Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Hypercholesterolemia as a Causal Risk Factor for COPD: Biomarker Discovery and Therapeutic Implications From NHANES Data
Authors Liu L, Wu C, Zhang Z, Jin X, Zou Q, Wang M, Huang B, Gan X, Tong J ![]()
Received 5 March 2025
Accepted for publication 11 October 2025
Published 12 November 2025 Volume 2025:20 Pages 3677—3696
DOI https://doi.org/10.2147/COPD.S526511
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Lu Liu,1,2,* Chaoling Wu,1,* Zhiyan Zhang,3 Xinxing Jin,4 Qi Zou,5 Mingxue Wang,1 Bo Huang,1 Xin Gan,2 Jianlin Tong1
1Department of Respiratory Medicine, Affiliated Hospital of Jiujiang University, Jiujiang, 332000, People’s Republic of China; 2Department of Respiratory and Critical Care, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi Province, 330006, People’s Republic of China; 3Jiangxi Institute of Respiratory Disease, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi Province, 330006, People’s Republic of China; 4Department of Respiratory and Critical Care, Gaoxin Hospital of the First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi Province, 330006, People’s Republic of China; 5Department of Geriatric Medicine, Affiliated Hospital of Jiujiang University, Jiujiang, 332000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xin Gan, Department of Respiratory and Critical Care, The First Affiliated Hospital of Nanchang University, 17 Yong Waizheng Street, Donghu District, Nanchang, Jiangxi Province, 330006, People’s Republic of China, Email [email protected] Jianlin Tong, Department of Respiratory medicine, Affiliated Hospital of Jiujiang University, No. 57 East Xunyang Road, Xunyang District, Jiujiang, 332000, People’s Republic of China, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD), the third leading cause of global mortality, imposes substantial socioeconomic burdens. Existing therapies, such as smoking cessation and non-invasive ventilation, primarily alleviate symptoms without arresting disease progression. Comorbidities, including cardiovascular disease and metabolic syndrome, exacerbate functional decline, yet the causal role of dyslipidemia in COPD pathogenesis remains unclear. This study seeks to establish a causal link between hypercholesterolemia and COPD while identifying potential biomarkers and therapeutic targets.
Methods: Leveraging cross-sectional data from the National Health and Nutrition Examination Survey (NHANES), we employed Mendelian randomization (MR) analysis using 71 single-nucleotide polymorphisms (SNPs) associated with hypercholesterolemia, integrated with bioinformatics tools for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Demographic variables (age, sex, body mass index [BMI]) and dietary factors were compared between COPD patients and controls.
Results: Univariate analyses identified significant differences in demographics and dietary patterns between COPD and non-COPD groups (P < 0.05). Multivariate logistic regression revealed: (1) a reduced risk of COPD with elevated total cholesterol levels (odds ratio [OR]: 0.815, 95% confidence interval [CI]: 0.721– 0.923, P = 0.001); and (2) increased COPD risk associated with higher age and BMI. Bioinformatics analyses pinpointed atorvastatin, fenofibrate, and pravastatin as candidate therapeutics. Gene interaction networks and pathway enrichment highlighted roles for lipid homeostasis and cholesterol metabolism.
Conclusion: Analysis of NHANES data demonstrates an inverse association between cholesterol levels and COPD prevalence, with MR confirming a causal relationship. These findings underscore targetable pathways and suggest repurposing statins and fibrates, meriting further mechanistic studies and clinical trials for validation.
Keywords: COPD, Mendelian randomization, NHANES, HMGCR
Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent global condition that significantly impairs patients’ quality of life and imposes a substantial burden on public health systems. According to the Global Burden of Disease Study, COPD ranks as the third leading cause of death, resulting in considerable economic and social impacts on patients and their families.1 Current treatment modalities, including pharmacotherapy, rehabilitation, and oxygen therapy, While current therapies like smoking cessation programs and non-invasive ventilation (NIV) improve symptoms and reduce exacerbations, these interventions still face limitations in enhancing patients’ quality of life and reducing mortality rates.2 Additionally, COPD patients often present with comorbidities such as coronary atherosclerosis and pulmonary embolism, comorbidities—particularly cardiovascular diseases (CVD), metabolic syndrome, and osteoporosis—synergistically accelerate functional decline in COPD, complicating disease management during stable periods.3,4 This situation underscores the necessity for in-depth research into the etiology of COPD and the development of novel therapeutic approaches.
Cholesterol has garnered increasing attention for its roles in both physiological and pathological states. Beyond being a vital component of cell membranes, cholesterol is involved in various biological processes, including cell signaling, hormone synthesis, and bile acid formation.5,6 In recent years, researchers have begun to explore the relationship between hypercholesterolemia and COPD. Elevated cholesterol levels are closely associated with the development of multiple diseases, particularly cardiovascular diseases and metabolic syndrome. Studies suggest that hypercholesterolemia may influence the pathogenesis of COPD, dysregulated lipid profiles, characterized by elevated low-density lipoprotein cholesterol (LDL-C) and reduced high-density lipoprotein cholesterol (HDL-C) levels, are commonly observed in patients with chronic obstructive pulmonary disease (COPD). This phenomenon is particularly pronounced among smokers. Studies indicate that smokers exhibit significantly higher LDL-C and lower HDL-C levels compared to non-smokers. These alterations are closely linked to smoking-induced oxidative stress and chronic inflammatory responses-key pathological mechanisms in COPD development.7,8 Furthermore, elevated triglyceride (TG) levels are implicated in COPD pathophysiology. Multiple studies have indicated that hypertriglyceridemia positively correlates with COPD severity and accelerated lung function decline compared to controls. These findings suggest disturbances in lipid metabolism may play a noteworthy role in COPD progression, potentially contributing to underlying disease mechanisms,7,9 offering new perspectives for its prevention and treatment.10 However, despite accumulating evidence indicating an association between the two, comprehensive studies on their causal relationship and underlying mechanisms remain insufficient, providing ample space for future research.
To investigate the connection between cholesterol levels and the incidence of COPD, we utilized the National Health and Nutrition Examination Survey (NHANES) dataset, employing a cross-sectional study design to analyze the relationship between the two variables. The richness and representativeness of the NHANES dataset enable this study to provide more universally applicable conclusions regarding the etiology of COPD. Considering the potential confounding factors and the possibility of reverse causality inherent in cross-sectional studies, we further employed Mendelian randomization (MR) analysis to identify single nucleotide polymorphisms (SNPs) associated with hypercholesterolemia and their impact on COPD. This approach not only offers more reliable causal inferences but also lays the groundwork for subsequent exploration of potential pathogenic mechanisms and therapeutic targets. Through this research design, we aim to elucidate the specific role of cholesterol metabolism in the pathogenesis of COPD and propose directions for future studies. This endeavor will not only enhance our understanding of the pathophysiological mechanisms of COPD but also provide a theoretical basis for developing new therapeutic strategies.
Methods
This study comprises three distinct parts. First, we examined the association between cholesterol levels and the incidence of COPD using cross-sectional data from the NHANES database. Second, we conducted a two-sample Mendelian randomization (MR) analysis to further investigate the causal relationship between hypercholesterolemia and COPD prevalence. Finally, we performed molecular function and pathway analyses based on genes identified through single nucleotide polymorphisms (SNPs), and predicted potential therapeutic targets and drugs.
Clinical Study Design
This part of the study is a cross-sectional analysis. All data used for analysis were freely available and obtained from NHANES 2009–2018, conducted by the National Center for Health Statistics (NCHS) of the Centers for Disease Control and Prevention (CDC). NHANES collects information on demographics, laboratory tests, health-related factors, and includes a 24-hour dietary recall assessment. From 2009 to 2018, NHANES included 49,693 participants. The inclusion and exclusion criteria of this study were as follows: Inclusion criteria: Participants who have been clearly diagnosed with or without COPD by a physician. Participants with documented total cholesterol levels. Exclusion criteria: Patients who answered “unknown” when asked about their COPD diagnosis. Patients with missing total cholesterol records. After excluding cases with undiagnosed COPD and missing total cholesterol test results, 15,420 participants were included, with 606 diagnosed with COPD (Figure 1). Missing values for diet and BMI were imputed. As NHANES is a publicly available dataset, this study did not require approval from an institutional review board. All participants provided informed consent.
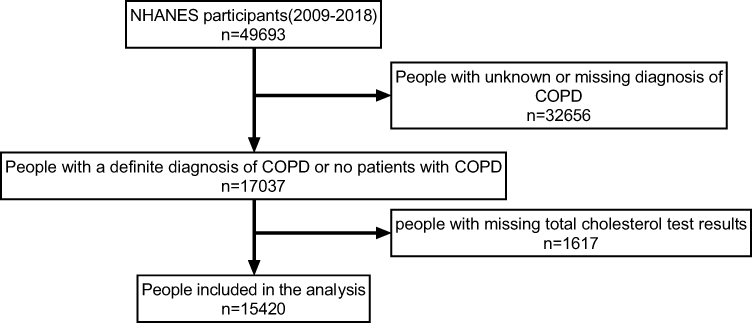 |
Figure 1 Flowchart of participant selection in the analysis. A total of 49,693 participants from the NHANES database (2009–2018) were initially considered. After excluding individuals with an unknown or missing diagnosis of COPD (n=32,656), 17,037 participants with a confirmed diagnosis of COPD were identified. Following the exclusion of 1,617 individuals with missing total cholesterol test results, 15,420 individuals were ultimately included in the analysis. |
COPD-Related Information
Self-reported COPD data were recorded in the “Medical Conditions” section of the NHANES interview. Participants who answered “yes” to the question “Have you ever been told by a doctor or other health professional that you have COPD or emphysema or chronic bronchitis?” were included in the case group; those who answered “no” were included in the control group.
Total Cholesterol Levels
Total cholesterol test results were recorded in the “Laboratory Tests” section of the NHANES database.
Other Relevant Factors
Considering that people of different BMI, age, and ethnic groups have different dietary structures, which can affect total cholesterol levels, we have included these variables in our study. Moreover, recent studies demonstrate that dietary intake of micronutrients including vitamins modulates type 2 airway inflammation through immunomodulatory pathways.11 We also collected the following covariates: age, gender, race, BMI, carotenoids, total energy intake, fats, carbohydrates, various vitamins, and niacin. Age, gender, and race data were obtained from the demographic section of the NHANES database; BMI data were obtained from the physical measurements section; and dietary-related data, including carotenoids, total energy intake, and fats, were obtained from the first-day dietary recall section. Missing values for covariates including BMI and carotenoids were addressed using mean imputation methodology.
MR Data Acquisition
The UK Biobank is a large biomedical database and research resource containing in-depth genetic and health information from 500,000 UK participants. The UK Biobank’s genome-wide association study (GWAS) summary dataset for cholesterol (ukb-b-10912) includes 462,933 European ancestry samples, and the GWAS summary dataset for COPD (ukb-a-67) includes 337,159 European ancestry samples. Both GWAS datasets are available at https://gwas.mrcieu.ac.uk/.
Gene Data Acquisition
We conducted MR analysis using a series of stringent steps to meticulously process exposure and outcome data. SNPs identified in the MR analysis were mapped to their corresponding genes using the NCBI database (https://www.ncbi.nlm.nih.gov/snp/).
Protein–Protein Interaction Network Construction and Hub Gene Identification
Protein–protein interaction (PPI) networks were constructed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (http://string-db.org/), selecting a confidence score of ≥0.4 as the threshold for PPI. The resulting PPI networks were visualized using Cytoscape software (https://www.cytoscape.org/). Subsequently, the Cytohubba plugin in Cytoscape was used to identify hub genes by applying the Maximum Clique Centrality (MCC) algorithm, selecting the top 10 hub genes from the PPI network.
Functional Clustering and Enrichment Analysis of Hub Genes and Chromosomal Localization
We performed Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using the R packages “clusterProfiler” and “org.Hs.eg.db”. GO enrichment analysis provides a standardized system for the functions of genes and proteins, including biological processes (BP), molecular functions (MF), and cellular components (CC). KEGG pathway analysis offers information on discovered pathways and their involved interactions. Enriched GO terms and KEGG pathways were selected with an adjusted p-value (p.adj) < 0.05. Chromosomal localization maps were generated using the “circlize” package in R.
Transcription Factor and Hub Gene Network Construction
The list of hub genes obtained was uploaded to NetworkAnalyst (https://www.networkanalyst.ca/) to construct transcription factor–gene networks using the JASPAR database.
Identification of Candidate Drugs and Molecular Docking of Drugs with Genes
Based on the selected hub genes, potential therapeutic drugs were predicted using the Gene–Drug Interaction Database (GBIdb) (https://dgidb.org/). The 3D structures of the predicted drugs were searched in the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), and protein structures were obtained from the UniProt database (https://www.uniprot.org/). Molecular docking was performed using the CB-Dock2 website (https://cadd.labshare.cn/cb-dock2/php/blinddock.php) to predict interaction patterns between protein active sites and ligands. The collected data were processed and visualized based on previously reported studies.
Statistical Analysis
Statistical analyses were performed using SPSS 25 and R 4.21. Due to NHANES’s stratified, multistage probability cluster sampling design, we included data from five cycles of NHANES (2009–2018) and applied appropriate weights for statistical analysis. For descriptive statistics, non-normally distributed continuous variables were presented as median (interquartile range), and binary variables as n (%). For univariate analysis, non-normally distributed continuous variables were analyzed using non-parametric tests, and categorical variables using chi-square tests. A p-value < 0.05 (two-tailed) was considered statistically significant. Variables with p < 0.05 in univariate logistic regression analysis were included in multivariate logistic regression analysis.
Mendelian randomization studies were conducted in R 4.21 using the “TwoSampleMR” package to explore the causal relationship between hypercholesterolemia and COPD. MR is a method that uses genetic variation as a natural experiment to investigate causal relationships between potentially modifiable risk factors and health outcomes. The effectiveness of MR relies on three key assumptions: 1) the genetic instrument is associated with the exposure; 2) the instrument is independent of confounders; 3) the instrument affects the outcome only through the exposure. To construct genetic instruments for hypercholesterolemia and COPD, we obtained reliable (P < 5×10−8) and independent (r2 < 0.001, kb = 10,000) SNPs associated with hypercholesterolemia, without using proxy SNPs. We then harmonized the effect alleles between exposure and outcome datasets. Random-effects models, including Inverse Variance Weighted (IVW), MR-Egger, and weighted median methods, were used to test for causality, with IVW as the primary analysis method. Cochrane’s Q test was used to assess heterogeneity, and MR-Egger intercept test to detect horizontal pleiotropy.
Results
NHANES Analysis Results
In the univariate analysis shown in Table 1, COPD patients differed significantly from the healthy population in terms of demographics such as age, gender, race, and BMI (P < 0.001, 0.009, < 0.001, 0.006). Furthermore, in the analysis of dietary patterns presented in Table 2, factors such as sugar, fiber, fat, vitamin E, beta-carotene, vitamin B2, vitamin K, and total cholesterol (TC) also showed significant differences between COPD patients and the healthy population, with all P values less than 0.05.
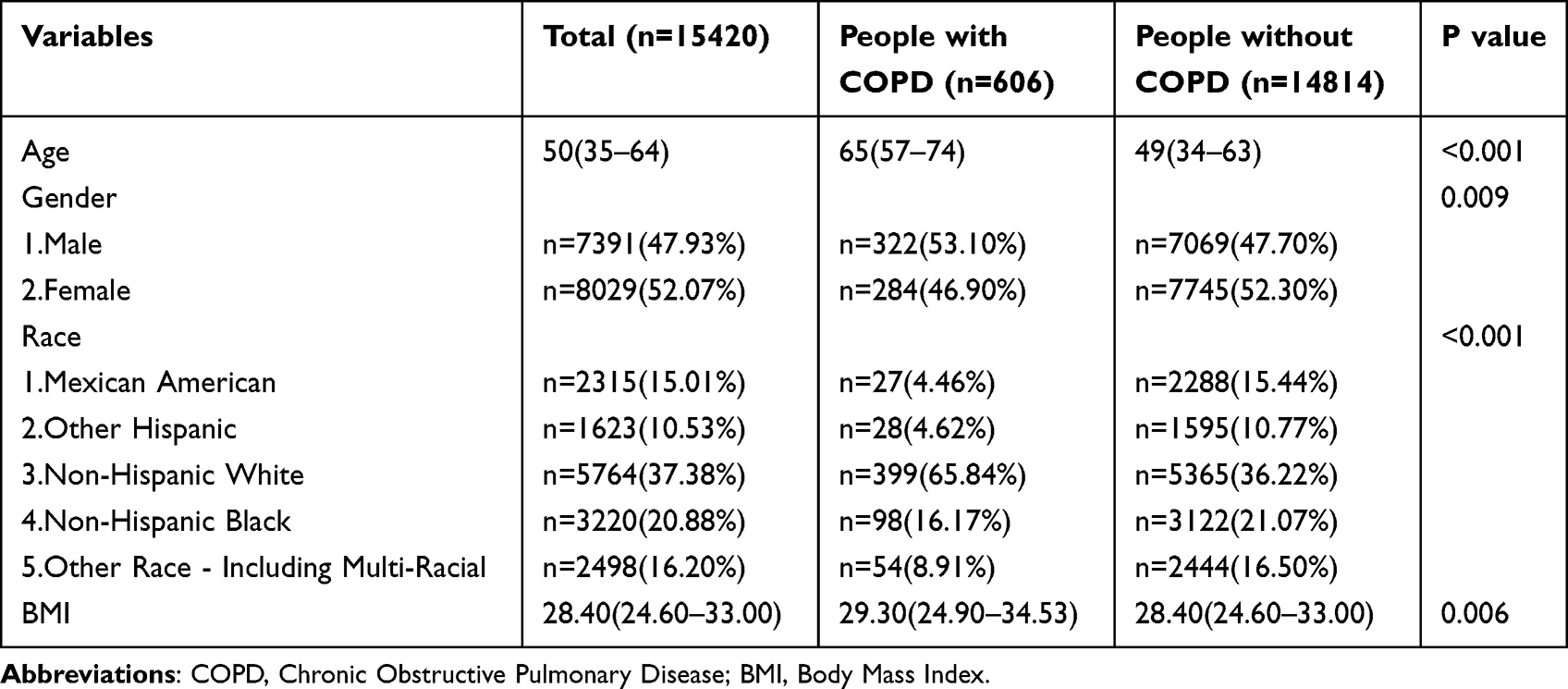 |
Table 1 The Demographic Characteristics of the Study Population |
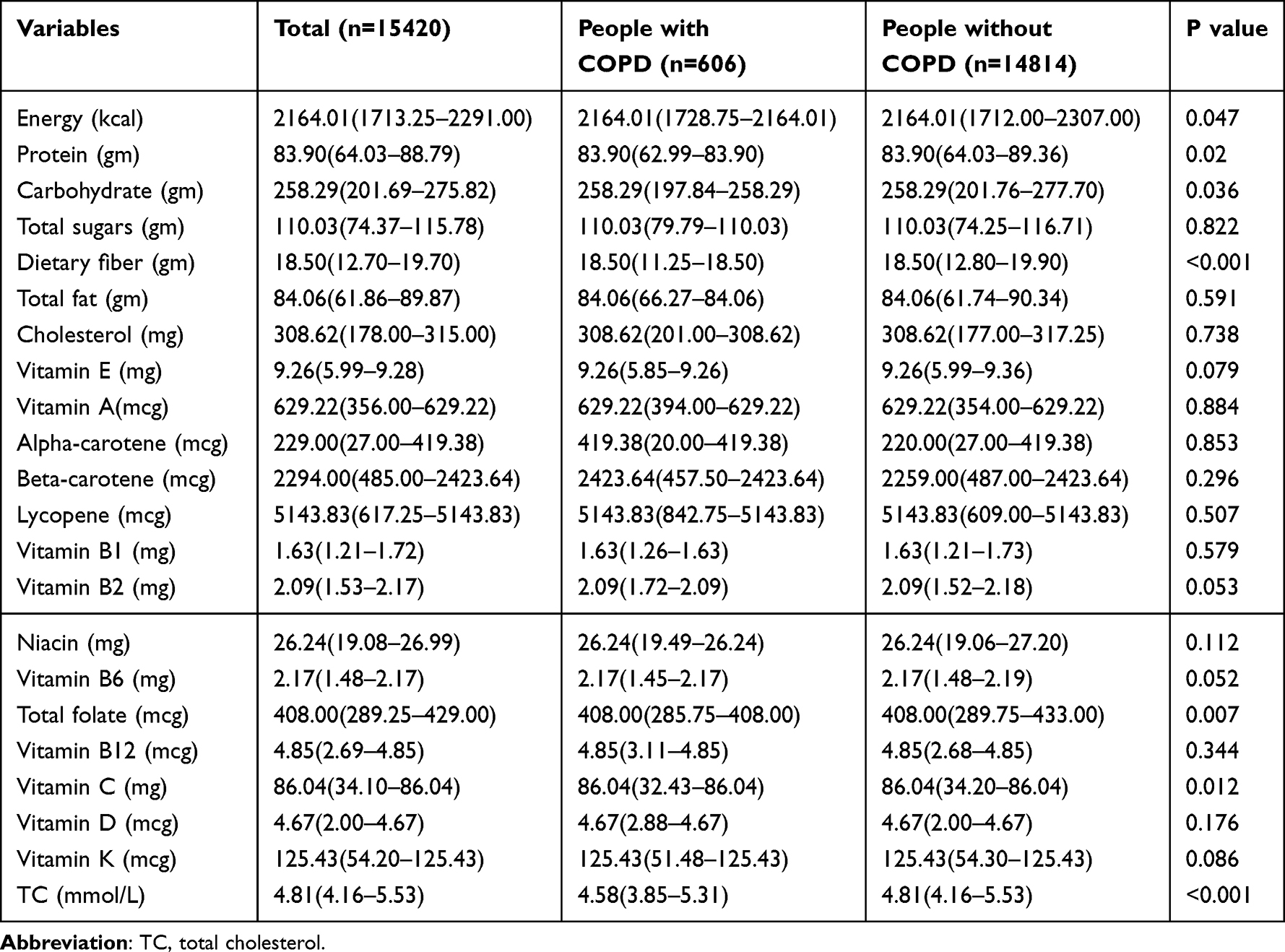 |
Table 2 The Dietary Pattern and TC of the Study Population |
Subsequently, variables with P < 0.05 in univariate analysis were included in multivariate logistic regression analysis. We found that age, BMI, race, and TC were associated with the incidence of COPD. Age and BMI were identified as risk factors for COPD (OR (95% CI): 1.06 (1.054–1.066), P < 0.001; OR (95% CI): 1.02 (1.002–1.038), P = 0.026). Regarding race, compared to Mexican Americans, individuals from other racial groups, except for Non-Hispanic Whites, had a higher prevalence of COPD (OR (95% CI): 3.666 (2.314–5.806), P < 0.001; OR (95% CI): 2.24 (1.369–3.665), P = 0.001; OR (95% CI): 4.047 (2.219–7.38), P < 0.001) (Table 3). However, higher total cholesterol (TC) levels were associated with a lower risk of COPD, suggesting that TC is a protective factor for COPD (OR (95% CI): 0.815 (0.721–0.923), P = 0.001). We then plotted a restricted cubic spline curve between TC and OR. Given the large sample size, we selected 10 nodes for the plot. The restricted cubic spline analysis confirmed no evidence of nonlinearity (P_nonlinear = 0.248), supporting the use of a linear model for the TC-COPD association, with a significant overall P-value (P_overall < 0.001; Figure 2).
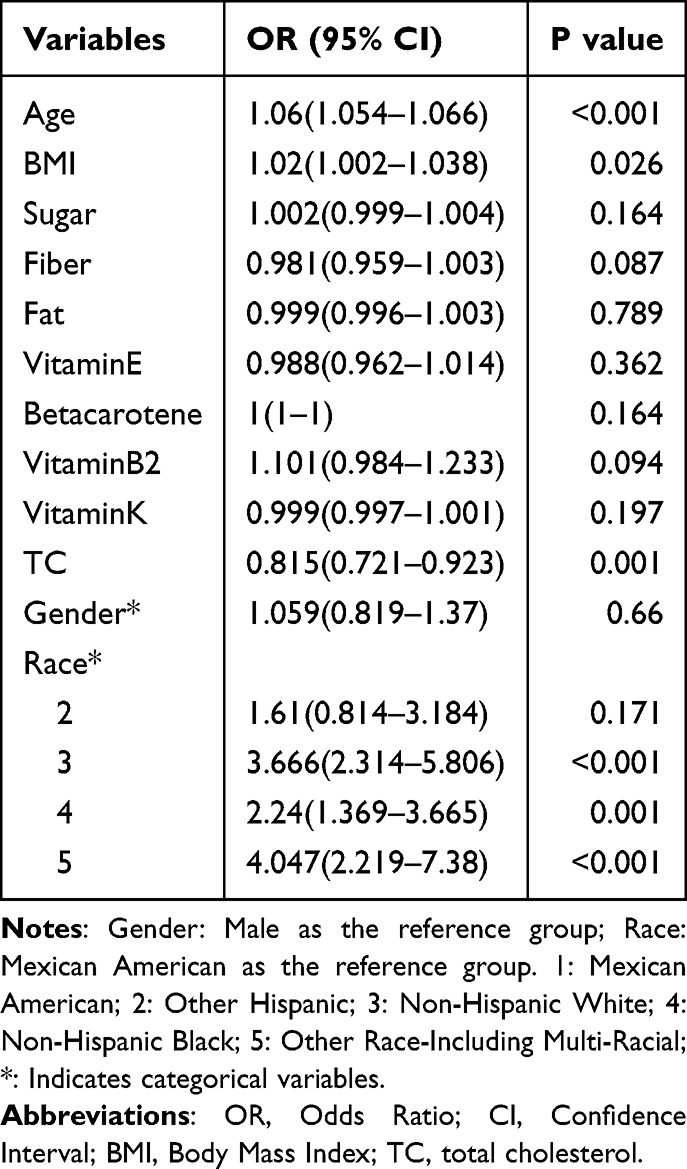 |
Table 3 Results of Multi Factor Logistics Regression Analysis |
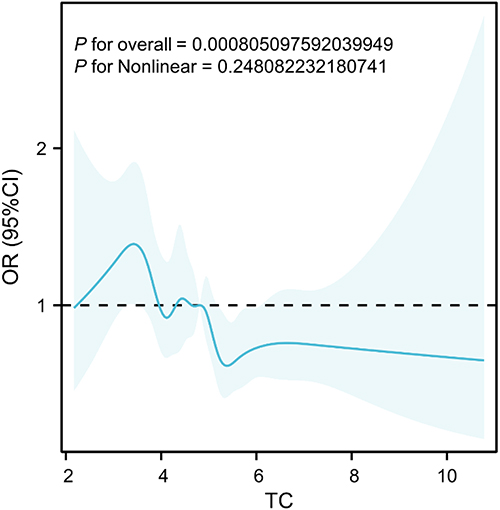 |
Figure 2 Association between total cholesterol (TC) and the odds ratio (OR) for COPD. The plot displays the relationship between TC levels and the odds of COPD, with the 95% confidence intervals (CI) shaded. The dashed horizontal line at OR = 1 indicates the null value, where no association is observed. The restricted cubic spline curve shows a statistically significant overall association (P_overall < 0.001), with no evidence of nonlinearity (P_nonlinear = 0.248). This suggests a linear relationship, where higher TC levels are generally associated with increased COPD risk, though the effect may appear modest across the observed range. |
MR Analysis Results
After removing linkage disequilibrium and harmonizing the exposure and outcome data based on effect alleles, 71 SNPs were ultimately included in the analysis, with detailed information on these SNPs provided in the Supplementary Table 1. The results revealed a causal relationship between hypercholesterolemia and COPD (OR < 1, P = 0.038) (Table 4), which was validated by both the MR-Egger and weighted median methods (MR Egger Q=59.73; P=0.784, IVW Q=64.52; P=0.663). Furthermore, the results from Cochrane’s Q test demonstrated that there was no heterogeneity in this study (Q = 59.57; P = 0.784) (Figure 3).
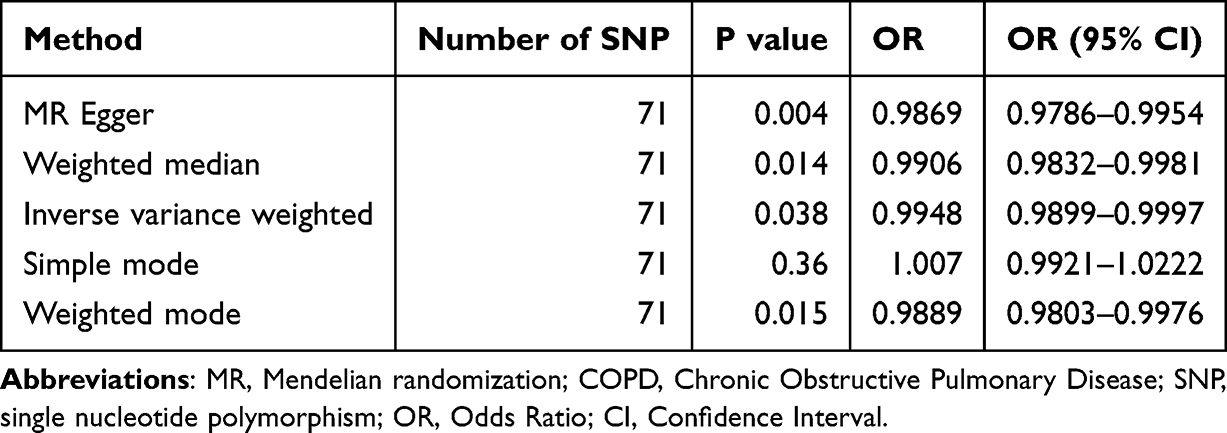 |
Table 4 MR for the Association Between Predicted Hypercholesterolemia and COPD |
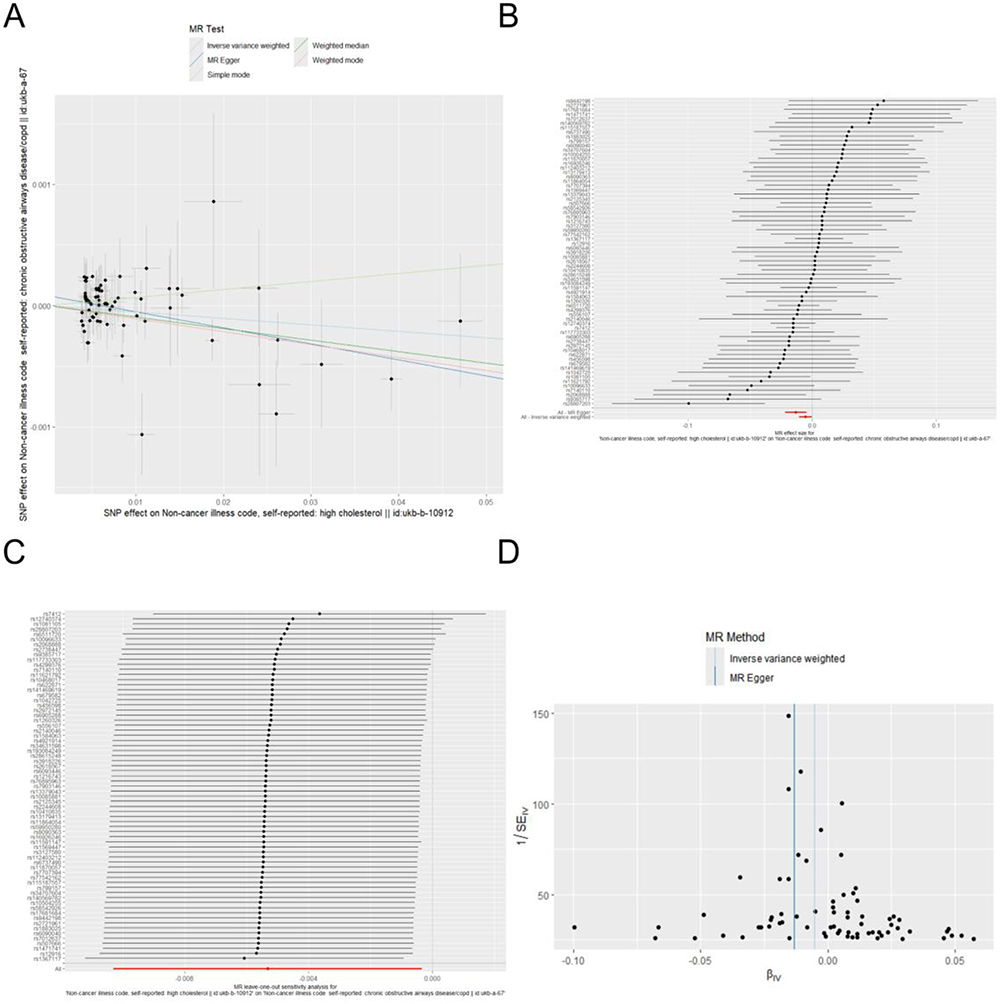 |
Figure 3 Mendelian randomization analysis examining the effect of TC on COPD. (A) Scatter plot of causal effect estimates depicting the casual effective between TC and COPD; (B) Forest of the TC on the odds ratio for COPD; (C) Leave-one-out sensitivity analysis chart between TC and COPD; (D) Funnel plot to assess heterogeneity and potential bias. |
GO and KEGG Enrichment Pathway Analysis
We searched each of the 71 SNPs obtained from the MR analysis in the NCBI database and identified 51 corresponding genes, with the gene details provided in the Supplementary Table 1. To better understand the biological functions of these identified genes and the potential pathogenesis of COPD, we performed GO and KEGG pathway enrichment analyses using the “clusterProfiler” package in R (Figure 4). The functional enrichment analysis revealed that the most significantly enriched GO terms were related to lipid homeostasis in biological processes (BP), endocytic vesicles in cellular components (CC), and lipoprotein particle binding, sterol transfer activity, and cholesterol transfer activity in molecular functions (MF). Additionally, the KEGG pathways primarily involved cholesterol metabolism, bile secretion, and fat digestion and absorption.
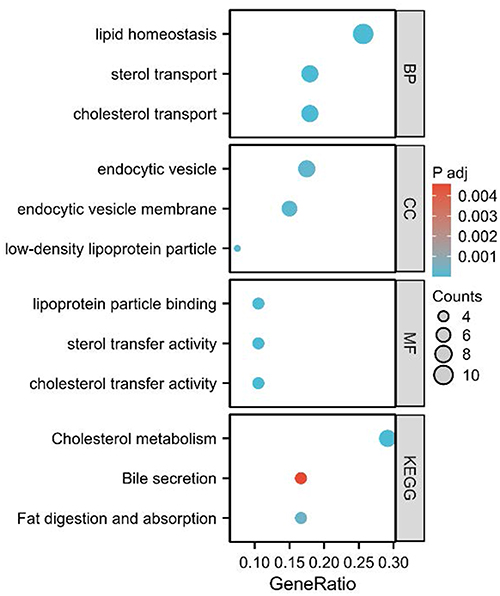 |
Figure 4 Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for cholesterol-related biological processes and functions. The figure displays the results of enrichment analysis categorized by biological processes (BP), cellular components (CC), molecular functions (MF), and KEGG pathways. The x-axis represents the GeneRatio, with the size of the circles corresponding to the number of genes involved in each pathway or process (Counts). The color intensity of the circles indicates the adjusted p-value (P adj), with darker blue representing more significant enrichment. Notably, the KEGG pathway “Bile secretion” is highlighted in red due to its significant association with cholesterol metabolism. |
Construction of PPI Network and Identification of Hub Genes
The gene-protein interaction (PPI) network was constructed using the STRING database to analyze the interactions between genes, with the number of interactions for each gene shown in Figure 5. To identify the central genes in the PPI network, we used the cytoHubba plugin of Cytoscape software. Based on the MCC algorithm, the top 10 genes were selected as potential hub genes (Figure 6), including NOS3, PCSK9, SCARB1, CELSR2, APOB, TM6SF2, ABCA1, MLXIPL, HMGCR, and APOE. Additionally, we used the “circlize” package in R software to map the chromosomal locations of the 10 hub genes (Figure 7).
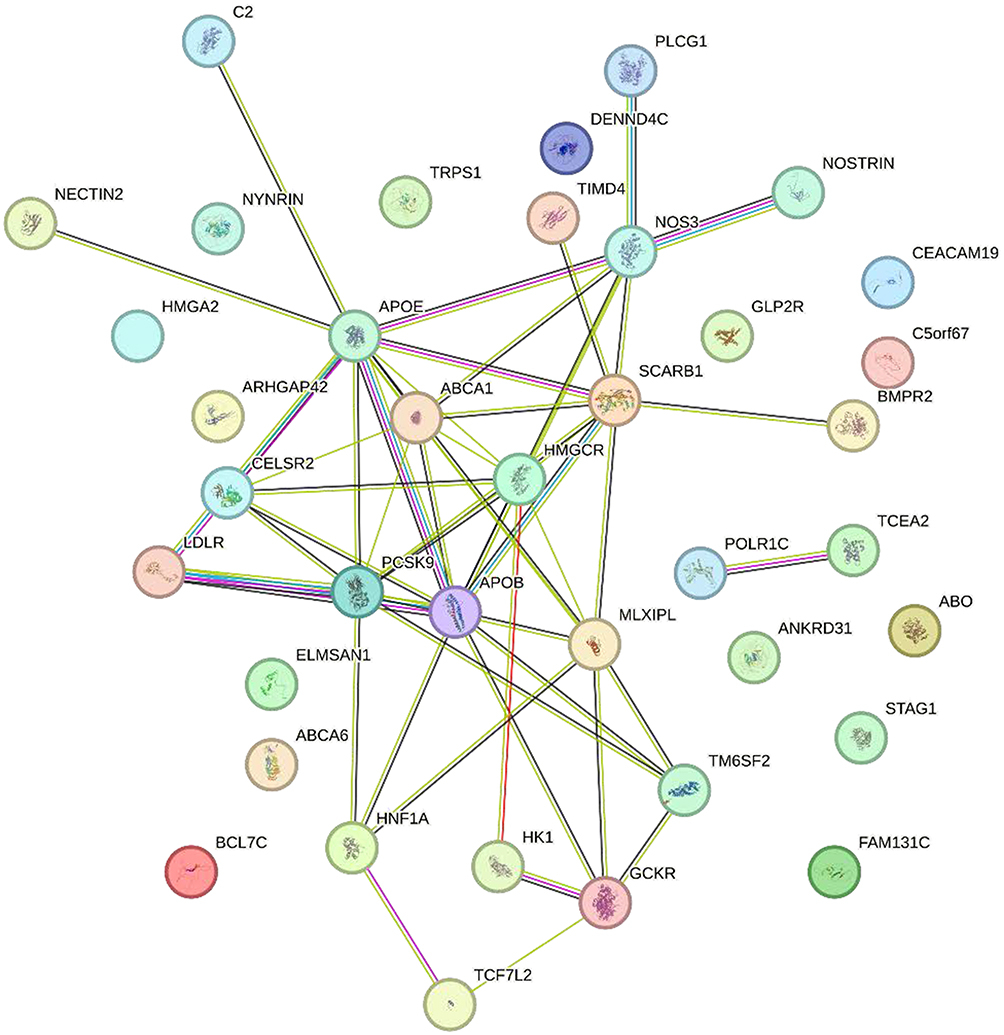 |
Figure 5 Protein-protein interaction (PPI) network of cholesterol-related genes. The network was constructed using known protein interactions, highlighting key genes associated with cholesterol metabolism and transport. Nodes represent individual proteins, and edges indicate interactions between them. Genes such as APOE, PCSK9, APOB, ABCA1, and LDLR are centrally positioned in the network, suggesting their crucial roles in cholesterol regulation. The color of the nodes corresponds to different functional categories, and the line colors represent the type of interaction (eg, physical, genetic). |
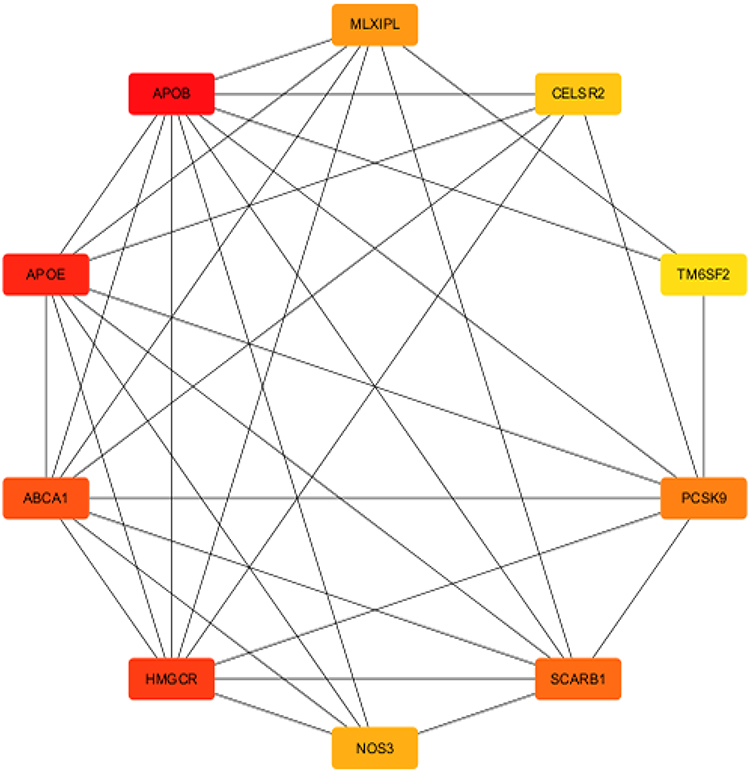 |
Figure 6 PPI network of cholesterol-related genes with highlighted key nodes. This network illustrates the interactions between genes associated with cholesterol metabolism and transport. The nodes represent individual proteins, with edges indicating their interactions. Key genes, including APOE, APOA1, HMGCR, and PCSK9, are marked in red, indicating their central roles in cholesterol regulation. Other important genes such as ABCA1, NOS3, and SCARB1 are shown in Orange. The color coding emphasizes the functional significance of these genes in lipid metabolism. |
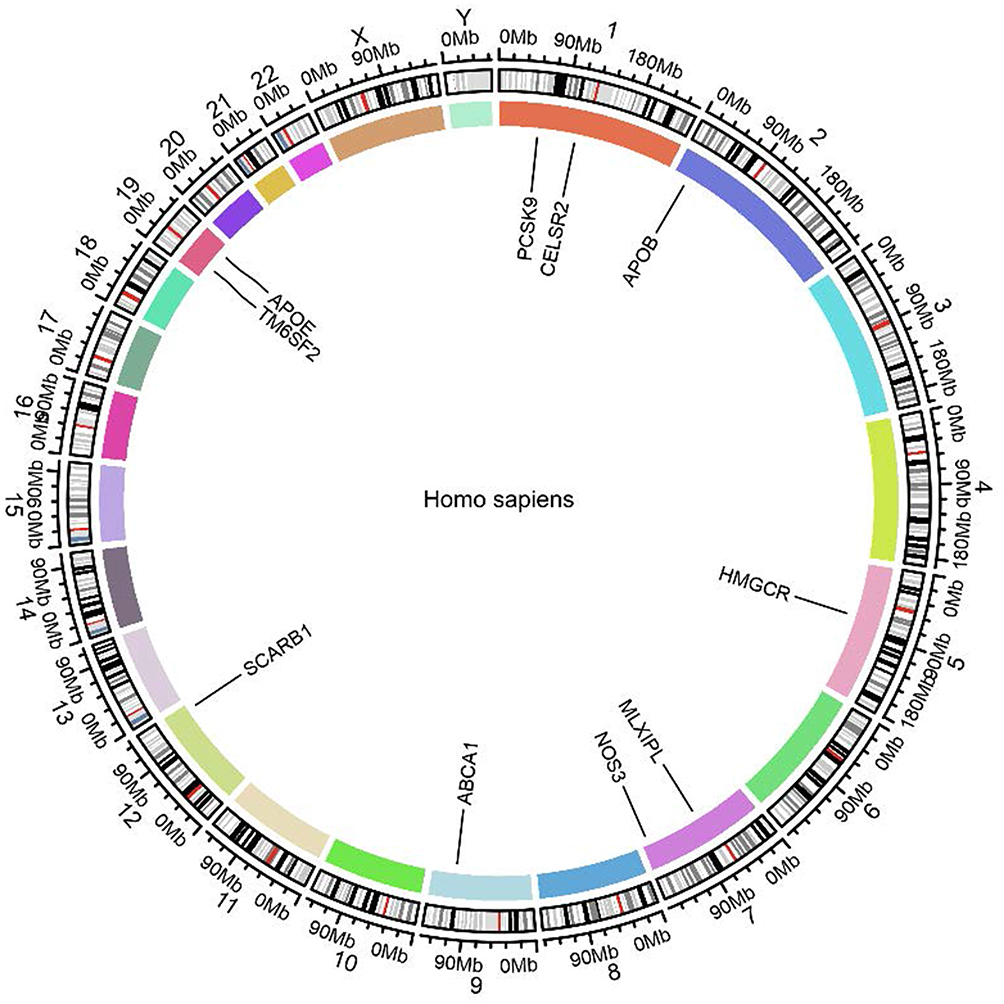 |
Figure 7 Genomic localization of cholesterol-related genes in the human genome. The circular plot shows the chromosomal positions of key genes involved in cholesterol metabolism and transport across the human genome. The genes APOE, PCSK9, CELSR2, APOB, ABCA1, NOS3, HMGCR, MLXIPL, and SCARB1 are highlighted, with their locations marked along the chromosomes. Each segment of the circle represents a specific chromosome, and the colored sections indicate the regions where these genes are located. |
TF‐Hub Genes Interaction
The NetworkAnalyst database was used to predict and visualize the transcription factor (TF)-gene interactions for the hub genes. The TF-gene interaction network included 45 nodes and 63 edges (Figure 8). Among the interactions, NOS3 was found to interact with 11 transcription factors, including SPIB, JUN, RELA, GATA3, YY1, NFIC, NFYA, HOXA5, SPF, STAT1, and USF2. Additionally, FOXC1 interacted with four hub genes, namely CELSR2, PCSK9, HMGCR, and MLXIPL. However, these findings require further validation.
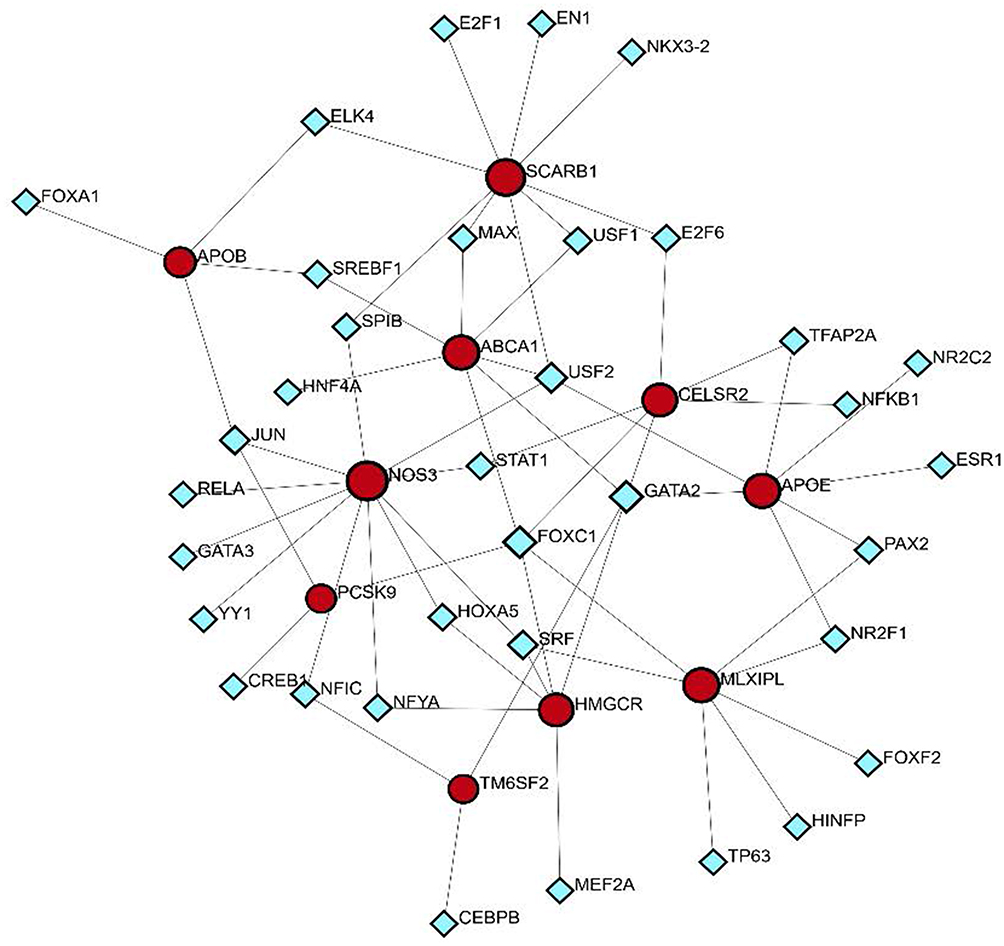 |
Figure 8 Gene regulatory network of cholesterol-related genes and transcription factors. This network diagram illustrates the interactions between cholesterol metabolism genes and associated transcription factors. The red nodes represent key cholesterol-related genes, including APOE, SCARB1, ABCA1, NOS3, PCSK9, HMGCR, and CELSR2, which are central to cholesterol regulation and lipid homeostasis. The blue diamond-shaped nodes represent transcription factors that interact with these genes. |
Results of Therapeutic Drug Molecules
SPIED and CMAP analyses were used to identify therapeutic drug molecules for the 10 hub genes. We successfully predicted therapeutic drug molecules for 7 hub genes, including APOB (Figure 9). To determine the most tightly connected drugs in the drug-gene interaction network, we used the cytoHubba plugin in Cytoscape software. Drugs with a drgee cutoff value greater than or equal to 2 were visualized, and the most tightly connected drugs were selected for subsequent molecular docking. Ultimately, three drugs were identified: Atorvastatin, Fenofibrate, and Pravastatin (Table 5).
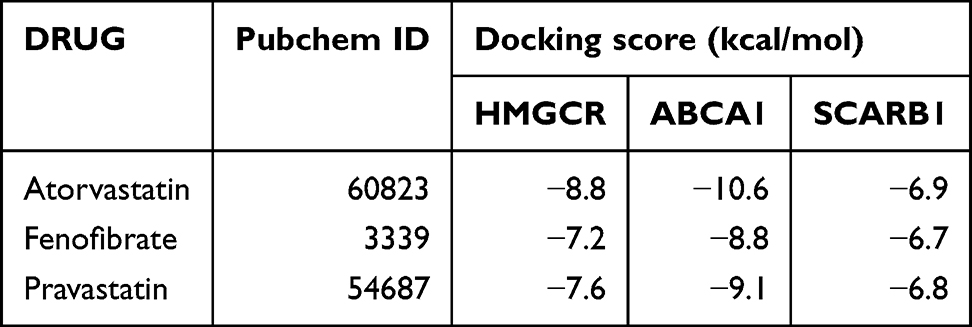 |
Table 5 The Result of Molecular Docking |
 |
Figure 9 Drug-gene interaction network related to cholesterol metabolism. This network diagram highlights the interactions between cholesterol-related genes and associated therapeutic agents. The blue nodes represent key cholesterol-related genes, including APOB, SCARB1, HMGCR, NOS3, APOE, ABCA1, and PCSK9, which play central roles in lipid regulation. The Orange nodes represent therapeutic drugs and compounds, such as Atorvastatin, Rosuvastatin, Fenofibrate, and Simvastatin, that are known to influence these cholesterol-related genes. |
Molecular Docking Simulation
Molecular docking simulations were conducted to explore the potential therapeutic mechanisms of these drugs. Except for the hub genes APOB, NOS3, APOE, and PCSK9, for which no relevant data were available, the remaining three hub genes were docked to evaluate their binding affinity with the three compounds as potential therapeutic targets. The binding energies between the proteins and molecules are shown in Table 4. The binding energies of Atorvastatin and Pravastatin were higher than those of Fenofibrate, suggesting that Atorvastatin and Pravastatin may have unexpected effects in the treatment of COPD, indicating that atorvastatin and pravastatin may have certain effects in preventing the occurrence of chronic obstructive pulmonary disease. The 3D docking structures are presented in Figure 10.
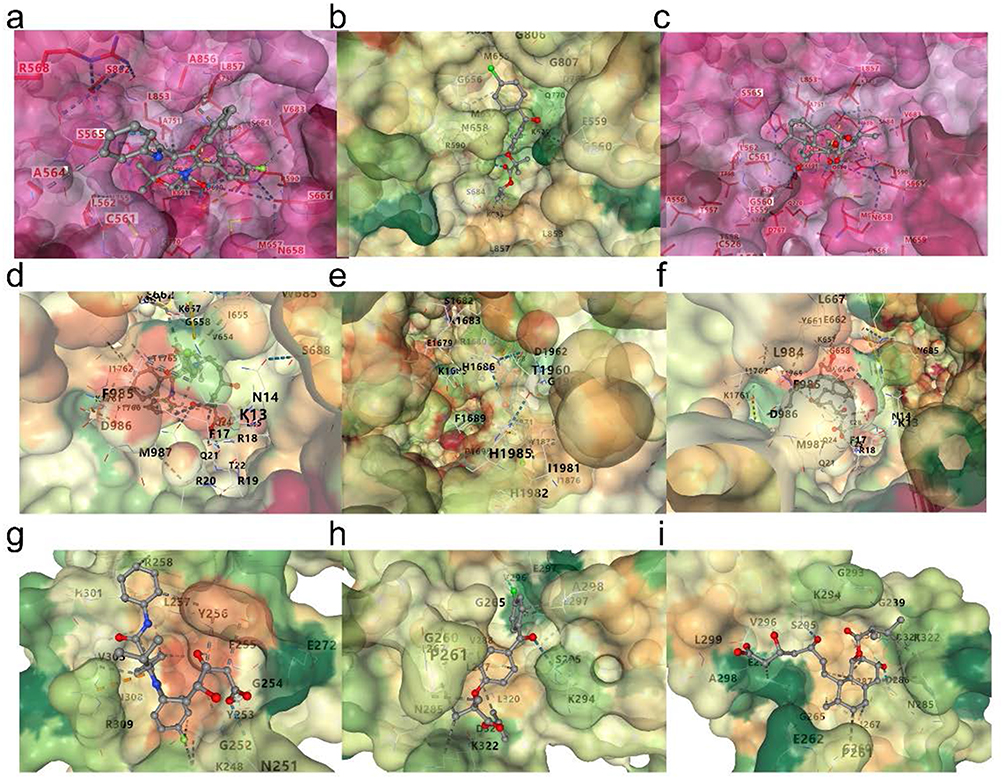 |
Figure 10 Docking simulations of hub gene-encoded proteins with small molecular compounds. (a) Docking interaction of HMGCR with Atorvastatin. (b) Docking interaction of HMGCR with Fenofibrate. (c) Docking interaction of HMGCR with Pravastatin. (d) Docking interaction of ABCA1 with Atorvastatin. (e) Docking interaction of ABCA1 with Fenofibrate. (f) Docking interaction of ABCA1 with Pravastatin. (g) Docking interaction of SCARB1 with Atorvastatin. (h) Docking interaction of SCARB1 with Fenofibrate. (i) Docking interaction of SCARB1 with Pravastatin. The protein surfaces are colored according to the electrostatic potential (red = negative, blue = positive), and key residues involved in ligand interactions are labeled. The docked ligands are shown in stick representation with atoms colored as follows: carbon (grey), oxygen (red), nitrogen (blue), and hydrogen (white). These results demonstrate the binding modes and potential interactions of the selected compounds with the target proteins. |
Discussion
Chronic obstructive pulmonary disease (COPD) is currently the third leading cause of death globally, severely impacting patients’ quality of life and imposing a substantial economic burden on society. The primary characteristics of the disease include persistent progressive airflow limitation and excessive mucus secretion caused by abnormal lung inflammation, leading to decreased lung function and symptoms such as dyspnea.2 Despite current treatments, which mainly include pharmacotherapy, rehabilitation, and oxygen therapy, there remain limitations in improving patients’ quality of life and reducing mortality rates.12,13 Therefore, in-depth research into the pathogenesis of COPD and the search for new therapeutic strategies are of paramount importance.
In the first part of this study, we used a cross-sectional study design with data from the National Health and Nutrition Examination Survey (NHANES) to analyze the relationship between cholesterol levels and COPD prevalence. Compared to previous studies by Ruan et al, our study included a larger sample size, making the results more generalizable. We found that COPD patients had lower cholesterol levels than healthy individuals, a result consistent with Ruan et al’s findings.14 Due to database constraints, our study precluded direct investigation of smoking’s impact on cholesterol levels. Nevertheless, existing research has demonstrated that smoking alters the expression of key rate-limiting enzymes (eg, 3-hydroxy-3-methylglutaryl-CoA reductase, HMGCR) and transcription factors (eg, sterol regulatory element-binding protein 2, SREBP2) in cholesterol metabolism, thereby inducing cholesterol dysregulation.15 Aldo et al demonstrated elevated total cholesterol levels in active smokers, with significant reductions observed following short-term smoking cessation.16 While Aldo et al’s cohort exclusively enrolled heavy smokers (≥20 pack-years) without history of lipid-modifying or bronchodilator therapy, some participants might not have met current GOLD criteria for COPD diagnosis. This suggests cholesterol metabolism may play distinct roles during the disease continuum from chronic bronchitis to established COPD. Contrasting prior reports, our study observed paradoxically lower total cholesterol levels in COPD patients versus controls. This divergence could potentially be attributed to post-diagnosis behavioral modifications—particularly smoking cessation. This may indicate that abnormalities in cholesterol metabolism play a significant role in the pathogenesis of COPD. Previous studies, such as those by Lee et al, have shown that reduced cholesterol levels are associated with decreased lung function, especially in forced expiratory volume (FEV1) and forced vital capacity (FVC).17 In preclinical studies, cholesterol metabolism has been found to be associated with inflammatory levels. Jundi et al observed that excessive cholesterol accumulation can lead to macrophage dysfunction, thereby promoting the release of inflammatory factors such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6).7 Abnormal cholesterol levels can also influence the infiltration and polarization of inflammatory cells, such as macrophages.18,19 Moreover, cholesterol regulates the intensity and duration of the inflammatory response by affecting membrane fluidity and signaling pathways. Studies have also shown that cholesterol metabolites, such as oxysterols and cholesterol esters, are involved in regulating immune responses, possibly by binding to specific receptors to enhance or inhibit the inflammatory response.20,21 In COPD patients, changes in cholesterol-metabolizing enzyme levels are also noteworthy. Yuan et al found that the key enzyme in the cholesterol synthesis pathway, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), was upregulated in COPD patients, which may be related to cholesterol accumulation and pulmonary inflammation.22 Kotlyarov’s research found that the expression of ATP-binding cassette transporter A1 (ABCA1) in the lung tissue of COPD patients also showed significant changes. Its role in cholesterol reverse transport may be impaired, thereby affecting lipid homeostasis and inflammation in the lungs.23 Elevated cholesterol levels upregulate ABCA1 expression, thereby enhancing reverse cholesterol transport and subsequently suppressing pro-inflammatory activity in macrophages.18 Therefore, further investigation into the relationship between cholesterol metabolism and pulmonary inflammation in COPD could provide new insights into the disease’s pathogenesis and offer potential therapeutic targets. In the preliminary analysis, we also found differences in COPD prevalence among different racial groups. Compared to the Mexican American population, the Other Race - Including Multi-Racial group had a higher prevalence of COPD, which may be attributed to differences in dietary habits and obesity rates among various racial groups.24 There is currently a lack of rigorous standards for dietary assessments in different racial backgrounds. These findings could help identify high-risk populations and provide a basis for early screening and intervention for COPD. Integrating these demographic features into clinical practice could support the development of personalized prevention and treatment strategies.
Mendelian randomization (MR) is a technique used to explore causal associations, with the assumption that the alleles of interest are randomly and uniformly distributed in the population, similar to randomized controlled trials, reducing the limitations of observational studies0.25 Considering that the relationship between cholesterol levels and COPD incidence may be influenced by confounding factors such as cardiovascular diseases and reverse causality, we performed a two-sample Mendelian randomization analysis to further investigate the association between cholesterol levels and COPD. Consistent with our observational findings, higher cholesterol levels were identified as a protective factor against COPD prevalence (OR < 1, P = 0.038).
To further understand the potential mechanisms linking cholesterol metabolism and COPD pathogenesis, we mapped the SNPs obtained from the MR analysis to their corresponding genes using the NCBI database and analyzed potential signaling pathways through GO and KEGG enrichment analyses. The functional enrichment analysis revealed that the most significantly enriched GO terms were: Biological Process (BP): lipid homeostasis, Cellular Component (CC): endocytic vesicle, and Molecular Function (MF): lipoprotein particle binding, sterol transfer activity, and cholesterol transfer activity. The primary KEGG pathways involved were cholesterol metabolism, bile secretion, and fat digestion and absorption, which further indicate the involvement of these genes in cholesterol metabolism. We constructed a protein-protein interaction (PPI) network using the STRING database and Cytoscape software, and identified the top 10 hub genes using the MCODE and cytoHubba plugins in Cytoscape (NOS3, PCSK9, SCARB1, CELSR2, APOB, TM6SF2, ABCA1, MLXIPL, HMGCR, APOE). Nitric oxide (NO) promotes cGMP generation via soluble guanylate cyclase (sGC), leading to smooth muscle relaxation, vasodilation, and reduced blood pressure, while also influencing lung remodeling through endothelial cell proliferation and migration.26,27 Reduced NOS3 activity, which catalyzes NO formation, is linked to lung function deterioration, and its interaction with pathways like PPARγ and NF-κB significantly affects lipid metabolism.28,29 Thus, regulating NOS3 may be a new therapeutic target for delaying lung remodeling.30 PCSK9 and SCARB1 have diverse roles beyond cholesterol metabolism, including inflammation and immune responses.31–33 Higham et al linked PCSK9 levels in COPD patients to disease severity and exacerbation frequency.34 SCARB1 expression correlates with pro-inflammatory cytokines, indicating its role in inflammation regulation and lipid metabolism in lung tissue, with dysfunction leading to cholesterol accumulation and exacerbated inflammation.33,35–37 CELSR2, a cadherin superfamily member, is vital for intercellular adhesion and signal transduction, influencing cell polarity and interactions.38 Liu et al found that CELSR2 regulates macrophage polarization and inflammatory factor release, worsening lung inflammation and COPD.39 It also affects cholesterol synthesis and levels via the SREBP pathway and LDLR interaction.38,40 APOB and APOE regulate lipid metabolism and inflammation, linking them to COPD onset.39 APOB is causally related to serum LDL-C levels and COPD, indicating its role in COPD pathogenesis.41 APOE polymorphisms may affect COPD susceptibility, with a study showing no significant link in Han Chinese but a notable interaction with smoking status.42 TM6SF2 encodes a liver membrane protein; the E167K mutation disrupts lipid metabolism, impacting hepatic lipid accumulation and VLDL secretion.43 Its expression relates to inflammatory mediators and immune function, regulated by oxLDL in macrophages, suggesting a role in COPD pathogenesis.44,45 ABCA1 is a membrane transporter that effluxes cholesterol and phospholipids, promoting HDL formation and is linked to lung inflammation and oxidative stress.21,23,46 Its expression is negatively correlated with pro-inflammatory factors like IL-1β and TNF-α, and smoking reduces its levels, suggesting it may help alleviate COPD inflammation.47 MLXIPL, a key transcription factor in the liver and adipose tissue, regulates glycolysis and lipogenesis, impacting energy metabolism and fat storage.48,49 It is linked to inflammation, with abnormal metabolism worsening lung inflammation and promoting COPD progression.50,51 HMGCR, a key enzyme in cholesterol synthesis, converts HMG-CoA to mevalonate.52 It may affect lung inflammation and COPD through lipid metabolism and oxidative stress.53
In the network analysis of transcription factor (TF) and hub gene interactions, we found that FOXC1 is associated with several hub genes. As a transcription factor, FOXC1 regulates the expression of target genes by binding to specific DNA sequences, influencing cell fate determination and tissue formation.54 FOXC1 plays a critical role in the inflammatory response by modulating cytokine expression. On one hand, the upregulation of FOXC1 is associated with increased expression of pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6), which play significant pro-inflammatory roles in chronic inflammatory diseases like COPD.55 On the other hand, FOXC1 can influence the activation status of macrophages and lymphocytes, thereby regulating immune responses. For example, FOXC1 regulates the polarization of macrophages, promoting their M1 polarization, which enhances the inflammatory response.56 In cholesterol metabolism, FOXC1 acts as a crucial transcriptional regulator that directly modulates HMGCR expression, fine-tunes SREBP-2 activity and nuclear translocation, and alters expression of multiple mevalonate pathway enzymes – collectively governing cholesterol homeostasis. Notably, these regulatory actions are mechanistically implicated in COPD pathogenesis.57,58 Moreover, FOXC1 compromises airway epithelial integrity by dysregulating lipid metabolism, thereby impairing barrier function. This pathogenic cascade is mechanistically linked to FOXC1-mediated transcriptional control of the ABCA1 transporter, which accelerates cholesterol efflux and disrupts membrane architecture.58
In subsequent studies, we predicted drug targets for the hub genes and used Cytoscape software to identify drugs closely connected to the genes, constructing a network diagram. We identified three drugs: Atorvastatin, Pravastatin, and Fenofibrate. Fenofibrate, a selective PPAR-α (peroxisome proliferator-activated receptor alpha) agonist, mainly regulates lipid metabolism, glucose metabolism, and the inflammatory response by activating PPAR-α.59 In chronic airway diseases, Fenofibrate can reduce the release of inflammatory mediators, such as TNF-α and IL-6, by inhibiting the NF-κB signaling pathway, thereby alleviating lung inflammation.60 Fenofibrate has also been shown to lower oxidative stress levels in the lungs by regulating lipid metabolism, further reducing inflammation.61 Atorvastatin and Pravastatin, as HMGCR inhibitors, not only regulate blood lipid levels but also exhibit anti-inflammatory and antioxidant effects by inhibiting the release of inflammatory mediators and modulating immune cell functions.62–64 In preclinical studies, Melo et al found that Atorvastatin could promote lung tissue repair in emphysema mice by inhibiting Nrf2 and MMP-12 production.65 Current evidence from randomized controlled trials demonstrates substantial heterogeneity in the therapeutic efficacy of statins for COPD management, as evidenced by divergent outcomes in landmark studies by Zhang et al versus Criner et al66–68 We postulate that this therapeutic heterogeneity originates from differential metabolic characteristics among enrolled COPD subpopulations: Statins may confer significant prognostic benefits in COPD patients with metabolic comorbidities (eg, hyperlipidemia or coronary heart disease) through dual lipid-modulating and anti-inflammatory effects, whereas limited clinical benefits are observed in pure COPD phenotypes lacking these underlying pathophysiological alterations. However, clinical studies have not provided conclusive evidence regarding Atorvastatin’s role in reducing COPD incidence. Kim et al’s study found that Atorvastatin did not reduce the incidence of COPD. Furthermore, the effectiveness of HMGCR inhibitors in reducing the frequency of acute exacerbations and hospitalizations in COPD patients remains controversial.67,69,70 Our study explored the potential targets of Atorvastatin and Pravastatin in COPD treatment, providing evidence regarding the effectiveness of these drugs.
The limitations of this study are primarily reflected in several aspects. First, although we analyzed large-scale population data, the lack of experimental validation may affect the generalizability and reliability of the results. Second, despite the relatively large sample size, genetic differences across different racial groups may lead to inconsistencies in the results, limiting the broader applicability of the conclusions. Third, a notable limitation is the exclusion of smoking status as a covariate due to its high missing data rate (approximately 80%) in the NHANES database, which precluded reliable adjustment; future studies utilizing more comprehensive datasets are warranted to confirm our findings. Furthermore, the study did not deeply explore the clinical relevance of the findings, which limits our understanding of the specific mechanisms linking cholesterol levels with COPD. Therefore, future research should consider incorporating laboratory validation and multi-center data collection to enhance the reliability and validity of the findings. In conclusion, through a systematic analysis of NHANES data and the application of Mendelian randomization, this study reveals the causal relationship between hypercholesterolemia and COPD and identifies potential therapeutic drugs. Our findings not only provide new research directions for the prevention and treatment of COPD but also lay the foundation for future clinical trials and personalized medical strategies. As our understanding of the pathogenesis of COPD deepens, the study of transcription factors and their interaction networks will provide important biomarkers for novel targeted therapies, advancing the management of chronic diseases.
Conclusion
Through real-world research using NHANES data, we identified an association between cholesterol levels and the prevalence of COPD. Additionally, from a genetic perspective, this study suggests a potential relationship between cholesterol levels and COPD. However, further research is needed to elucidate the potential mechanisms underlying the relationship between cholesterol and COPD. Additionally, incorporating routine cholesterol monitoring into the management of COPD in clinical practice may be beneficial, particularly for patients who have quit smoking or have not responded well to non-invasive ventilation (NIV) treatment. Defining a cholesterol threshold for COPD risk stratification, along with coordinated management of cardiovascular disease and osteoporosis comorbidities in patients with COPD and hypercholesterolemia, may help address the complex interplay between heart and lung disease.
Ethical Approval
Our research is in compliance with the relevant national legislation. Specifically, the study is exempt from Institutional Review Board (IRB) approval under Article 32, Items 1 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects (February 18, 2023, China) (https://www.gov.cn/zhengce/zhengceku/2023-02/28/content_5743658.htm). Therefore, no IRB approval is required for this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
2. Theo Vos, Christine Allen, Megha Arora et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602. doi:10.1016/S0140-6736(16)31678-6
3. Bertoletti L, Couturaud F, Sanchez O, Jimenez D. Pulmonary embolism and chronic obstructive pulmonary disease. Semin Thromb Hemost. 2023;49(8):809–815. doi:10.1055/s-0042-1756190
4. Kotlyarov S. Analysis of differentially expressed genes and signaling pathways involved in atherosclerosis and chronic obstructive pulmonary disease. Biomol Concepts. 2022;13(1):34–54. doi:10.1515/bmc-2022-0001
5. Xia W, Wang H, Zhou X, et al. The role of cholesterol metabolism in tumor therapy, from bench to bed. Front Pharmacol. 2023;14:928821. doi:10.3389/fphar.2023.928821
6. Benito-Vicente A, Uribe KB, Jebari S, Galicia-Garcia U, Ostolaza H, Martin C. Familial hypercholesterolemia: the most frequent cholesterol metabolism disorder caused disease. Int J Mol Sci. 2018;19(11):3426. doi:10.3390/ijms19113426
7. Jundi B, Ahmed H, Reece J, Geraghty P. The relationship of cholesterol responses to mitochondrial dysfunction and lung inflammation in chronic obstructive pulmonary disease. Medicina. 2023;59(2). doi:10.3390/medicina59020253
8. Freyberg J, Landt EM, Afzal S, Nordestgaard BG, Dahl M. Low-density lipoprotein cholesterol and risk of COPD: copenhagen General Population Study. ERJ Open Res. 2023;9(2):00496–2022. doi:10.1183/23120541.00496-2022
9. Wang Y, Long X, Tan M, Song X. Associations of platelet to high-density lipoprotein cholesterol ratio with chronic obstructive pulmonary disease: a cross-sectional study from the US national health and nutrition examination survey. Int J Chronic Obstr. 2024;19:2321–2332. doi:10.2147/COPD.S481197
10. Zhang C, Dai W, Yang S, Wu S, Kong J. Resistance to cholesterol gallstone disease: hepatic cholesterol metabolism. J Clin Endocrinol Metab. 2024;109(4):912–923. doi:10.1210/clinem/dgad528
11. Zhang P, Lopez R, Arrigain S, Rath M, Khatri SB, Zein JG. Dietary patterns in patients with asthma and their relationship with asthma-related emergency room visits: NHANES 2005-2016. J Asthma. 2022;59(10):2051–2059. doi:10.1080/02770903.2021.1984529
12. Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. doi:10.1038/nrdp.2015.76
13. Zhong N, Wang C, Yao W, et al. Prevalence of chronic obstructive pulmonary disease in China: a large, population-based survey. Am J Respir Crit Care Med. 2007;176(8):753–760. doi:10.1164/rccm.200612-1749OC
14. Ruan W, Yan C, Zhu H, et al. Downregulated level of insulin in COPD patients during AE; role beyond glucose control? Int J Chronic Obstr. 2019;14:1559–1566. doi:10.2147/COPD.S197164
15. Ozkan-Nikitaras T, Grzesik DJ, Romano LEL, Chapple JP, King PJ, Shoulders CC. N-SREBP2 provides a mechanism for dynamic control of cellular cholesterol homeostasis. Cells. 2024;13(15). doi:10.3390/cells13151255
16. Pezzuto A, Ricci A, D’Ascanio M, et al. Short-term benefits of smoking cessation improve respiratory function and metabolism in smokers. Int J Chronic Obstr. 2023;18:2861–2865. doi:10.2147/COPD.S423148
17. Lee C, Cha Y, Bae SH, Kim YS. Association between serum high-density lipoprotein cholesterol and lung function in adults: three cross-sectional studies from US and Korea National Health and Nutrition Examination Survey. BMJ Open Resp Res. 2023;10(1):e001792. doi:10.1136/bmjresp-2023-001792
18. Kotlyarov S, Bulgakov A. Lipid metabolism disorders in the comorbid course of nonalcoholic fatty liver disease and chronic obstructive pulmonary disease. Cells. 2021;10(11):2978. doi:10.3390/cells10112978
19. Chen H, Li Z, Dong L, Wu Y, Shen H, Chen Z. Lipid metabolism in chronic obstructive pulmonary disease. Int J Chronic Obstr. 2019;14:1009–1018. doi:10.2147/COPD.S196210
20. Bauer R, Brüne B, Schmid T. Cholesterol metabolism in the regulation of inflammatory responses. Front Pharmacol. 2023;14:1121819. doi:10.3389/fphar.2023.1121819
21. Zhu WT, Li CH, Dai TT, et al. Effects of allyl isothiocyanate on the expression, function, and its mechanism of ABCA1 and ABCG1 in pulmonary of COPD rats. Int Immunopharmacol. 2021;101(Pt B):108373. doi:10.1016/j.intimp.2021.108373
22. Yuan H, Wu H, Cheng J, Xiong J. SIAH1 ubiquitination-modified HMGCR inhibits lung cancer progression and promotes drug sensitivity through cholesterol synthesis. Can Cell Inter. 2023;23(1):71. doi:10.1186/s12935-023-02914-w
23. Kotlyarov S. Participation of ABCA1 transporter in pathogenesis of chronic obstructive pulmonary disease. Int J Mol Sci. 2021;22(7):3334.
24. Shan Z, Rehm CD, Rogers G, et al. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999-2016. JAMA. 2019;322(12):1178–1187. doi:10.1001/jama.2019.13771
25. Sanderson E. Multivariable Mendelian randomization and mediation. Cold Spring Harbor Perspectives Med. 2021;11(2):a038984. doi:10.1101/cshperspect.a038984
26. Man AWC, Zhou Y, Reifenberg G, et al. Deletion of adipocyte NOS3 potentiates high-fat diet-induced hypertension and vascular remodelling via chemerin. Cardiovasc Res. 2023;119(17):2755–2769. doi:10.1093/cvr/cvad164
27. Huang P, Xiang T, Wang Q, et al. Protective effect of Xixin-Ganjiang herb pair for warming the lungs to dissolve phlegm in chronic obstructive pulmonary disease rats based on integrated network pharmacology and metabolomics. Biomed Chromatograp. 2024;38(6):e5851. doi:10.1002/bmc.5851
28. Wu CY, Cilic A, Pak O, et al. CEACAM6 as a novel therapeutic target to boost HO-1-mediated antioxidant defense in COPD. Am J Respir Crit Care Med. 2023;207(12):1576–1590. doi:10.1164/rccm.202208-1603OC
29. Zhong Y, Xu Y, Tan Y, et al. Lipidomics of the erythrocyte membrane and network pharmacology to explore the mechanism of mangiferin from Anemarrhenae rhizoma in treating type 2 diabetes mellitus rats. J Pharmaceut Biomed Anal. 2023;230:115386. doi:10.1016/j.jpba.2023.115386
30. Shih YM, Chang YJ, Cooke MS, et al. Alkylating and oxidative stresses in smoking and non-smoking patients with COPD: implications for lung carcinogenesis. Free Radic Biol Med. 2021;164:99–106. doi:10.1016/j.freeradbiomed.2020.12.442
31. Ghalali A, Alhamdan F, Upadhyay S, et al. Contrasting effects of intracellular and extracellular human PCSK9 on inflammation, lipid alteration and cell death. Commun Biol. 2024;7(1):985. doi:10.1038/s42003-024-06674-9
32. Chen Z, Shao W, Li Y, et al. Inhibition of PCSK9 prevents and alleviates cholesterol gallstones through PPARα-mediated CYP7A1 activation. Metabolism. 2024;152:155774. doi:10.1016/j.metabol.2023.155774
33. Zhou L, Ji S, Xue R, et al. Comparative analysis of Scarb1 and Cd36 in grass carp (Ctenopharyngodon idellus): implications for DHA uptake. Comp Biochem Physiol B. 2025;275:111025. doi:10.1016/j.cbpb.2024.111025
34. Higham A, Dungwa J, Jackson N, Singh D. Relationships between airway remodeling and clinical characteristics in COPD patients. Biomedicines. 2022;10(8):1992. doi:10.3390/biomedicines10081992
35. Guan Y, Liu X, Yang Z, et al. PCSK9 promotes LDLR degradation by preventing SNX17-mediated LDLR recycling. Circulation. 2025;151(21):1512–1526. doi:10.1161/CIRCULATIONAHA.124.072336
36. Azhar NAA, Chua YA, Nawawi H, Jusoh SA. Structural dynamics of LDL receptor interactions with E498A and R499G variants of PCSK9. J Mol Modeling. 2025;31(6):161. doi:10.1007/s00894-025-06380-1
37. Bitetto G, Lopez G, Ronchi D, et al. SCARB1 downregulation in adrenal insufficiency with Allgrove syndrome. Orphanet J Rare Dis. 2023;18(1):152. doi:10.1186/s13023-023-02763-w
38. Tan J, Che Y, Liu Y, et al. CELSR2 deficiency suppresses lipid accumulation in hepatocyte by impairing the UPR and elevating ROS level. FASEB J. 2021;35(10):e21908. doi:10.1096/fj.202100786RR
39. Liu A, Yu L, Li X, et al. Celsr2-mediated morphological polarization and functional phenotype of reactive astrocytes in neural repair. Glia. 2023;71(8):1985–2004. doi:10.1002/glia.24378
40. Bandesh K, Freeland K, Traurig M, et al. Pleiotropic effects of an eQTL in the CELSR2/PSRC1/SORT1 cluster that associates with LDL-C and resting metabolic rate. J Clin Endocrinol Metab. 2025;110(2):480–488. doi:10.1210/clinem/dgae498
41. Huang P, Zhao Y, Wei H, et al. Causal relationships between blood lipid levels and chronic obstructive pulmonary disease: a mendelian randomization analysis. Int J Chronic Obstr. 2025;20:83–93. doi:10.2147/COPD.S476833
42. Zhang Y, Li XH, Zhou YT, et al. The association study of Apolipoprotein E polymorphisms and chronic obstructive pulmonary disease in the Chinese population: a case-control study. Medicine. 2020;99(49):e23442. doi:10.1097/MD.0000000000023442
43. Li ZY, Wu G, Qiu C, et al. Mechanism and therapeutic strategy of hepatic TM6SF2-deficient non-alcoholic fatty liver diseases via in vivo and in vitro experiments. World J Gastroenterol. 2022;28(25):2937–2954. doi:10.3748/wjg.v28.i25.2937
44. Zhu W, Liang W, Lu H, et al. Myeloid TM6SF2 deficiency inhibits atherosclerosis. Cells. 2022;11(18):2877. doi:10.3390/cells11182877
45. Zhang Y, Xie M, Wen J, et al. Hepatic TM6SF2 activates antitumour immunity to suppress metabolic dysfunction-associated steatotic liver disease-related hepatocellular carcinoma and boosts immunotherapy. Gut. 2025;74(4):639–651. doi:10.1136/gutjnl-2024-333154
46. Choi HY, Choi S, Iatan I, Ruel I, Genest J. Biomedical advances in ABCA1 transporter: from bench to bedside. Biomedicines. 2023;11(2):561. doi:10.3390/biomedicines11020561
47. Kotlyarov S, Kotlyarova A. Bioinformatic analysis of ABCA1 gene expression in smoking and chronic obstructive pulmonary disease. Membranes. 2021;11(9):674. doi:10.3390/membranes11090674
48. Hehl L, Creasy KT, Vitali C, et al. A genome-first approach to variants in MLXIPL and their association with hepatic steatosis and plasma lipids. Hepatol Commun. 2024;8(5). doi:10.1097/HC9.0000000000000427.
49. Wang H, Cao Y, Shu L, et al. Long non-coding RNA (lncRNA) H19 induces hepatic steatosis through activating MLXIPL and mTORC1 networks in hepatocytes. J Cell Mol Med. 2020;24(2):1399–1412. doi:10.1111/jcmm.14818
50. Wang W, Mei A, Qian H, et al. The role of glucagon-like peptide-1 receptor agonists in chronic obstructive pulmonary disease. Int J Chronic Obstr. 2023;18:129–137. doi:10.2147/COPD.S393323
51. Holz O, DeLuca DS, Roepcke S, et al. Smokers with COPD show a shift in energy and nitrogen metabolism at rest and during exercise. Int J Chronic Obstr. 2020;15:1–13. doi:10.2147/COPD.S217474
52. Mistry H, Richardson CD, Higginbottom A, et al. Relationships of brain cholesterol and cholesterol biosynthetic enzymes to Alzheimer’s pathology and dementia in the CFAS population-derived neuropathology cohort. Neurosci Res. 2024;204:22–33. doi:10.1016/j.neures.2024.01.003
53. Liu J, Ma H, Meng L, et al. Construction and external validation of a ferroptosis-related gene signature of predictive value for the overall survival in bladder cancer. Front Mol Biosci. 2021;8:675651. doi:10.3389/fmolb.2021.675651
54. Xue H, Liu F, Ai Z, et al. FOXC1 downregulates Nanog expression by recruiting HDAC2 to its promoter in F9 cells treated by retinoic acid. Int J Mol Sci. 2021;22(5):2255. doi:10.3390/ijms22052255
55. Wang J, Wang Y, Zhang H, et al. Identification of a novel microRNA-141-3p/Forkhead box C1/β-catenin axis associated with rheumatoid arthritis synovial fibroblast function in vivo and in vitro. Theranostics. 2020;10(12):5412–5434. doi:10.7150/thno.45214
56. He T, Shang J, Gao C, et al. A novel SIRT6 activator ameliorates neuroinflammation and ischemic brain injury via EZH2/FOXC1 axis. Acta pharmaceutica Sinica B. 2021;11(3):708–726. doi:10.1016/j.apsb.2020.11.002
57. Sunata K, Miyata J, Kawashima Y, et al. Inflammatory profile of eosinophils in asthma-COPD overlap and eosinophilic COPD: a multi-omics study. Front Immunol. 2024;15:1445769. doi:10.3389/fimmu.2024.1445769
58. Gu Y, Bi X, Liu X, et al. Roles of ABCA1 in chronic obstructive pulmonary disease. COPD. 2025;22(1):2493701. doi:10.1080/15412555.2025.2493701
59. Balakumar P, Sambathkumar R, Mahadevan N, et al. Molecular targets of fenofibrate in the cardiovascular-renal axis: a unifying perspective of its pleiotropic benefits. Pharmacol Res. 2019;144:132–141. doi:10.1016/j.phrs.2019.03.025
60. Alhirmizi IAO, Uysal F, Arslan SO, et al. Fenofibrate attenuates asthma features in an ovalbumin-induced mouse model via suppressing NF-κB binding activity. Respir Physiol Neurobiol. 2023;314:104083. doi:10.1016/j.resp.2023.104083
61. Rojas DA, Coronado K, Pérez-Reytor D, Karahanian E. Reduction of alcohol-dependent lung pathological features in rats treated with fenofibrate. Int J Mol Sci. 2024;25(23):12814. doi:10.3390/ijms252312814
62. Liu Q, Dong S, Zhou X, et al. Effects of long-term intervention with losartan, aspirin and atorvastatin on vascular remodeling in juvenile spontaneously hypertensive rats. Molecules. 2023;28(4):1844.
63. Lu RA, Zeki AA, Ram-Mohan S, et al. Inhibiting airway smooth muscle contraction using pitavastatin: a role for the mevalonate pathway in regulating cytoskeletal proteins. Front Pharmacol. 2020;11:469. doi:10.3389/fphar.2020.00469
64. Cai G, Liu J, Cai M, Shao L. Exploring the causal effect between lipid-modifying drugs and idiopathic pulmonary fibrosis: a drug-target Mendelian randomization study. Lipids Health Dis. 2024;23(1):237. doi:10.1186/s12944-024-02218-6
65. Melo AC, Cattani-Cavalieri I, Barroso MV, et al. Atorvastatin dose-dependently promotes mouse lung repair after emphysema induced by elastase. Biomed Pharmacothe. 2018;102:160–168. doi:10.1016/j.biopha.2018.03.067
66. Schenk P, Spiel AO, Hüttinger F, et al. Can simvastatin reduce COPD exacerbations? A randomised double-blind controlled study. Europ Resp J. 2021;58(1):2001798. doi:10.1183/13993003.01798-2020
67. Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. New Engl J Med. 2014;370(23):2201–2210. doi:10.1056/NEJMoa1403086
68. Zhang W, Zhang Y, Li CW, Jones P, Wang C, Fan Y. Effect of statins on COPD: a meta-analysis of randomized controlled trials. Chest. 2017;152(6):1159–1168. doi:10.1016/j.chest.2017.08.015
69. Kim JH, Choi HG, Kwon MJ, et al. The influence of prior statin use on the prevalence and exacerbation of chronic obstructive pulmonary disease in an adult population. Front Med. 2022;9:842948. doi:10.3389/fmed.2022.842948
70. Cao C, Wu Y, Xu Z, et al. The effect of statins on chronic obstructive pulmonary disease exacerbation and mortality: a systematic review and meta-analysis of observational research. Sci Rep. 2015;5:16461. doi:10.1038/srep16461
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exploring the Causal Relationship Between Frailty and Chronic Obstructive Pulmonary Disease: Insights From Bidirectional Mendelian Randomization and Mediation Analysis
Cheng Z, Wu J, Xu C, Yan X
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:193-205
Published Date: 25 January 2025
A Bidirectional Mendelian Randomization Study Investigating the Causal Relationship Between Ankylosing Spondylitis and Chronic Obstructive Pulmonary Disease
Pan D, Dai X, Li P, Xue L
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:259-271
Published Date: 8 February 2025
Low-Density Lipoprotein Cholesterol as a Protective Factor in COPD and Implications for Statin Therapy: A Multi-Omics Genetic Epidemiology Study
Du T, Cao J, Dai Z, Xie X, Zhang G, Li Y, Chen B, Xu T, Feng J
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:2409-2422
Published Date: 14 July 2025
Observational and Genetic Evidence Reveals the Effect of Serum Lipid Levels on COPD Risk
Jia G, Guo T, Liu L, He C
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:2705-2714
Published Date: 5 August 2025
Effects of Diabetes Mellitus and Glycemic Traits on COPD and Pulmonary Function Traits: Insights From Mendelian Randomization and NHANES
Wang N, Wang G, Liu T, Ji W, Li M, Li T, Hu T, Shi Z
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:3575-3590
Published Date: 4 November 2025