back to Archived Journals » Integrated Blood Pressure Control » Volume 12
Hyperuricemia and Hypertension: Links and Risks
Authors Stewart DJ ![]() , Langlois V, Noone D
, Langlois V, Noone D
Received 3 October 2019
Accepted for publication 27 November 2019
Published 24 December 2019 Volume 2019:12 Pages 43—62
DOI https://doi.org/10.2147/IBPC.S184685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Douglas J Stewart,1 Valerie Langlois,1,2 Damien Noone1,2
1Division of Nephrology, The Hospital for Sick Children, Toronto, Ontario M5G 1X8, Canada; 2Department of Paediatrics, University of Toronto, Toronto, Ontario M5G 1X8, Canada
Correspondence: Damien Noone
Email [email protected]
Abstract: Hyperuricemia has long been recognized to be associated with increased cardiovascular risk, including risk of developing hypertension. Epidemiological findings suggest that the link with hypertension is stronger in children and adolescents. Uric acid acts as a strong antioxidant compound in the extracellular environment but has pro-inflammatory effects within the intracellular setting. A chronic phase of microvascular injury is known to occur after prolonged periods of hyperuricemia. This is proposed to contribute to afferent arteriolopathy and elevation of blood pressure that may become unresponsive to uric acid-lowering therapies over time. Studies have struggled to infer direct causality of hyperuricemia due to a vast number of confounders including body mass index. The aim of this review is to present the available data and highlight the need for large scale prospective randomized controlled trials in this area. At present, there is limited evidence to support a role for uric acid-lowering therapies in helping mitigate the risk of hypertension.
Keywords: hyperuricemia, hypertension, urate, cardiovascular, chronic kidney disease
Introduction
Hypertension affects an estimated 1.13 billion people worldwide and the World Health Organization reports that it is implicated in 13% of deaths globally.1 They have set a target of reducing its prevalence by 25% by the year 2025 (from the 2010 baseline). The etiology of essential hypertension is multifactorial and complex, with an increasing number of contributory factors being appreciated in recent decades. The relationship between uric acid (UA) and blood pressure (BP) is certainly not new and has been recognized for over a century.2 Multiple studies have demonstrated an independent association between hyperuricemia and hypertension, although the issue of direct causality is still hotly debated.
The aim of this review is to present the available evidence on the links between hyperuricemia and hypertension with an emphasis on the potential role of UA-lowering therapies in helping mitigate this risk.
Uric Acid Homeostasis
Uric acid (C5H4N4O3), or 2,6,8-trihydroxypurine, is a heterocyclic carbon-based compound with a molecular weight of 168 Da. It is a weak acid with pKa ≈ 5.8 in the blood and ≈ 5.35 in the urine. It exists predominantly in its monovalent deprotonated ionic form, the urate anion, within the physiological arterial blood pH of 7.40. As such, the following equation is shifted far to the right:
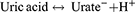
UA is the end-product of purine mononucleotide catabolism. Purines can be synthesized endogenously or can be derived from dietary sources. Purines perform many vital functions within the body. Notably, adenosine triphosphate (ATP) provides the energy to drive intracellular reactions, and the purine nucleobases, adenine and guanine, are integral components of ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). The catabolism of the purine mononucleotides adenylic acid (AMP), guanylic acid (GMP) and inosinic acid (IMP) to UA involves several enzymatic steps (Figure 1). An intermediate step is the production of the purine bases guanine and hypoxanthine. These are then converted to xanthine and irreversibly oxidized to UA by the two interconvertible isoforms of xanthine oxidoreductase (XOR) - xanthine dehydrogenase (XDH) and xanthine oxidase (XO). The latter isoform produces reactive oxygen species (ROS) and is generated under conditions of physiological stress and ischemia. A salvage pathway exists that allows for purine reutilization involving the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT).
 |
Figure 1 Urate production pathways. De novo synthesis of urate starts with the generation of phosphoribosyl pyrophosphate (PRPP) from ribose 5-phosphate (ribose 5-P) and adenosine triphosphate (ATP), leading to the formation of the purine mononucleotide inosine monophosphate (IMP). IMP is indirectly converted to hypoxanthine, a purine base. Adenosine monophosphate (AMP) and guanosine monophosphate (GMP) are involved in the production of the other purine bases adenine and guanine respectively. Enzymatic salvage pathways exist that allow for further generation of AMP, IMP and GMP from these purine bases. The conversion of fructose to fructose 1-phosphate by fructokinase generates AMP. Under conditions of ischemia, the conversion of hypoxanthine to xanthine, and subsequently to the urate anion, generates superoxide (•O₂⁻) and thus stimulates the release of ROS. Allopurinol and febuxostat act as xanthine oxidase (XO) inhibitors to block the formation of urate. Rasburicase and pegloticase are recombinant versions of uricase that promote the breakdown of uric acid (UA) into allantoin. Abbreviations: ADP, adenosine diphosphate; GTP, guanosine triphosphate; 5’NT, 5’nucleotidase; APRT, adenine phosphoribosyl transferase; HPRT, hypoxanthine-guanine phosphoribosyl transferase; PNP, purine nucleotide phosphorylase; XDH, xanthine dehydrogenase; NAD⁺, nicotinamide adenine dinucleotide (oxidized form); NADP, nicotinamide adenine dinucleotide (reduced form). |
UA homeostasis is governed by the balance between the rate of UA generation (determined by purine catabolism), renal excretion, and intestinal secretion. UA is derived mainly from the breakdown of purines in the liver and bowel, as well as the kidneys, muscle and vascular endothelium. The exogenous supply of UA is derived from dietary sources of purines including meat, seafood and alcohol. Purine-free formula diets have been associated with approximately a 40% reduction in urinary UA excretion suggesting that diet provides a significant source of urate precursors, despite there being few dietary sources of UA alone.3
UA metabolism in the tissues is minimal and most elimination occurs via the kidneys and intestine. Intestinal excretion of UA is followed by metabolism by gut bacteria in a process called intestinal uricolysis. This accounts for 25–35% of UA elimination, and renal excretion accounts for the remaining 65–75%.4 The majority of circulating urate is free with less than 5% albumin-bound. Most urate is therefore readily filtered by the glomeruli, although up to 90% may then be reabsorbed.5
Multiple urate transporters have been identified that play a role in renal tubular reabsorption and secretion of urate, thus helping regulate homeostasis (Figure 2) and maintain plasma levels within a range.6 The main transporters identified to date are:
- URAT1 (urate transporter 1) – which belongs to the organic acid transporter (OAT) family and is encoded by the SLC22A12 (solute carrier family 22 member 12) gene on chromosome 11. It was the first urate transporter described in detail and plays a role in urate reabsorption at the apical surface of the proximal tubular epithelium.7 It reabsorbs UA in exchange for monovalent anions such as acetoacetate, hydroxybutyrate, lactate and nicotinoate. Certain drugs including benzbromarone, probenecid and losartan, are known to exert a uricosuric effect via inhibition of URAT1.8
- GLUT9 (glucose transporter 9) – another major urate reabsorptive transporter. This is a hexose transporter and is encoded by the SLC2A9 gene on chromosome 4. GLUT9 reabsorbs urate at the basolateral membrane of the proximal tubule and has been demonstrated to be the single most important genetic determinant of serum UA (SUA) levels and hyperuricemia in a large human genome-wide study of 28,141 individuals.9,10
- ABCG2 (ATP-binding cassette subfamily G member 2; encoded by the ABCG2 gene) – is known to have effects on both intestinal and renal excretion of urate. It acts at the apical surface of the proximal tubular epithelium as a urate extrusion pump. Polymorphisms of ABCG2 may contribute to a “pseudo-overproduction” phenotype due to reduced UA intestinal excretion.11 ABCG2 expression has been found to be upregulated in partially nephrectomized rats, and SUA levels were unaltered by the change in renal function.12 This suggests that SUA homeostasis may be maintained in the context of renal impairment by means of upregulated intestinal excretion via ABCG2.
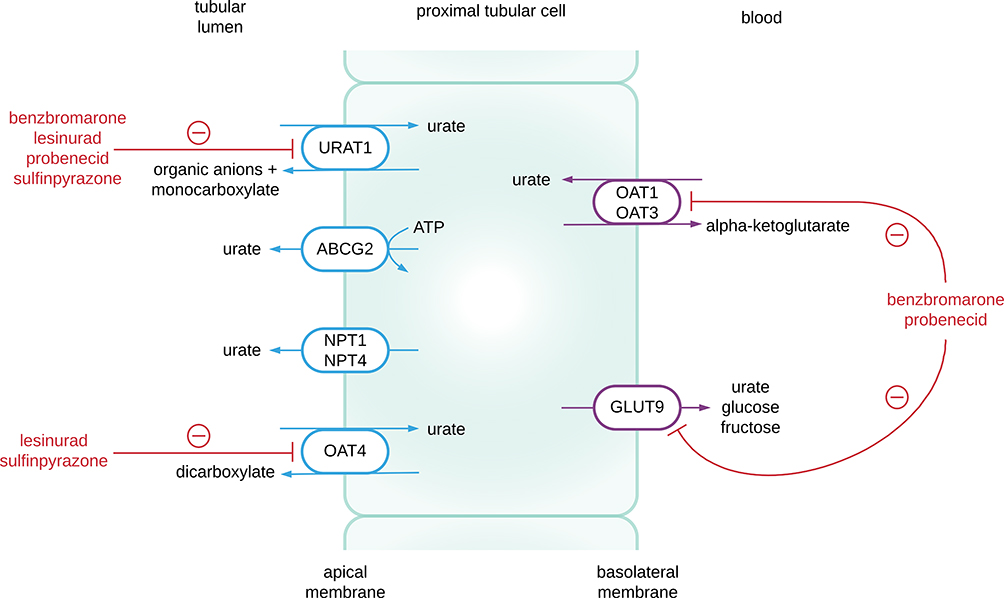 |
Figure 2 Renal urate transporters. Urate reabsorption is mediated by the proximal tubular apical transporters URAT 1 and OAT4, and the basolateral transporter GLUT 9. Urate secretion is mediated by the apical transporters ABCG2, NPT1, NPT4, and the basolateral transporters OAT1 and OAT3. Benzbromarone and probenecid act as inhibitors of URAT 1, OAT1, OAT 3 and GLUT9. Lesinurad and sulfinpyrazone inhibit URAT 1 and OAT4. |
Our understanding of urate homeostasis is still evolving as multiple other urate transporters have been identified in recent years, such as NPT1 (nicotinate phosphoribosyltransferase; encoded by the SLC17A1 gene).13
Definition of Hyperuricemia
Age- and sex-related normative ranges for serum urate exist, although cut-off values do vary among studies, thus hindering comparison of results. Hyperuricemia is typically defined as serum urate concentration > 6.0 mg/dL (> 360 µmol/L) in women, > 7.0 mg/dL (> 415 µmol/L) in men and > 5.5 mg/dL (> 330 µmol/L) in children and adolescents.14 The fractional excretion of filtered urate (FEU) is higher in children (15–30%) compared to adults (10%).15 This accounts for a linear increase in serum urate levels up to the age of 15 years. The uricosuric effect of estrogens leads to lower serum urate levels in premenopausal women. Following menopause, however, urate levels tend to be more comparable to those of males of similar age.
A serum urate concentration of 7.0 mg/dL (415 µmol/L) is defined as the upper limits of normal, as it approaches the limits of urate solubility within water. Urate is more soluble in plasma than urine, and crystal deposits may form in the soft tissues and joints when urate exceeds solubility limits, often at concentrations exceeding 10.0 mg/dL (600 µmol/L). These urate crystals form as monosodium urate (MSU). Multiple factors have been identified that may contribute to an increased risk of MSU crystallization.16
Most humans excrete urine that is supersaturated with UA. As the urinary pH is more variable than that of blood, the ratio of UA to urate varies considerably. At lower urinary pH, there will be a greater proportion of un-dissociated UA compared to urate. In the context of urinary acidification, there is a predisposition for UA urinary crystallization to occur that may contribute to the development of nephrolithiasis.17
Biological Actions of Uric Acid
The majority of mammalian species have low levels of circulating UA as it is converted by the enzyme uricase (urate oxidase) to the highly soluble compound allantoin. Uricase is situated in the hepatocyte peroxizomes. Humans, and some of the other higher primates, lack the ability to produce uricase due to the presence of two truncating mutations in the uricase gene that introduce premature stop codons.18 Humans are therefore exposed to relative hyperuricemia.
In the extracellular environment, UA is one of the strongest antioxidant compounds alongside ascorbate.19 Indeed, UA plays a role in preventing lipid peroxidation although ascorbate must be present for this to occur.20 Consequently, reduced oxidative stress is a hypothesized evolutionary advantage to the relative hyperuricemia present in humans.21 In experimental models, UA has been found to act as a peroxynitrite scavenger in neuronal cells, diminishing the transport of leukocytes across the blood brain barrier.22 Hyperuricemia may offer a neuroprotective advantage in neurodegenerative diseases such as Alzheimer’s disease,23 Parkinson’s disease,24 and multiple sclerosis.25 In rats, UA has been associated with reduced production of the free radical nitrotyrosine by hepatocytes during hemorrhagic shock, and its administration leads to attenuated liver tissue injury.26 High SUA levels in patients with atherosclerosis have been correlated with elevated total serum antioxidant capacity, and are proposed to play a compensatory role in reducing vascular oxidative damage.27 UA is present in the respiratory epithelial lining fluid where it contributes to antioxidant defenses. After accounting for potential confounders, hypouricemia in smokers has been attributed to higher rates of chronic obstructive pulmonary disease (COPD) and lung cancer.28 Urate may also play an immunological role by acting as a “danger signal” when released from dying cells. In doing so, it may help activate CD8 cells and facilitate apoptotic recognition by dendritic cells.29 Furthermore, it has been theorized that the uricase mutation acts to create an evolutionary advantageous “thrifty gene” that maintains BP in the setting of low dietary salt intake, and may help boost body fat stores when faced with food scarcity.30,31 Increased proximal tubular reabsorption of sodium has been demonstrated in hyperuricemic individuals, suggestive of the metabolic changes seen in those with hyperinsulinism.32
Despite these proposed benefits, urate is known to act as a pro-oxidant and pro-inflammatory mediator within the intracellular environment. Multiple in vitro biochemical effects have been described including activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.33 The resultant superoxide generation by phagocytes stimulates the release of ROS which contribute to oxidative stress. Other effects include upregulation of pro-inflammatory transcription factors, vasoconstrictive factors and chemokines, which may induce proximal tubular dysfunction34,35 as well as vascular smooth muscle proliferation.36,37 Urate acts to induce both mitochondrial and endothelial dysfunction.38,39 The infusion of UA into humans has been associated with increased interleukin 6 (IL-6) levels in response to an oral lipid challenge which may help explain increased cardiovascular risk in those with subclinical hyperuricemia.40 Furthermore, it has been associated with impaired endothelial nitric oxide (eNO) release and impaired acetylcholine-induced forearm vasodilation.41 Conversely, evidence suggests that hypouricemia may be associated with deleterious physiological effects. Extremely low levels of urate (e.g. < 1.0 mg/dL [< 60 µmol/L]) have been linked with increased risk of cardiovascular events in a J-shaped distribution in adults.42–44
The hypothesis regarding the antagonistic pro- and antioxidant effects of UA has been described in the literature as the “oxidant-antioxidant paradox”.45 This may help explain the mixed and contradictory results of studies whereby hyperuricemia is induced in vivo via UA infusion. Under certain conditions, particularly the hydrophilic environment, the protective effects of UA are manifested. Whether extracellular UA is always associated with beneficial effects requires further research. What remains clear is that UA demonstrates different physiological properties within different biological systems and biochemical environments.
Etiology of Hyperuricemia
A range of factors have been identified that may contribute to increased hyperuricemia risk. These can be broadly classified into factors associated with increased purine synthesis and/or urate production, versus factors that inhibit urate excretion and clearance. These are summarized in Table 1. The latter is a more common cause of secondary hyperuricemia and can be exacerbated by volume depletion and the use of diuretic medications (both loop and thiazide diuretics), as hypovolemia leads to compensatory UA proximal tubular reabsorption.
 |
Table 1 Etiology of Hyperuricemia |
A small number of individuals with hyperuricemia have inherited defects resulting in primary overproduction of UA. Genetic deficiency of HPRT is associated with Lesch-Nyhan syndrome, an X-linked recessive disorder characterized by hyperuricemia and self-mutilating behaviours. Other monogenic causes of hyperuricemia include phosphoribosylpyrophosphate synthetase (PRS) superactivity and adenine phosphoribosyltransferase (APRT) deficiency.46
The exogenous pool of UA is determined by dietary intake and may be boosted by purine rich foods. However, as stated above, dietary purines only contribute to a small proportion of total body UA generation.3,47 Evidence suggests that total protein intake does not correlate with hyperuricemia risk.48 Furthermore, intake of dairy,48 caffeine49,50 and vitaminC51,52 may be inversely associated with hyperuricemia risk.
The percentage increase in the consumption of fructose in the Westernized diet over the past four decades, particularly from high-fructose corn syrup added to sugar-sweetened beverages, has been linked to an increased incidence of hyperuricemia, as well as obesity.53–55 Although fruits contain natural fructose, their ability to induce hyperuricemia is believed to be offset by a combination of other intrinsic properties of the fruit, including the presence of fibre (which slows intestinal absorption of UA), vitamin C (which provides an antioxidant effect and enhances urinary UA excretion), catechins and flavonoids (which both offer further antioxidant effects).56 Fructose is known to activate the hepatocyte fructokinase pathway. The depletion of ATP in this pathway leads to increased purine turnover.48 XO-mediated disposal of purines occurs, and UA is generated as a terminal product. Intracellular accumulation of UA contributes to mitochondrial oxidative stress, leading to fructose generation via the aldose reductase pathway, and further fructokinase upregulation. There is controversy as to whether GLUT9 may function as both a fructose and urate transporter, thus being responsible for urate reabsorption in the context of increased fructose intake.57,58
Evidence suggests that hyperuricemia risk may be partially programmed in utero. Preeclampsia is associated with increased maternal and fetal SUA levels, believed to occur secondary to endothelial dysfunction and reduced glomerular filtration rate (GFR).59,60 Maternal hyperuricemia during pregnancy is known to contribute to placental dysfunction, leading to intrauterine growth restriction (IUGR), increased risk of salt-sensitive hypertension, vascular disease, reduced nephron number, and increased risk of hyperuricemia in the offspring.30,61 The risk of these outcomes is particularly increased in the context of gestational hypertension and proteinuria.62 Twin studies and genetic epidemiological techniques estimate that there is considerable heritability with regards to SUA levels, ranging from 40% to 73%.63–65 Genome wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) at 28 genomic loci implicated in UA regulation, accounting for an estimated 6.0–7.7% of SUA variability.66,67
Epidemiology of Hyperuricemia
United States epidemiological data from 2007 to 2008 suggest that the prevalence of hyperuricemia in the adult population is as high as 21.4% and this continues to increase over time.68 Within the US population, the prevalence is marginally higher in African Americans than those from other ethnic groups (25.7% versus 22.1%). Italian data also suggest increasing prevalence between the years 2005 to 2009 (from 8.5% to 11.9% of the population respectively) using a cut-off SUA value of 6.0 mg/dL (360 µmol/L).69 The highest prevalence of hyperuricemia worldwide has been described in the Taiwanese aboriginal population with rates of 41.4%.70 Factors that may be contributing to increased hyperuricemia prevalence include the aforementioned change in dietary patterns, increased life expectancy, increased rates of obesity, and the increased availability of certain medications (particularly diuretics).
Hyperuricemia and Its Relationship with Disease
The relationship between hyperuricemia and disease is evidenced most clearly with gout71 and nephrolithiasis, but epidemiologic associations with heart failure (both with and without preserved ejection fraction),72,73 insulin resistance and components of the metabolic syndrome,74,75 atherosclerosis,76 endothelial dysfunction,74 acute and chronic renal impairment, and hypertension have been reported, despite an inability to infer direct causality. The specific links with hypertension will be discussed in detail later in this review.
Hyperuricemia and Overall Cardiovascular Risk
Longitudinal data from the National Health and Nutrition Examination Survey (NHANES) demonstrates that total and cardiovascular mortality correlates with SUA levels in both sexes over 10 years of follow up. From the lowest to the highest SUA quartile, there was a significant increase in cardiovascular mortality rates from 3.1 to 7.8 per 1000 person-years.77 This association remained statistically significant even after adjustment for multiple comorbidities. Evidence from the European Study on Cardiovascular Risk Prevention and Management in Daily Practice (EURIKA) study suggests that SUA levels are positively associated with 10-year risk of cardiovascular mortality in patients without evidence of cardiovascular disease but with at least one cardiovascular risk factor (i.e. dyslipidemia, hypertension, smoking, diabetes or obesity).78 Even SUA levels within the upper normal range have been attributed to increased cardiovascular risk.79,80 Studies have looked at the role of allopurinol in reducing cardiovascular risk in those with hyperuricemia. A meta-analysis by Higgins and colleagues identified favorable improvement in markers of vascular health for those treated with XO inhibition.81 Furthermore, allopurinol use has been associated with reduction in left ventricular mass.82 Overall, there is a wealth of data to suggest SUA acts an independent risk factor for cardiovascular disease whilst also acting as a risk factor for other factors that may influence cardiovascular risk such as hypertension, obesity and insulin resistance. Indeed, the Pressioni Arteriose Monitorate E Loro Associazioni (PAMELA) study demonstrated that the effect of SUA on total and cardiovascular mortality outweighed that of age or BP.83
Hyperuricemia and Kidney Disease
Elevated SUA is recognized not just as a biomarker for cardiovascular, but also for renal, morbidity. The relationship with chronic kidney disease (CKD) is complicated by the fact that reduced GFR is, in turn, associated with increased SUA levels secondary to reduced renal clearance. In experimental models, this has been somewhat counter-balanced by a compensatory increase in intestinal excretion of UA in the setting of renal insufficiency.12,84
Hyperuricemia can be implicated in the development of acute kidney injury (AKI), either via crystal-dependent or other independent mechanisms. The former is typically a complication of tumour lysis syndrome whereby tubular obstruction occurs due to crystal deposition within the tubular lamina.85 AKI in this context is associated with significant mortality, and prophylactic rasburicase (recombinant uricase) may help mitigate this risk.86 Studies have demonstrated that in post-operative patients, elevated SUA levels increase the risk of AKI in a mechanism that is crystal-independent.87,88 The proposed mechanisms by which UA may induce both renal and cardiovascular injury will be explored later but include a pro-inflammatory response and endothelial dysfunction.
Large scale trials including the German Chronic Kidney Disease (GCKD) study89 and NHANES90 showed that the age-standardized prevalence of hyperuricemia and gout increases with decline in GFR. In those with established CKD, there is limited evidence to suggest that SUA levels predict progression of disease except in certain groups including those with diabetic nephropathy and IgA nephropathy.91,92 However, SUA levels have been demonstrated to predict the development of CKD in those with normal renal function, as summarized by Johnson and colleagues.93 Furthermore, a large scale prospective trial of 21,475 healthy individuals followed for a median of 7 years demonstrated that SUA levels in the range 7.0–8.9 mg/dL (415–535 µmol/L) were associated with a doubling of the risk of incident CKD (estimated GFR [eGFR] < 60 mL/min/1.73m2), and levels ≥ 9.0 (≥ 540 µmol/L) were associated with a tripling of the risk, after adjusting for baseline eGFR.94 This study supports an independent association between hyperuricemia and the subsequent development of new onset kidney disease.
As will be discussed later in this review, hyperuricemia is a known independent risk factor for hypertension, as well as the metabolic syndrome and glomerular proteinuria.95 These are risk factors which may, in turn, lead to progression of CKD.96 Pediatric data has demonstrated that, in those with an eGFR < 60 mL/min/1.73m2, the prevalence of hyperuricemia is 70%, compared with 34% in a control group with eGFR > 60 mL/min/1.73m2. There was noted to be close correlation between SUA and BP in the group with stage 3–5 CKD, and this group were more likely to be hypertensive.97 Data from the CKiD group has also highlighted that, in children and adolescents, hyperuricemia may act as a risk factor for faster progression of CKD.98 Studies have suggested potential roles for UA-lowering therapies (allopurinol99 and febuxostat100) in slowing GFR decline. However, the use of febuxostat in adults with asymptomatic hyperuricemia and stage 3 CKD failed to demonstrate a protective effect on progressive renal impairment compared with placebo.101
The Link Between Hyperuricemia and Hypertension
Historical Observations
An association between hyperuricemia and hypertension was postulated as far back as 1879 where a relationship between “gouty families” and elevated BP was noted.2 Not long after, Haig made the link between SUA and multiple comorbidities including hypertension.102 However, due to lack of data to suggest a direct association, SUA was widely believed to act as a surrogate marker for comorbidities such as diabetes, kidney disease and obesity. The clinical relevance of SUA was therefore largely disregarded.
In the 1960s, the link between SUA and BP was revisited and slowly gained greater recognition. Studies noted that 25–47% of adults with untreated hypertension were hyperuricemic.103,104 This figure rose to 75% in those with malignant hypertension.103 In 1972, the Israeli Heart Trial demonstrated that for young males (aged 17 to 25 years) within the highest tertile for SUA measurement, there was a two-fold increase in risk of hypertension after 5 years of follow up.105 This association has since been described worldwide across different ethnicities including African American, Asian American and Japanese populations.106–111
Epidemiological Studies Demonstrating Association
Over the past three decades, there has been an increasing appreciation that hyperuricemia is a strong independent predictor of hypertension and may indeed be causative. In 1994, Jossa and colleagues reported an independent association between SUA levels and the development of hypertension in 619 Italian males over 12 years of follow up; where age, BMI, serum cholesterol and serum triglyceride levels were controlled for.112 In more recent years, a host of clinical trials have demonstrated comparable findings.113–117
In 2011, Grayson and colleagues published a systematic review of 55,607 patients in 18 prospective cohort studies.118 They identified that for every 1 mg/dL (60 µmol/L) increase in UA levels, the pooled relative risk for incident hypertension was 1.13 (95% CI 1.06–1.20) after accounting for potential confounders. This risk was more pronounced in females and younger individuals. Another cohort study of 15,143 subjects identified a similar risk for incident prehypertension with a 9% increased risk for those with SUA levels in the fourth quintile of the population, and 17% risk for those with levels in the fifth quintile, compared with those in the bottom quintile.119 Cicero and colleagues selected 619 individuals from the Brisighella Heart Study and found a significant incremental increase in risk of hypertension and metabolic syndrome, along with an increase of carotid intima media thickness (CIMT), in those progressing from the second to fourth quartile of SUA measurement (from 23% to 56.3% for hypertension prevalence).120 These were otherwise healthy individuals (mostly adults) without known cardiovascular disease. In participants of the Generation 3 Framingham cohort with mean age of 40 years, SUA was significantly associated with measures of arterial vascular stiffness, including carotid-femoral and carotid-radial pulse wave velocities (CF-PWV and CR-PWV).121 This data was supported by a more recent study showing SUA to be an independent associating factor for CF-PWV after adjustment for SBP/DBP, BMI and gender.122 A retrospective cohort study looking at 3,584 prehypertensive Japanese adults found that the cumulative risk of progressing from prehypertension to hypertension over a 5 year period was significantly higher in those with hyperuricemia (30.7% versus 24.0%).123 This study, however, only looked at single assessment of office BP and did not assess repeat measurements or 24 hr ambulatory BP. In 2045 adult participants of the PAMELA study, a large Italian longitudinal epidemiological study on blood pressure, SUA was found to independently predict both new onset home and ambulatory hypertension. The risk increased 34% and 29% respectively for an increase in SUA of 1 mg/dL (60 µmol/L).83 As alluded to previously, this SUA increase was associated with a fully adjusted increase in risk of cardiovascular and all-cause mortality (22% and 12% respectively).
With regards to an association with age, hyperuricemia (> 6.8 mg/dL [410 µmol/L]) has been found to predict refractory hypertension in females ≥ 65 years (odds ratio 3.11, 95% CI 1.06–9.1) independent of CKD, although this association was not found in males.124 Lee and colleagues assessed the relationship between SUA, BP and age in a cohort of 45,908 Korean adults who had never been on either UA-lowering or antihypertensive therapies. The association was not significant in those ≥ 60 years of age, but was present below this age threshold, and was noted to be stronger in females.125 In men < 60 years, hyperuricemia increased the relative risk of hypertension by approximately 30%, and in women < 40 years, this risk was elevated 2.6 fold. More recent data from the same group showed a positive association between SUA and incident hypertension (specifically increase in DBP) over a mean follow up of 3.3 years in those < 55 years of age (relative risk 1.74 per 1.0 mg/dL [60 µmol/L] of SUA) compared with those ≥ 55 years.126 Similar findings were reported by Forman and colleagues in 745 males from the Health Professionals’ Follow-up Study where the relative risk of incident hypertension for 1-SD increase in SUA (i.e. 1.25 mg/dL [75 µmol/L] increase) was 1.38 in those younger than 60 years and 0.9 in those ≥ 60 years.127 Epidemiological data thereby suggests an age-dependent weakening of the association between SUA and BP.
Multiple studies support a stronger association between SUA and hypertension in those of younger age. The Moscow Children’s Hypertension Study and the Hungarian Children’s Health Study were the first to describe this association in detail, despite much lower incidence of hyperuricemia in this younger cohort.128,129 These trials found that hyperuricemia strongly correlated with hypertension but they did not control for diagnoses independent of UA levels, such as known renal, cardiovascular and endocrine disease. This therefore impeded the generalization of results. In the former study, hyperuricemia (> 8.0 mg/dL [> 480 µmol/L]) was present in 9.5% of normotensive adolescents, 49% of those with borderline hypertension, and 73% of those with moderate to severe hypertension. The latter study followed 17,634 children over 13 years and found that hyperuricemia, early sexual maturity and elevated heart rate were all risk factors for developing hypertension.
Essential hypertension has been reported in 89% of children and adolescents with UA levels > 5.5 mg/dL (> 330 µmol/L), compared with 30% in those with secondary hypertension and 0% in healthy controls or those diagnosed with white-coat hypertension.79 These differences in SUA could not be accounted for by variability in GFR. The relationship between SUA levels and both SBP and DBP was continuous, and SBP relationship was stronger than that reported in adults (r=0.8053).130 A study of 501 children and adolescents (aged 6–18 years) referred for cardiovascular risk assessment found that for each 1 mg/dL (60 µmol/L) increase in SUA level, the risk of having prehypertension or hypertension increased by at least 50% over normotensive children.131 This study controlled for factors including puberty, gender, BMI and renal function. The National Survey 1999–2006 demonstrated that among 6036 US adolescents (aged 12–17 years), 34% had SUA levels > 5.5 mg/dL (≥ 330 µmol/L) and these individuals had a twofold increased risk for hypertension (defined as SBP/DBP ≥ 95% for age, sex and height) compared with those in a lower SUA range.132 The Bogalusa Heart Study followed 577 child and adolescent patients, divided into white and black ethnicity, over an average of 12 years and found that elevated childhood UA levels were associated with increased BP beginning in childhood, with higher BP persisting into adulthood. This trend was noted in both ethnic groups and in both genders.113 A Korean longitudinal birth cohort looked at a much younger cohort of pre-school children. They followed 449 children and identified that SUA levels at 3 years of age were associated with SUA levels at 5 and 7 years. Those with elevated SUA (above the median) at 3 years of age had a significant increase in SBP and DBP compared with those who had SUA levels below the median. Additionally, elevated SUA levels at both 3 and 5 years of age had a cumulative effect in causing a significant elevation of SBP at 7 years of age.133 SUA and purine metabolites were measured in 246 children from the Dutch KOALA Birth Cohort Study. Higher DBP was related to higher ratios of UA/xanthine and xanthine/hypoxanthine, which can be interpreted as proxy markers of UA production.134 The conclusion from this study is that SUA production may have a causal role in raising DBP, an indicator of increased systemic vascular resistance and a risk factor for cardiovascular disease in younger individuals.135 In 539 adolescents with type 2 diabetes mellitus (T2DM) recruited to the TODAY study, elevated SUA (≥ 6.8 mg/dL [≥ 410 µmol/L]) at baseline was present in a large proportion (25.6%) of the cohort and it was an independent risk factor for incident hypertension and albuminuria over 7 years of follow up, after controlling for baseline eGFR and insulin sensitivty.136
In adults with essential hypertension, SUA has been found to be a strong independent predictor of a nocturnal non-dipping BP profile.137 Non-dipping of BP overnight by the normal 10–20% seen in normotensive individuals, as assessed by ambulatory BP monitoring (ABPM), is known to be associated with greater risk of cardiovascular, cerebrovascular and renal disease.138,139 Additionally, increased SUA levels were independently associated with abnormal circadian BP variability (BPV), also assessed by ABPM, in 300 patients with untreated essential hypertension.140 BPV, which includes concepts such as “white-coat hypertension” or “masked hypertension”, is recognized as a risk factor for end organ damage, as well as cardiovascular and cerebrovascular morbidity and mortality.141–143 The conclusion from these studies is that SUA may therefore predict a more pathogenic BP profile in those with established hypertension. In 6984 adults (mean age 50.3) with treated hypertension, no relationship was found between SUA levels and long-term BP control, although higher UA levels were associated with longitudinal decline in GFR, and with increased cardiovascular risk in women.144
Han and colleagues recently sought to address the temporal relationship between hyperuricemia and hypertension.145 They conducted a longitudinal study of 17,044 Chinese adults and, using a novel statistical approach in the form of a cross-lagged path analysis, surmised that hyperuricemia leads to hyperinsulinism which then contributes to the development of hypertension. Despite this conclusion, the relationship demonstrated was still an association without evidence of causality, and multiple sources of confounding were identified.146 Several studies have demonstrated a possible role for prehypertensive or hypertensive states in inducing hyperuricemia, and have sought to explain the mechanism by which this may occur. Early hypertension is known to reduce renal blood flow via the development of microvascular damage. Evidence suggests that this impairs renal excretion of urate and increases UA synthesis secondary to local tissue ischemia.147–149 This has been supported by evidence that angiotensin II and norepinephrine infusions in normotensive subjects induce hyperuricemia, reduce renal perfusion and raise BP, with reversibility observed upon discontinuation of the infusions.150
Genetic Studies
Mendelian randomization analysis in two Danish cohorts sought to investigate genetic polymorphisms in the SLC2A9 gene (responsible for urate reabsorption via GLUT9), the primary genetic determinant of SUA levels. This study’s results contrasted with observational findings and could not identify a causal association between hyperuricemia secondary to SLC2A9 variants and the risk of developing hypertension or ischemic heart disease.151 Body mass index (BMI) was, however, implicated as a potential confounder and was independently associated with cardiovascular risk.
A meta-analysis of GWAS analysed the results of 28,283 Caucasian individuals and identified SNPs at 8 genetic loci (ABCG2, GCKR, PDZK1, R3HDM2-INHBC region, RREB1, SLC17A1, SLC22A11, SLC2A9) demonstrating statistically significant genome-wide association with SUA levels, and 2 of these loci (ABCG1 and SLC2A9) achieved significant association with gout risk. A “genetic urate score” was created based on the effect of the most significant SNP at each locus. This was strongly associated with SUA levels and gout (odds ratio 12.4 per 1.68 mg/dL [100 µmol/L]). However, there was found to be no association with BP, fasting glucose, eGFR, CKD (defined as eGFR < 60 mL/min/1.73m2) or coronary heart disease (CHD) in this cohort.66 These results were reproduced in the Women’s Genome Health Study (WGHS) cohort.
A smaller study in a Japanese cohort assessed polymorphisms within the XDH gene and found that these may contribute to a phenotype predisposed to hypertension, atherosclerosis and CKD. This effect is postulated to be secondary to interference with circulating nitric oxide reductase which may in turn affect levels of ROS and thereby increase cardiovascular morbidity.152
Genetic data suggest a complex interplay of factors between UA expression and the development of cardiovascular disease. The inability to establish direct causality between hyperuricemia secondary to genetic polymorphisms and the risk of hypertension does, however, need to be interpreted with caution as epigenetic factors may play an important role in gene expression over the course of a lifetime, and alternative biochemical pathways, such as fructose metabolism, affect UA levels independent of the genetic expression of urate transporters. GWAS have characteristically looked at SUA levels and cardiovascular risk in adult populations but have excluded children and adolescents. Results may not be generalizable to the general population given that they typically look at cohorts of shared ethnicity. Furthermore, many of the cardiovascular outcomes in these cohorts are dependent on patient self-reporting, and data on the use of UA-lowering treatment within cohorts are also inconsistent.
Pre-Clinical Studies
Although the extrapolation of results from animal and in vitro models poses multiple challenges, an array of studies have sought to explore the pathogenic etiology for hypertension secondary to hyperuricemia. In 1913, Desgrez demonstrated that the injection of UA into rabbits led to a direct increase in BP.153 More recently, animal studies using rats have established that inducing mild hyperuricemia using oxonic acid, a uricase inhibitor, can induce hypertension after a short period of 3 weeks.154 Concurrent treatment with either allopurinol (a XO inhibitor) or benziodarone (a uricosuric agent) lowered SUA levels and prevented the development of hypertension. This mechanism was independent of urate crystal formation as renal biopsies were normal by light microscopy.
A two-step model, divided into acute and chronic phases, has been recognized as a potential explanation for the development of elevated BP due to hyperuricemia:
- Acute phase - hypertension in these experimental models has been attributed to an initial acute UA-dependent phase with an increase in juxtaglomerular renin release and a decrease in macula densa NO synthase production within the kidney. UA has been demonstrated to impair phosphorylation of endothelium-derived nitric oxide (eNO) via activation of UA transporters.155 The resultant increase in circulating ROS, and increased oxidative stress in the macula densa, leads to an ischemic-type endothelial injury identified on renal biopsy. This injury is independent of crystal formation.154,156,157 This phase results in renal vasoconstriction that is independent of salt intake and reversible with the use of UA-lowering medications and renin-angiotensin-aldosterone system (RAAS) blockade. Notably, losartan has a role as both an angiotensin II blocker and a uricosuric agent.158 Benzbromarone, irbesartan and losartan have all been found to diminish the effect of UA on eNO suppression.155
- Chronic phase - after prolonged hyperuricemia, a chronic phase of injury occurs that is associated with inflammatory microvascular changes predominantly affecting the afferent arterioles. There is uptake of UA into vascular smooth muscle cells (VSMC) via URAT1, resulting in activation of proinflammatory mediators such as monocyte chemoattractant protein-1 (MCP-1) and increased cyclooxygenase-2 (COX-2) mRNA expression.159 The resultant VSMC injury leads to progressive afferent arteriolopathy and interstitial inflammation, believed to be independent of UA levels. It mimics the progression of salt-sensitive hypertension that is seen in humans and is unresponsive to UA-lowering treatment.160 Murine knockout models have demonstrated that loss of uricase and SLC2A9 can contribute to hypertension and metabolic syndrome, as well as premature mortality.161,162 However, as humans lack the uricase enzyme, the extrapolation of these results should be interpreted with caution.
Another animal model using knockout mice with deletion of hepatocyte-specific GLUT9 found that induced hyperuricemia (with the use of inosine) was not linked with hypertension despite the development of mild renal impairment.163 In hepatocytes, GLUT9 acts as a urate transporter to provide urate to the hepatocyte enzyme uricase. This study suggested a lack of direct causality between urate and BP in mice. Again, the lack of uricase in humans leads us to query the direct applicability of such findings.
Rat models have linked high fructose-containing diets with insulin resistance, hypertension, hypertriglyceridemia, and fatty liver disease,164–166 and similar findings have been demonstrated in humans.167 In rats, fructose is noted to cause an elevation in BP at the time of consumption but this effect is not sustained and this may be due to the effect of the uricase enzyme which helps to metabolize SUA.168 Fructose’s role in inducing metabolic syndrome and hypertension has been addressed by Nakagawa and colleagues.169 They found that allopurinol and benzbromarone were both effective in preventing or reversing features of metabolic syndrome in rats within an 8 week period, and allopurinol successfully prevented fructose-induced systolic hypertension, as well as hyperinsulism, hypertriglyceridemia and weight gain.
Therapeutic Management of Hyperuricemia and Effects on BP Control
In both younger and older age groups, UA-lowering therapies have been used to test the association between SUA levels and BP. These agents include XO inhibitors (e.g. allupurinol), non–purine analogue inhibitors of XO (e.g. febuxostat) and uricosuric agents (e.g. benzbromarone, benziodarone, ethebencid, probenecid, sulfinpyrazone, zoxazolamine). Rasburicase is licensed for use in the treatment of acute hyperuricemia such as that induced by chemotherapy. A list of these medications is summarized in Table 2.
 |
Table 2 Uric Acid Lowering Therapies |
A number of trials have demonstrated beneficial effects of allopurinol, possibly independent of its UA-lowering properties. Allopurinol has been associated with improvement in vascular endothelial function, as measured by forearm venous occlusion plethysmography, in elderly patients with chronic heart failure,170 in heavy smokers171 and in type 2 diabetic patients.172 These effects have been attributed to reduction in vascular oxidative stress with observed improvement in forearm blood flow. Probenecid use did not induce similar effects, thus raising the question as to whether urate itself actually plays a direct role in endothelial dysfunction.170 It is proposed that the beneficial effects of allopurinol may be related to a reduction in ROS production by the blocking of XO, and that urate itself may not be the direct causative agent for hypertension. Indeed, in healthy controls, UA infusion was not associated with acute evidence of endothelial dysfunction173 and has also been found to improve endothelial function in patients with type 1 diabetes and smokers, suggesting that elevated UA concentrations in vivo may be protective for those with pre-established cardiovascular risk.174 A significant short-term lowering of SUA with uricase in patients with T2DM was not found to have a direct vascular effect thus suggesting that there is no causal link between elevated SUA concentrations and endothelial dysfunction.175 However, uricase depletes UA via conversion to allantoin and, in doing so, increases the production of ROS which would be expected to have a pro-inflammatory effect, and would diminish extracellular UA levels thus limiting the antioxidant effects of UA.
With regards to BP control, allopurinol has been found to lower brachial and central BP and reduce the risk of progressive CIMT in patients without hyperuricemia following ischemic stroke and transient ischemic attack (TIA).176 However, there was no observed difference in markers of endothelial dysfunction, such as von Willebrand factor (vWF) and soluble intercellular adhesion molecule 1 (s-ICAM-1), in those treated with allopurinol versus placebo. In those with modestly elevated SUA concentrations (6.0–6.2 mg/dL [360–370 µmol/L]), allopurinol has been demonstrated to lower SBP and DBP in middle-aged overweight and prehypertensive adults over a period of 8 weeks.177 Kanbay and colleagues demonstrated a reduction in 24 hr systolic ambulatory BP, along with an increase in eGFR, in patients with asymptomatic hyperuricemia treated with allopurinol for 4 months.178 These patients did, however, experience significant reduction in SUA levels. Recent RCTs have been unable to find associations between allopurinol use and change in endothelial-dependent vasodilation,179 kidney-specific or systemic RAAS activity (measured as change in renal blood flow response to captopril, and plasma renin and angiotensin II activity),180 and an improvement in BP with adjuvant use of allopurinol in those already on thiazide diuretics as first-line treatment for hypertension.181 A meta-analysis looking at 738 individuals, most of whom were adults, found that allopurinol was responsible for a small reduction in both SBP (3.3 mmHg) and DBP (1.3 mmHg) compared with controls.182 Evidence suggests that older hypertensive adults (> 65 years) require higher doses of allopurinol to achieve a greater reduction in BP,183 and those treated with higher doses also have significantly lower risk of cardiac and cerebrovascular events.184 However, recent studies looking at cohorts with mean age of 64185 and 67186 years respectively have demonstrated contradictory findings and have not found significant changes in BP control with the use of allopurinol, febuxostat or benzbromarone. In the LIFE study, losartan’s use as an antihypertensive agent was associated with lower risk of cardiovascular morbidity and mortality compared with atenolol, and this may be secondary to its adjunctive role as a uricosuric agent.187 The use of pegloticase, a recombinant uricase conjugated to polyethylene glycol, has been explored for the treatment of gout refractory to standard treatment or where standard treatment is contraindicated. Significant reductions in SBP and DBP were found in the group receiving pegloticase on a biweekly basis who maintained SUA levels < 6.0 mg/dL (< 360 µmol/L) throughout the study period and these effects were independent of alterations in eGFR.188 Of note, pegloticase does not inhibit XO and this is therefore contradictory evidence to suggest that SUA may have a direct effect on BP control.
On the basis of the aforementioned experimental studies, it is postulated that subjects with hyperuricemia who have not had hypertension long enough to develop secondary VSMC arteriolar injury could benefit most from UA-lowering therapy. Feig and colleagues performed a crossover trial in 30 adolescents (aged 11–17 years) with newly diagnosed hypertension and SUA levels ≥ 6.0 mg/dL (≥ 360 µmol/L).189 Allopurinol was compared with placebo for 4 weeks in each arm of the trial and they found that 20 of the 30 adolescents achieved normal BP by casual and ambulatory criteria whilst taking allopurinol compared with 1 participant taking placebo. Soletsky and colleagues looked at 4-week treatment with either allopurinol or probenecid compared with placebo and found a BP reduction in prehypertensive obese adolescents (aged 11 to 17 years). There was a significant reduction in both SBP and DBP, by 10.2 mmHg and 9.0 mmHg respectively, compared with placebo.190 In a randomized open-label trial of 42 adolescents with essential hypertension aged 12–19 years, allopurinol used in combination with enalapril was found to be more effective than allopurinol alone in reducing BP and reducing SUA levels. All patients had baseline SUA levels ≥ 5.5 mg/dL (≥ 330 µmol/L) and 37% of the cohort were overweight or obese.191
A Cochrane review in 2017192 looking to determine whether the use of UA-lowering therapies lowers BP in hypertensive patients identified only 3 randomized controlled trials (RCTs) that met inclusion criteria, including the aforementioned trials by Feig and Soletsky.189,190 The third multicenter trial included 109 adults (aged ≥ 18 years) with primary hypertension.193 The use of febuxostat, compared with placebo, was evaluated over 6 weeks. No significant difference between the febuxostat and placebo groups was found in ambulatory daytime or nighttime BP, or clinic SBP or DBP, from baseline to week 6. There was however a reduction in SBP in the subgroup analysis of hypertensive and hyperuricemic patients with normal renal function. The conclusion of this systematic review was that there is low quality of evidence to establish that UA-lowering therapies reduce BP. When assessing BP via 24 hr ABPM, there was no significant reduction in either SBP (−6.2 mmHg, 95% CI −12.8 to 0.5) or DBP (−3.9 mmHg, 95% CI −9.2 to 1.4). This was despite high quality evidence showing that these therapies do effectively reduce SUA levels (by 3.1 mg/dL [185 µmol/L] [95% CI 2.4 to 3.8]).
Data from both NHANES 1999–2004194 and 2003–2006195 support an epidemiological association between fructose intake and hypertension in the US population. Fructose is associated with acute elevation of BP in healthy young adults and this effect was not observed with glucose intake.196 Reduction of SBP and DBP has been demonstrated prospectively in those who reduced their consumption of sugar-sweetened beverages.197 In adult males receiving 200 g of fructose per day for 2 weeks, there was an increase in fasting SUA levels with elevation of both SBP and DBP obtained via 24 hr ABPM recordings. Allopurinol was found to lower SUA and prevent the increase in daytime SBP and DBP that was seen in the control group.167
Whether the management of hyperuricemia may represent a new frontier in the optimization of BP control has still not been fully expounded and large-scale RCTs are lacking. The use of UA lowering therapies, particularly in younger age groups, requires further evaluation. At present, the side effect profile of UA-lowering agents limits their widespread use. In particular, allopurinol is associated with dose-related adverse effects that range from a relatively common rash to rarer effects such as aplastic anemia and severe hypersensitivity reactions (reported in 0.38% of patients).198 Renal insufficiency may predispose to the build-up of oxypurinol, the major metabolite of allopurinol, which increases the risk of potentially life-threatening reactions.199 Concurrent use of ACE inhibitors and allopurinol may also raise the risk of potentially lethal skin reactions such as toxic epidermal necrolysis.200 Probenecid can affect the renal excretion of other drugs (such as captopril, NSAIDs and penicillins) and its uricosuric effect may induce nephrolithiasis.201 Furthermore, there is no current evidence to suggest that any of the UA-lowering therapies alone are more effective at lowering BP than traditional antihypertensive agents.
Conclusion
SUA is an independent risk factor for incident hypertension but there still remains a lack of concrete evidence to support the therapeutic lowering of UA in order to treat or prevent hypertension, or to reduce the risk of cardiovascular, cerebrovascular or renal disease. It is postulated that XO inhibitors would be of greater benefit than uricosuric agents in reducing this risk as they both lower SUA and block the production of pro-inflammatory ROS. In the management of hyperuricemia, there is also a role for the development of novel therapies that could target the multiple urate transporters that have been identified to date. At present, we lack a true understanding of the temporality of the relationship between SUA and BP, and the specific effect of SUA on endothelial function versus that of ROS generated by purine metabolism. Genetic studies suggest factors, notably BMI, may act as confounders and that causality cannot be inferred.
Large scale, prospective, double blind RCTs are required to look at specific UA-lowering therapies to assess if they can be used to treat hypertension, or if they can alter the progression from prehypertensive to hypertensive states. There is considerable evidence to suggest that the relationship between SUA and hypertension is lost with increasing age and with duration of hypertension. The role of UA-lowering therapies in different age groups needs to be evaluated fully as those of younger age may benefit the most if the two-step pathogenic hypothesis is to be believed.
Disclosure
The authors report no other conflicts of interest in this work.
References
1. Stevens G. Global health risks: mortality and burden of disease attributable to selected major risks. Bull World Health Organ. 2009.
2. Mahomed FA. On chronic Bright’s disease, and its essential symptoms. Lancet. 1879;1:389–404.
3. Griebsch A, Zöllner N. Effect of ribomononucleotides given orally on uric acid production in man. Adv Exp Med Biol. 1974;41:443–449. doi:10.1007/978-1-4757-1433-3_9
4. Roch-Ramel F, Guisan B. Renal transport of urate in humans. News Physiol Sci. 1999;Apr(14):80–84.
5. Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19(6):358–371. doi:10.1053/j.ackd.2012.07.009
6. Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Annu Rev Physiol. 2015;77:323–345. doi:10.1146/annurev-physiol-021113-170343
7. Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature. 2002;417(6887):447–452. doi:10.1038/nature742
8. Hamada T, Ichida K, Hosoyamada M, et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens. 2008;21(10):1157–1162. doi:10.1038/ajh.2008.245
9. Vitart V, Rudan I, Hayward C, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40(4):437–442. doi:10.1038/ng.106
10. Kolz M, Johnson T, Sanna S, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6):e1000504. doi:10.1371/journal.pgen.1000504
11. Ichida K, Matsuo H, Takada T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3(3):764.
12. Yano H, Tamura Y, Kobayashi K, Tanemoto M, Uchida S. Uric acid transporter ABCG2 is increased in the intestine of the 5/6 nephrectomy rat model of chronic kidney disease. Clin Exp Nephrol. 2014;18(1):50–55. doi:10.1007/s10157-013-0806-8
13. Iharada M, Miyaji T, Fujimoto T, et al. Type 1 sodium-dependent phosphate transporter (SLC17A1 protein) is a Cl(-)-dependent urate exporter. J Biol Chem. 2010;285(34):26107–26113. doi:10.1074/jbc.M110.122721
14. Kasper D, Hauser S, Longo D, Jameson JL, Loscalzo J. Harrison’s Principles of Internal Medicine. McGraw-Hill Education. 19th Edition 2015.
15. Sanchez Bayle M, Vazquez Martul M, Ecija Peiro JL, Garcia Vao C, Ramo Mancheño C. Renal handling of uric acid in normal children by means of the pyrazinamide and sulfinpyrazone tests. Int J Pediatr Nephrol. 1987;8(1):5–8.
16. Martillo MA, Nazzal L, Crittenden DB. The crystallization of monosodium urate. Curr Rheumatol Rep. 2014;16(2):400. doi:10.1007/s11926-013-0400-9
17. Sakhaee K, Adams-Huet B, Moe OW, Pak CYC. Pathophysiologic basis for normouricosuric uric acid nephrolithiasis. Kidney Int. 2002;62(3):971–979. doi:10.1046/j.1523-1755.2002.00508.x
18. Wu XW, Lee CC, Muzny DM, Caskey CT. Urate oxidase: primary structure and evolutionary implications. Proc Natl Acad Sci U S A. 1989;86(23):9412–9416. doi:10.1073/pnas.86.23.9412
19. Simic MG, Jovanovic SV. Antioxidation mechanisms of uric acid. J Am Chem Soc. 1989;111:5778–5782. doi:10.1021/ja00197a042
20. Frei B, Stocker R, Ames BN. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad Sci U S A. 1988;85(24):9748–9752. doi:10.1073/pnas.85.24.9748
21. Hooper DC, Spitsin S, Kean RB, et al. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci. 2002;95(2):675–680. doi:10.1073/pnas.95.2.675
22. Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci. 2006;78(11):6858–6862. doi:10.1073/pnas.78.11.6858
23. Spitsin S, Koprowski H. Role of uric acid in Alzheimer’s disease. J Alzheimer’s Dis. 2010;19(4):1337–1338. doi:10.3233/JAD-2010-1336
24. Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson’s disease. Am J Epidemiol. 2007;166(5):561–567. doi:10.1093/aje/kwm127
25. Rentzos M, Nikolaou C, Anagnostouli M, et al. Serum uric acid and multiple sclerosis. Clin Neurol Neurosurg. 2006;108(6):527–531. doi:10.1016/j.clineuro.2005.08.004
26. Tsukada K, Hasegawa T, Tsutsumi S, et al. Effect of uric acid on liver injury during hemorrhagic shock. Surgery. 2000;127(4):439–446. doi:10.1067/msy.2000.104486
27. Nieto FJ, Iribarren C, Gross MD, Comstock GW, Cutler RG. Uric acid and serum antioxidant capacity: a reaction to atherosclerosis? Atherosclerosis. 2000;148(1):131–139. doi:10.1016/s0021-9150(99)00214-2
28. Horsfall LJ, Nazareth I, Petersen I. Serum uric acid and the risk of respiratory disease: a population-based cohort study. Thorax. 2014;69(11):1021–1026. doi:10.1136/thoraxjnl-2014-205271
29. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425(6957):516–521. doi:10.1038/nature01991
30. Watanabe S, Kang DH, Feng L, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension. 2002;40(3):355–360. doi:10.1161/01.HYP.0000028589.66335.AA
31. Johnson RJ, Andrews P. Fructose, uricase, and the Back-to-Africa hypothesis. Evol Anthropol. 2010;19(6):250–257. doi:10.1002/evan.20266
32. Cappuccio FP, Strazzullo P, Farinaro E, Trevisan M. Uric acid metabolism and tubular sodium handling: results from a population-based study. J Am Med Assoc. 1993;270(3):354–359. doi:10.1001/jama.1993.03510030078038
33. Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Physiol. 2007;293(2):C584–C598. doi:10.1152/ajpcell.00600.2006
34. Kang D-H, Park S-K, Lee I-K, Johnson RJ. Uric acid–induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005;16(12):3553–3562. doi:10.1681/ASN.2005050572
35. Xiao J, Zhang XL, Fu C, et al. Soluble uric acid increases NALP3 inflammasome and interleukin-1β expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int J Mol Med. 2015;35(5):1347–1354. doi:10.3892/ijmm.2015.2148
36. Rao GN, Corson MA, Berk BC. Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression. J Biol Chem. 1991;266:8604–8608.
37. Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino H, Tuck ML. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J Hypertens. 2008;26:269–275. doi:10.1097/HJH.0b013e3282f240bf
38. Zharikov S, Krotova K, Hu H, et al. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Physiol. 2008;295(5):C1183–C1190. doi:10.1152/ajpcell.00075.2008
39. Sánchez-Lozada LG, Lanaspa MA, Cristóbal-García M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron - Exp Nephrol. 2012;121(3–4):e71–e87. doi:10.1159/000345509
40. Tanaka T, Milaneschi Y, Zhang Y, Becker KG, Zukley L, Ferrucci L. A double blind placebo controlled randomized trial of the effect of acute uric acid changes on inflammatory markers in humans: a pilot study. PLoS One. 2017;12(8):e0181100. doi:10.1371/journal.pone.0181100
41. Waring WS, Webb DJ, Maxwell SR. Effect of local hyperuricemia on endothelial function in the human forearm vascular bed. Br J Clin Pharmacol. 2000;49:511.
42. Iso T, Kurabayashi M. Extremely low levels of serum uric acid are associated with endothelial dysfunction in humans. Circ J. 2015;79(5):978–980. doi:10.1253/circj.CJ-15-0232
43. De Leeuw PW, Thijs L, Birkenhäger WH, et al. Prognostic significance of renal function in elderly patients with isolated systolic hypertension: results from the Syst-Eur trial. J Am Soc Nephrol. 2002;13(9):2213–2222. doi:10.1097/01.ASN.0000027871.86296.92
44. Verdecchia P, Schillaci G, Reboldi GP, Santeusanio F, Porcellati C, Brunetti P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension: the PIUMA study. Hypertension. 2000;36(6):1072–1078. doi:10.1161/01.HYP.36.6.1072
45. Sautin YY, Johnson RJ. Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids. 2008;27(6):608–619. doi:10.1080/15257770802138558
46. Cameron JS, Moro F, Simmonds HA. Gout, uric acid and purine metabolism in paediatric nephrology. Pediatr Nephrol. 1993;7(1):105–118. doi:10.1007/BF00861588
47. Yu KH, See LC, Huang YC, Yang CH, Sun JH. Dietary factors associated with hyperuricemia in adults. Semin Arthritis Rheum. 2008;37(4):243–250. doi:10.1016/j.semarthrit.2007.04.007
48. Choi HK, Liu S, Curhan G. Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the third National Health and Nutrition Examination Survey. Arthritis Rheum. 2005;52(1):283–289. doi:10.1002/(ISSN)1529-0131
49. Choi HK, Curhan G. Coffee, tea, and caffeine consumption and serum uric acid level: the third National Health and Nutrition Examination Survey. Arthritis Care Res. 2007;57(5):816–821. doi:10.1002/(ISSN)1529-0131
50. Pham NM, Yoshida D, Morita M, et al. The relation of coffee consumption to serum uric acid in Japanese men and women aged 49–76 years. J Nutr Metab. 2010;2010:930757. doi:10.1155/2010/930757
51. Choi HK, Gao X, Curhan G. Vitamin C intake and the risk of gout in men: a prospective study. Arch Intern Med. 2009;169(5):502–507. doi:10.1001/archinternmed.2008.606
52. Huang HY, Appel LJ, Choi MJ, et al. The effects of vitamin C supplementation on serum concentrations of uric acid: results of a randomized controlled trial. Arthritis Rheum. 2005;52(6):1843–1847. doi:10.1002/art.21105
53. Rho YH, Zhu Y, Choi HK. The epidemiology of uric acid and fructose. Semin Nephrol. 2011;31(5):410–419. doi:10.1016/j.semnephrol.2011.08.004
54. Chen J, Muntner P, Hamm LL, et al. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med. 2004;140(3):167–174. doi:10.7326/0003-4819-140-3-200402030-00007
55. Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD. Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2011;6(10):2364–2373. doi:10.2215/CJN.02180311
56. Nakagawa T, Lanaspa MA, Johnson RJ. The effects of fruit consumption in patients with hyperuricaemia or gout. Rheumatology. 2019;58(7):1133–1141. doi:10.1093/rheumatology/kez128
57. Manolescu AR, Augustin R, Moley K, Cheeseman C. A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol Membr Biol. 2007;24(5–6):455–563. doi:10.1080/09687680701298143
58. Ebert K, Ludwig M, Geillinger KE, et al. Reassessment of GLUT7 and GLUT9 as putative fructose and glucose transporters. J Membr Biol. 2017;250(2):171–182.
59. Kang DH, Finch J, Nakagawa T, et al. Uric acid, endothelial dysfunction and pre-eclampsia: searching for a pathogenetic link. J Hypertens. 2004;22:229–235. doi:10.1097/00004872-200402000-00001
60. Sagen N, Haram K, Nilsen ST. Serum urate as a predictor of fetal outcome in severe pre‐eclampsia. Acta Obstet Gynecol Scand. 1984;63:71–75. doi:10.3109/00016348409156277
61. Feig DI, Nakagawa T, Karumanchi SA, et al. Hypothesis: uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int. 2004;66(1):281–287. doi:10.1111/j.1523-1755.2004.00729.x
62. Roberts JM, Bodnar LM, Lain KY, et al. Uric acid is as important as proteinuria in identifying fetal risk in women with gestational hypertension. Hypertension. 2005;46:1263–1269. doi:10.1161/01.HYP.0000188703.27002.14
63. Kalousdian S, Fabsitz R, Havlik R, Christian J, Rosenman R, Rao DC. Heritability of clinical chemistries in an older twin cohort: the NHLBI Twin Study. Genet Epidemiol. 1987;4:1–11. doi:10.1002/gepi.v4:1
64. Rice T, Vogler GP, Perry TS, Laskarzewski PM, Province MA, Rao DC. Heterogeneity in the familial aggregation of fasting serum uric acid level in five North American populations: the Lipid Research Clinics Family Study. Am J Med Genet. 1990;36:219–225. doi:10.1002/(ISSN)1096-8628
65. Wilk JB, Djousse L, Borecki I, et al. Segregation analysis of serum uric acid in the NHLBI Family Heart Study. Hum Genet. 2000;106:355–359. doi:10.1007/s004390051050
66. Yang Q, Köttgen A, Dehghan A, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet. 2010;3(6):523–530.
67. Köttgen A, Albrecht E, Teumer A, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45(2):145–154. doi:10.1038/ng.2500
68. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141. doi:10.1002/art.30520
69. Trifirò G, Morabito P, Cavagna L, et al. Epidemiology of gout and hyperuricaemia in Italy during the years 2005–2009: a nationwide population-based study. Ann Rheum Dis. 2013;72(5):694–700. doi:10.1136/annrheumdis-2011-201254
70. Chou CT, Lai JS. The epidemiology of hyperuricaemia and gout in Taiwan aborigines. Br J Rheumatol. 1998;37(3):258–262. doi:10.1093/rheumatology/37.3.258
71. Campion EW, Glynn RJ, Delabry LO. Asymptomatic hyperuricemia. Risks and consequences in the normative aging study. Am J Med. 1987;82(3):421. doi:10.1016/0002-9343(87)90441-4
72. Ndrepepa G. Uric acid and cardiovascular disease. Clin Chim Acta. 2018;484:150–163. doi:10.1016/j.cca.2018.05.046
73. Gu J, Fan YQ, Zhang HL, Zhang JF, Wang CQ. Serum uric acid is associated with incidence of heart failure with preserved ejection fraction and cardiovascular events in patients with arterial hypertension. J Clin Hypertens. 2018;20(3):560–567. doi:10.1111/jch.13210
74. Johnson RJ, Bakris GL, Borghi C, et al. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: report of a scientific workshop organized by the National Kidney Foundation. Am J Kidney Dis. 2018;7(6):851–865. doi:10.1053/j.ajkd.2017.12.009
75. Yoo TW, Sung KC, Shin HS, et al. Relationship between serum uric acid concentration and insulin resistance and metabolic syndrome. Circ J. 2005;69(8):928–933. doi:10.1253/circj.69.928
76. Borghi C, Rosei EA, Bardin T, et al. Serum uric acid and the risk of cardiovascular and renal disease. J Hypertens. 2015;33(9):1729–1741.
77. Stack AG, Hanley A, Casserly LF, et al. Independent and conjoint associations of gout and hyperuricaemia with total and cardiovascular mortality. QJM. 2013;106(7):647–658. doi:10.1093/qjmed/hct083
78. Borghi C, Rodriguez-Artalejo F, De Backer G, et al. Serum uric acid levels are associated with cardiovascular risk score: a post hoc analysis of the EURIKA study. Int J Cardiol. 2018;253:167–173. doi:10.1016/j.ijcard.2017.10.045
79. Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension. 2003;42(3):247–252. doi:10.1161/01.HYP.0000085858.66548.59
80. Niskanen LK, Laaksonen DE, Nyyssönen K, et al. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study. Arch Intern Med. 2004;164(14):1546–1551. doi:10.1001/archinte.164.14.1546
81. Higgins P, Dawson J, Lees KR, Mcarthur K, Quinn TJ, Walters MR. Xanthine oxidase inhibition for the treatment of cardiovascular disease: a systematic review and meta-analysis. Cardiovasc Ther. 2012;30(4):217–226. doi:10.1111/cdr.2012.30.issue-4
82. Rekhraj S, Gandy SJ, Szwejkowski BR, et al. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. 2013;5(61):926–932.
83. Bombelli M, Ronchi I, Volpe M, et al. Prognostic value of serum uric acid: new-onset in and out-of-office hypertension and long-term mortality. J Hypertens. 2014;32(6):1237–1244. doi:10.1097/HJH.0000000000000161
84. Vaziri ND, Freel RW, Hatch M. Effect of chronic experimental renal insufficiency on urate metabolism. J Am Soc Nephrol. 1995;6(4):1313–1317.
85. Shimada M, Johnson RJ, May WS, et al. A novel role for uric acid in acute kidney injury associated with tumour lysis syndrome. Nephrol Dial Transplant. 2009;24(10):2960–2964. doi:10.1093/ndt/gfp330
86. Coiffier B, Mounier N, Bologna S, et al. Efficacy and safety of rasburicase (recombinant urate oxidase) for the prevention and treatment of hyperuricemia during induction chemotherapy of aggressive non-Hodgkin’s lymphoma: results of the GRAAL1 (Groupe d’Etude des Lymphomes de l’Adulte Trial on R. J Clin Oncol. 2003;21(23):4402–4406. doi:10.1200/JCO.2003.04.115
87. Ejaz NS, Shields AR, Alloway RR, et al. Randomized controlled pilot study of B cell-targeted induction therapy in HLA sensitized kidney transplant recipients. Am J Transplant. 2013;13(12):3142–3154.
88. Ejaz AA, Mu W, Kang DH, et al. Could uric acid have a role in acute renal failure? Clin J Am Soc Nephrol. 2007;2:16–21. doi:10.2215/CJN.00350106
89. Jing J, Kielstein JT, Schultheiss UT, et al. Prevalence and correlates of gout in a large cohort of patients with chronic kidney disease: the German Chronic Kidney Disease (GCKD) study. Nephrol Dial Transplant. 2015;30(4):613–621. doi:10.1093/ndt/gfu352
90. Juraschek SP, Kovell LC, Miller ER, Gelber AC. Association of kidney disease with prevalent gout in the United States in 1988–1994 and 2007–2010. Semin Arthritis Rheum. 2013;42(6):551–561. doi:10.1016/j.semarthrit.2012.09.009
91. Altemtam N, Russell J, El Nahas M. A study of the natural history of diabetic kidney disease (DKD). Nephrol Dial Transplant. 2012;27(5):1847–1854. doi:10.1093/ndt/gfr561
92. Shi Y, Chen W, Jalal D, et al. Clinical outcome of hyperuricemia in IgA nephropathy: a retrospective cohort study and randomized controlled trial. Kidney Blood Press Res. 2012;35(3):153–160.
93. Johnson RJ, Nakagawa T, Jalal D, Sánchez-Lozada LG, Kang DH, Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transplant. 2013;28(9):2221–2228. doi:10.1093/ndt/gft029
94. Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19(12):2407–2413. doi:10.1681/ASN.2008010080
95. Jalal DI, Rivard CJ, Johnson RJ, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transplant. 2010;25(6):1865–1869.
96. Nashar K, Fried LF. Hyperuricemia and the progression of chronic kidney disease: is uric acid a marker or an independent risk factor? Adv Chronic Kidney Dis. 2012;19(6):386–391. doi:10.1053/j.ackd.2012.05.004
97. Noone DG, Marks SD. Hyperuricemia is associated with hypertension, obesity, and albuminuria in children with chronic kidney disease. J Pediatr. 2013;162(1):128–132. doi:10.1016/j.jpeds.2012.06.008
98. Rodenbach KE, Schneider MF, Furth SL, et al. Hyperuricemia and progression of CKD in children and adolescents: the Chronic Kidney Disease in Children (CKiD) cohort study. Am J Kidney Dis. 2015;66(6):984–992. doi:10.1053/j.ajkd.2015.06.015
99. Sezer S, Karakan S, Atesagaoglu B, Acar F. Allopurinol reduces cardiovascular risks and improves renal function in pre-dialysis chronic kidney disease patients with hyperuricemia. Saudi J Kidney Dis Transplant. 2014;25(2):316–320. doi:10.4103/1319-2442.128520
100. Sircar D, Chatterjee S, Waikhom R, et al. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: a 6-month, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2015;66(6):945–950. doi:10.1053/j.ajkd.2015.05.017
101. Kimura K, Hosoya T, Uchida S, et al. Febuxostat therapy for patients with stage 3 CKD and asymptomatic hyperuricemia: a randomized trial. Am J Kidney Dis. 2018;72(6):798–810. doi:10.1053/j.ajkd.2018.06.028
102. Haig A. The connecting link between the high-tension pulse and albuminuria. Br Med J. 1890;1:65–68. doi:10.1136/bmj.1.1515.65
103. Cannon PJ, Stason WB, Demartini FE, Sommers SC, Laragh JH. Hyperuricemia in primary and renal hypertension. N Engl J Med. 1966;275(9):457–464. doi:10.1056/NEJM196609012750902
104. Kinsey D, Walther R, Sise H, Whitelaw G, Smithwick R. Incidence of hyperuricemia in 400 hypertensive subjects. Circulation. 1961;24:972–973.
105. Kahn HA, Medalie JH, Neufeld HN, Riss E, Goldbourt U. The incidence of hypertension and associated factors: the Israel ischemic heart disease study. Am Heart J. 1972;84(2):171–182. doi:10.1016/0002-8703(72)90331-6
106. Dyer AR, Liu K, Walsh M, Kiefe C, Jacobs DR, Bild DE. Ten-year incidence of elevated blood pressure and its predictors: the CARDIA Study. J Hum Hypertens. 1999;13(1):13–21.
107. Imazu M, Yamamoto H, Toyofuku M, et al. Hyperinsulinemia for the development of hypertension: data from the Hawaii-Los Angeles-Hiroshima Study. Hypertens Res. 2002;24(5):531–536. doi:10.1291/hypres.24.531
108. Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42(4):474–480. doi:10.1161/01.HYP.0000091371.53502.D3
109. Nagahama K, Inoue T, Iseki K, et al. Hyperuricemia as a predictor of hypertension in a screened cohort in Okinawa, Japan. Hypertens Res. 2004;27(11):835–841. doi:10.1291/hypres.27.835
110. Nakanishi N, Okamoto M, Yoshida H, Matsuo Y, Suzuki K, Tatara K. Serum uric acid and risk for development of hypertension and impaired fasting glucose or type II diabetes in Japanese male office workers. Eur J Epidemiol. 2003;18(6):523–530. doi:10.1023/A:1024600905574
111. Taniguchi Y, Hayashi T, Tsumura K, Endo G, Fujii S, Okada K. Serum uric acid and the risk for hypertension and type 2 diabetes in Japanese men: the Osaka health survey. J Hypertens. 2001;19(7):1209–1215. doi:10.1097/00004872-200107000-00005
112. Jossa F, Farinaro E, Panico S, et al. Serum uric acid and hypertension: the Olivetti heart study. J Hum Hypertens. 1994;8(9):677–681.
113. Alper AB, Chen W, Yau L, Srinivasan SR, Berenson GS, Hamm LL. Childhood uric acid predicts adult blood pressure: the Bogalusa Heart Study. Hypertension. 2005;45(1):34–38. doi:10.1161/01.HYP.0000150783.79172.bb
114. Hunt SC, Stephenson SH, Hopkins PN, Williams RR. Predictors of an increased risk of future hypertension in Utah. A screening analysis. Hypertension. 1991;17(6 Pt 2):969–976. doi:10.1161/01.HYP.17.6.969
115. Perlstein TS, Gumieniak O, Williams GH, et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031–1036. doi:10.1161/01.HYP.0000248752.08807.4c
116. Sundström J, Sullivan L, D’Agostino RB, Levy D, Kannel WB, Vasan RS. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 2005;45(1):28–33. doi:10.1161/01.HYP.0000150784.92944.9a
117. Ouppatham S, Bancha S, Choovichian P. The relationship of hyperuricemia and blood pressure in the Thai army population. J Postgr Med. 2008;54(4):259–262. doi:10.4103/0022-3859.43509
118. Grayson PC, Young Kim S, Lavalley M, Choi HK. Hyperuricemia and incident hypertension: a systematic review and meta-analysis. Arthritis Care Res. 2011;63(1):102–110. doi:10.1002/acr.20344
119. Sun S, Yang H, Liu L, et al. Serum uric acid is an independent predictor for developing prehypertension: a population-based prospective cohort study. J Hum Hypertens. 2017;31(2):116–120. doi:10.1038/jhh.2016.48
120. Cicero AFG, Salvi P, D’Addato S, Rosticci M, Borghi C. Association between serum uric acid, hypertension, vascular stiffness and subclinical atherosclerosis: data from the Brisighella Heart Study. J Hypertens. 2014;32(1):57–64. doi:10.1097/HJH.0b013e328365b916
121. Mehta T, Nuccio E, McFann K, Madero M, Sarnak MJ, Jalal D. Association of uric acid with vascular stiffness in the Framingham Heart Study. Am J Hypertens. 2015;28(7):877–883. doi:10.1093/ajh/hpu253
122. Liu J, Wang K, Liu H, et al. Relationship between carotid-femoral pulse wave velocity and uric acid in subjects with hypertension and hyperuricemia. Endocr J. 2019;66(7):629–636. doi:10.1507/endocrj.EJ18-0570
123. Kuwabara M, Hisatome I, Niwa K, et al. Uric acid is a strong risk marker for developing hypertension from prehypertension: a 5-year Japanese cohort study. Hypertension. 2018;71(1):78–86. doi:10.1161/HYPERTENSIONAHA.117.10370
124. Mazza A, Lenti S, Schiavon L, et al. Asymptomatic hyperuricemia is a strong risk factor for resistant hypertension in elderly subjects from general population. Biomed Pharmacother. 2017;86:590–594. doi:10.1016/j.biopha.2016.11.104
125. Lee JJ, Ahn J, Hwang J, et al. Relationship between uric acid and blood pressure in different age groups. Clin Hypertens. 2015;21:14. doi:10.1186/s40885-015-0022-9
126. Lee SW, Kim HC, Nam C, et al. Age-differential association between serum uric acid and incident hypertensione. Hypertens Res. 2019;42(3):428–437. doi:10.1038/s41440-018-0168-4
127. Forman JP, Choi H, Curhan GC. Plasma uric acid level and risk for incident hypertension among men. J Am Soc Nephrol. 2007;18:287–292. doi:10.1681/ASN.2006080865
128. Rovda I, Kazakova LM, Plaksina E. [Parameters of uric acid metabolism in healthy children and in patients with arterial hypertension]. Pediatriia. 1990;8:19–22.
129. Török E, Gyárfás I, Csukás M. Factors associated with stable high blood pressure in adolescents. J Hypertens Suppl. 1985;3(3):S389–S390.
130. Brand FN, Mcgee DL, Kannel WB, Stokes J, Castelli WP. Hyperuricemia as a risk factor of coronary heart disease: the Framingham study. Am J Epidemiol. 1985;121(1):11–18. doi:10.1093/oxfordjournals.aje.a113972
131. Viazzi F, Antolini L, Giussani M, et al. Serum uric acid and blood pressure in children at cardiovascular risk. Pediatrics. 2013;132:e93–e99. doi:10.1542/peds.2013-0047
132. Loeffler LF, Navas-Acien A, Brady TM, Miller ER, Fadrowski JJ. Uric acid level and elevated blood pressure in US adolescents: National Health and Nutrition Examination Survey, 1999–2006. Hypertension. 2012;59:811–817. doi:10.1161/HYPERTENSIONAHA.111.183244
133. Park B, Lee HA, Lee SH, et al. Association between serum levels of uric acid and blood pressure tracking in childhood. Am J Hypertens. 2017;30(7):658–660. doi:10.1093/ajh/hpx037
134. Scheepers LEJM, Boonen A, Pijnenburg W, et al. Associations of plasmauric acid and purine metabolites with blood pressure in children: the KOALA Birth Cohort Study. J Hypertens. 2017;75(Suppl2):
135. Li Y, Wei FF, Wang S, Cheng YB, Wang JG. Cardiovascular risks associated with diastolic blood pressure and isolated diastolic Hypertension. Curr Hypertens Rep. 2014;16(11):489. doi:10.1007/s11906-014-0489-x
136. Bjornstad P, Laffel L, Lynch J, et al. Elevated serum uric acid is associated with greater risk for hypertension and diabetic kidney diseases in obese adolescents with type 2 diabetes: an observational analysis from the treatment options for Type 2 Diabetes in Adolescents and Youth (TODAY) Stu. Diabetes Care. 2019;42(6):1120–1128. doi:10.2337/dc18-2147
137. Turak O, Özcan F, Tok D, et al. Serum uric acid, inflammation, and nondipping circadian pattern in essential hypertension. J Clin Hypertens. 2013;15(1):7–13.
138. Verdecchia P, Porcellati C, Schillaci G, et al. Ambulatory blood pressure: an independent predictor of prognosis in essential hypertension. Hypertension. 1994;24(6):793–801. doi:10.1161/01.HYP.24.6.793
139. De La Sierra A, Redon J, Banegas JR, et al. Prevalence and factors associated with circadian blood pressure patterns in hypertensive patients. Hypertension. 2009;53(3):466–472. doi:10.1161/HYPERTENSIONAHA.108.124008
140. Çağlı K, Turak O, Canpolat U, et al. Association of serum uric acid level with blood pressure variability in newly diagnosed essential hypertension. J Clin Hypertens (Greenwich). 2015;17:929–935. doi:10.1111/jch.12641
141. Mancia G, Parati G, Hennig M, et al. Relation between blood pressure variability and carotid artery damage in hypertension: baseline data from the European Lacidipine Study on Atherosclerosis (ELSA). J Hypertens. 2001;19:1981–1989. doi:10.1097/00004872-200111000-00008
142. Tatasciore A, Renda G, Zimarino M, et al. Awake systolic blood pressure variability correlates with target-organ damage in hypertensive subjects. Hypertension. 2007;50:325–332. doi:10.1161/HYPERTENSIONAHA.107.090084
143. Hansen TW, Thijs L, Li Y, et al. Prognostic value of reading-to-reading blood pressure variability over 24 hours in 8938 subjects from 11 populations. Hypertension. 2010;55:1049–1057. doi:10.1161/HYPERTENSIONAHA.109.140798
144. Dawson J, Jeemon P, Hetherington L, et al. Serum uric acid level, longitudinal blood pressure, renal function, and long-term mortality in treated hypertensive patients. Hypertension. 2013;62(1):105–111. doi:10.1161/HYPERTENSIONAHA.113.00859
145. Han T, Meng X, Shan R, et al. Temporal relationship between hyperuricemia and obesity, and its association with future risk of type 2 diabetes. Int J Obes. 2018;42(7):1336–1344. doi:10.1038/s41366-018-0074-5
146. Dawson J, Wyss A. Chicken or the egg? Hyperuricemia, insulin resistance, and hypertension. Hypertension. 2017;70(4):698–699. doi:10.1161/HYPERTENSIONAHA.117.09685
147. Messerli FH, Frohlich ED, Dreslinski GR, Suarez DH, Aristimuno GG. Serum uric acid in essential hypertension: an indicator of renal vascular involvement. Ann Intern Med. 1980;93(6):617–621. doi:10.7326/0003-4819-93-6-817
148. Puig JG, Ruilope LM. Uric acid as a cardiovascular risk factor in arterial hypertension. J Hypertens. 1999;17(7):869–872. doi:10.1097/00004872-199917070-00001
149. Friedl HP, Till GO, Trentz O, Ward PA. Role of oxygen radicals in tourniquet-related ischemia-reperfusion injury of human patients. Klin Wochenschr. 1991;69(21–23):1109–1112. doi:10.1007/bf01645168
150. Ferris TF, Gorden P. Effect of angiotensin and norepinephrine upon urate clearance in man. Am J Med. 1968;44(3):359–365. doi:10.1016/0002-9343(68)90107-1
151. Palmer TM, Nordestgaard BG, Benn M, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ. 2013;347:f4262. doi:10.1136/bmj.f4262
152. Yang J, Kamide K, Kokubo Y, et al. Associations of hypertension and its complications with variations in the xanthine dehydrogenase gene. Hypertens Res. 2008;31(5):931–940. doi:10.1291/hypres.31.931
153. Desgrez A. Influence de la constitution des corps puriques sure leur action vis-a-vis de la pression arterielle. Comptes Rendus l’Academie Des Sci. 1913;156:93–94.
154. Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38(5):1101–1106. doi:10.1161/hy1101.092839
155. Mishima M, Hamada T, Maharani N, et al. Effects of uric acid on the NO production of HUVECs and its restoration by urate lowering agents. Drug Res (Stuttg). 2016;66(5):270–274. doi:10.1055/s-00023610
156. Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Ren Physiol. 2002;282(6):F991–F997.
157. Sanchez-Lozada LG, Soto V, Tapia E, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol. 2008;295(4):F1134–F1141. doi:10.1152/ajprenal.00104.2008
158. Wolff ML, Cruz JL, Vanderman AJ, Brown JN. The effect of angiotensin II receptor blockers on hyperuricemia. Ther Adv Chronic Dis. 2015;6(6):339–346. doi:10.1177/2040622315596119
159. Kanellis J, Watanabe S, Li JH, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41(6):1287–1293. doi:10.1161/01.HYP.0000072820.07472.3B
160. Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodríguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med. 2002;21(346):913–923. doi:10.1056/NEJMra011078
161. Lu J, Hou X, Yuan X, et al. Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int. 2018;93(1):69–80. doi:10.1016/j.kint.2017.04.031
162. Debosch BJ, Kluth O, Fujiwara H, Schürmann A, Moley K. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun. 2014;7(5):4642. doi:10.1038/ncomms5642
163. Preitner F, Pimentel A, Metref S, et al. No development of hypertension in the hyperuricemic liver-Glut9 knockout mouse. Kidney Int. 2015;87(5):940–947. doi:10.1038/ki.2014.385
164. Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose-induced insulin resistance and hypertension in rats. Hypertension. 1987;10(5):512–516. doi:10.1161/01.HYP.10.5.512
165. Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One. 2012;7(10):e47948. doi:10.1371/journal.pone.0047948
166. Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005;1(2):80–86. doi:10.1038/ncpneph0019
167. Perez-Pozo SE, Schold J, Nakagawa T, Sánchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes. 2010;34(3):454–461. doi:10.1038/ijo.2009.259
168. Johnson RJ, Sanchez-Lozada LG, Nakagawa T. The effect of fructose on renal biology and disease. J Am Soc Nephrol. 2010;21:2036–2039. doi:10.1681/ASN.2010050506
169. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Physiol. 2006;290(3):F625–F631. doi:10.1152/ajprenal.00140.2005
170. George J, Carr E, Davies J, Belch JJF, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114(23):2508–2516. doi:10.1161/CIRCULATIONAHA.106.651117
171. Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107(3):416–421. doi:10.1161/01.CIR.0000046\448.26751.58
172. Butler R, Morris AD, Belch JJF, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35(3):746–751. doi:10.1161/01.HYP.35.3.746
173. Waring WS. Hyperuricaemia does not impair cardiovascular function in healthy adults. Heart. 2004;90(2):155–159. doi:10.1136/hrt.2003.016121
174. Waring WS, McKnight JA, Webb DJ, Maxwell SRJ. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes. 2006;55(11):3127–3132. doi:10.2337/db06-0283
175. Waring WS, McKnight JA, Webb DJ, Maxwell SRJ. Lowering serum urate does not improve endothelial function in patients with type 2 diabetes. Diabetologia. 2007;50:2572–2579. doi:10.1007/s00125-007-0817-7
176. Higgins P, Walters MR, Murray HM, et al. Allopurinol reduces brachial and central blood pressure, and carotid intima-media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Heart. 2014;100(14):1085–1092. doi:10.1136/heartjnl-2014-305683
177. Madero M, Rodríguez Castellanos FE, Jalal D, et al. A pilot study on the impact of a low fructose diet and allopurinol on clinic blood pressure among overweight and prehypertensive subjects: a randomized placebo controlled trial. J Am Soc Hypertens. 2015;9(11):837–844. doi:10.1016/j.jash.2015.07.008
178. Kanbay M, Huddam B, Azak A, et al. A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin J Am Soc Nephrol. 2011;6(8):1887–1894. doi:10.2215/CJN.11451210
179. Borgi L, McMullan C, Wohlhueter A, Curhan GC, Fisher ND, Forman JP. Effect of uric acid-lowering agents on endothelial function: a randomized, double-blind, placebo-controlled trial. Hypertension. 2017;69(2):243–248. doi:10.1161/HYPERTENSION\AHA.116.08488
180. McMullan CJ, Borgi L, Fisher N, Curhan G, Forman J. Effect of uric acid lowering on renin-angiotensin-system activation and ambulatory BP: a randomized controlled trial. Clin J Am Soc Nephrol. 2017;12(5):807–816. doi:10.2215/CJN.10771016
181. Segal MS, Srinivas TR, Mohandas R, et al. The effect of the addition of allopurinol on blood pressure control in African Americans treated with a thiazide-like diuretic. J Am Soc Hypertens. 2015;9(8):610–619. doi:10.1016/j.jash.2015.05.009
182. Agarwal V, Hans N, Messerli FH. Effect of allopurinol on blood pressure: a systematic review and meta-analysis. J Clin Hypertens. 2013;15(6):435–442. doi:10.1111/jch.2013.15.issue-6
183. Beattie CJ, Fulton RL, Higgins P, et al. Allopurinol initiation and change in blood pressure in older adults with hypertension. Hypertension. 2014;64(5):1102–1107. doi:10.1161/HYPERTENSIONAHA.114.03953
184. MacIsaac RL, Salatzki J, Higgins P, et al. Allopurinol and cardiovascular outcomes in adults with hypertension. Hypertension. 2016;67(3):535–540. doi:10.1161/HYPERTENSIONAHA.115.06344
185. Ohta Y, Ishizuka A, Arima H, et al. Effective uric acid-lowering treatment for hypertensive patients with hyperuricemia. Hypertens Res. 2017;40(3):259–263. doi:10.1038/hr.2016.139
186. Kohagura K, Ohya Y, Yamazato M, et al. Effects of xanthine oxidase inhibitors on renal function and blood pressure in hypertensive patients with hyperuricemia. Hypertens Res. 2016;39(8):593–597. doi:10.1038/hr.2016.37
187. Høieggen A, Alderman MH, Kjeldsen SE, et al. The impact of serum uric acid on cardiovascular outcomes in the LIFE study. Kidney Int. 2004;65(3):1041–1049. doi:10.1111/j.1523-1755.2004.00484.x
188. Johnson RJ, Choi HK, Yeo AE, Lipsky PE. Pegloticase treatment significantly decreases blood pressure in patients with chronic gout. Hypertension. 2019;75:95–101. doi:10.1161/HYPERTENSIONAHA.119.12727
189. Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. J Am Med Assoc. 2008;300(8):924–932. doi:10.1001/jama.300.8.924
190. Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60(5):1148–1156. doi:10.1161/HYPERTENSIONAHA.112.196980
191. Assadi F. Allopurinol enhances the blood pressure lowering effect of enalapril in children with hyperuricemic essential hypertension. J Nephrol. 2014;27(1):51–56. doi:10.1007/s40620-013-0009-0
192. Gois PHF, de Moraes Souza ER. Pharmacotherapy for hyperuricemia in hypertensive patients. Cochrane Database Syst Rev. 2017;13(4):CD008652.
193. Gunawardhana L, McLean L, Punzi HA, et al. Effect of febuxostat on ambulatory blood pressure in subjects with hyperuricemia and hypertension: a phase 2 randomized placebo-controlled study. J Am Heart Assoc. 2017;6(11):e006683. doi:10.1161/JAHA.117.006683
194. Nguyen S, Choi HK, Lustig RH, Yuan HC. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr. 2009;154(6):807–813. doi:10.1016/j.jpeds.2009.01.015
195. Jalal DI, Smits G, Johnson RJ, Chonchol M. Increased fructose associates with elevated blood pressure. J Am Soc Nephrol. 2010;21(9):1543–1549. doi:10.1681/ASN.2009111111
196. Brown CM, Dulloo AG, Yepuri G, Montani J-P. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Integr Comp Physiol. 2008;294(3):R730–R737. doi:10.1152/ajpregu.00680.2007
197. Chen L, Caballero B, Mitchell DC, et al. Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: a prospective study among United States Adults. Circulation. 2010;121(22):2398–2406. doi:10.1161/CIRCULATIONAHA.109.911164
198. McInnes GT, Lawson DH, Jick H. Acute adverse reactions attributed to allopurinol in hospitalised patients. Ann Rheum Dis. 1981;40(3):245–249. doi:10.1136/ard.40.3.245
199. Kantor GL. Toxic epidermal necrolysis, azotemia, and death after allopurinol therapy. J Am Med Assoc. 1970;212(3):478–479. doi:10.1001/jama.1970.03170160066019
200. Feinstein J, Dai D, Zhong W, Freedman J, Feudtner C. Potential drug-drug interactions in infant, child, and adolescent patients in children’s hospitals. Pediatrics. 2015;135(1):e99–e108. doi:10.1542/peds.2014-2015
201. Cunningham RF, Israili ZH, Dayton PG. Clinical pharmacokinetics of probenecid. Clin Pharmacokinet. 1981;6(2):135–151. doi:10.2165/00003088-198106020-00004
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.