Back to Journals » Journal of Blood Medicine » Volume 6
Hydroxyurea decreases hospitalizations in pediatric patients with Hb SC and Hb SB+ thalassemia
Authors Lebensburger J, Patel R, Palabindela P, Bemrich-Stolz C, Howard T, Hilliard L
Received 30 September 2015
Accepted for publication 21 October 2015
Published 15 December 2015 Volume 2015:6 Pages 285—290
DOI https://doi.org/10.2147/JBM.S97405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Jeffrey D Lebensburger, Rakeshkumar J Patel, Prasannalaxmi Palabindela, Christina J Bemrich-Stolz, Thomas H Howard, Lee M Hilliard
Division of Pediatric Hematology Oncology, University of Alabama at Birmingham, Birmingham, AL, USA
Purpose: Patients with hemoglobin SC (Hb SC) and hemoglobin SB+ (Hb SB+) thalassemia suffer from frequent hospitalizations yet strong evidence of a clinical benefit of hydroxyurea (HU) in this population is lacking. Patients with recurrent hospitalizations for pain crisis are offered HU at our institution based on small cohort data and anecdotal benefit. This study identifies outcomes from a large cohort of patients with Hb SC and SB+ thalassemia who were treated with HU for 2 years.
Materials and methods: A retrospective review was conducted of 32 patients with Hb SC and SB+ thalassemia who were treated with HU. We reviewed the number, and reasons for hospitalization in the 2 years prior to, and 2 years post-HU treatment as well as laboratory changes from baseline, over 1 year.
Results: Patients with Hb SC and SB+ thalassemia started on HU for frequent pain, had a significant reduction in hospitalizations over 2 years as compared to the 2 years prior to HU initiation (mean total hospitalizations/year: pre-HU: 1.6 vs post-HU 0.4 hospitalizations, P<0.001; mean pain hospitalizations/year: pre-HU 1.5 vs post-HU 0.3 hospitalizations, P<0.001). Patients demonstrated hematologic changes including an increase in percent fetal hemoglobin (%HbF) pre–post HU (4.5% to 7.7%, P=0.002), mean corpuscular volume (74 to 86 fL, P<0,0001), and decrease in absolute neutrophil count (5.0 to 3.2×109/L, P=0.007). Patients with higher doses of HU demonstrated the greatest reduction in hospitalizations but this was unrelated to absolute neutrophil count.
Conclusion: This cohort of patients with Hb SC and SB+ thalassemia provides additional support for using HU in patients with recurrent hospitalizations for pain. A large randomized multicenter trial of HU to reduce pain admissions should be conducted to confirm these data and provide much needed evidence based recommendations for this population.
Keywords: sickle cell disease, hydroxyurea, outcomes research, vaso-occlusive pain crisis
Introduction
While the overall morbidity and mortality of patients with hemoglobin SS (Hb SS) and hemoglobin SB0 thalassemia is greater than patients with Hb SC and Hb SB+ thalassemia, individuals with these “milder” phenotypes can suffer from their disease and require therapeutic interventions.1–4 No randomized clinical trials in Hb SC and SB+ thalassemia have been completed to guide evidence based interventions; therefore, understanding the role for therapeutic interventions including hydroxyurea (HU) for pain is limited by low quality evidence.5,6 One National Institutes of Health (NIH) funded multicenter clinical trial (CHAMPS trial, NCT00532883) attempted to evaluate the role of HU and magnesium, in patients with Hb SC. Unfortunately, this critical trial was terminated early due to lack of enrollment.7 This randomized trial, along with several smaller cohort studies in patients with Hb SC, suggest that HU will increase the mean corpuscular volume (MCV) and % HbF while lowering the reticulocyte count.8–12 One prior cohort study of 15 patients demonstrated a potential clinical benefit in pain events per year in patients with Hb SC, but this finding was not replicated in the 38 randomized subjects in CHAMPS as no difference among treatment groups in the 15 serious adverse events and 111 adverse events for pain crisis was identified.7,13
Without stronger evidence based guidelines to direct management of patients with Hb SC and SB+ thalassemia, our institution offers HU to patients with Hb SC and SB+ thalassemia who suffer from multiple pain events. We discuss the risks and benefits of HU including the basis for offering HU based on anecdotal and small cohort data sets. With this dearth of literature in the field, we performed a retrospective review of our pediatric cohort of patients with Hb SC and SB+ thalassemia to attempt to confirm prior hematologic changes in this population and provide further evidence that HU may clinically benefit patients suffering from frequent hospitalizations for pain.
Materials and methods
With approval from the Institutional Review Board of the University of Alabama at Birmingham, we conducted a retrospective review of patients with Hb SC and SB+ thalassemia prescribed HU. We followed the principles outlined in the Declaration of Helsinki in the conduct of this research. As some patients prescribed HU were not adherent to their medication, we included in this review only patients on HU for a minimum of 2 years. Patients admitted to the hospital at Children’s of Alabama/University of Alabama at Birmingham, (COA/UAB) had full chart reviews of their hospitalization records while UAB satellite clinic patients (UAB satellite clinics conducted in Montgomery, Tuscaloosa, Huntsville, and Opelika, AL) had their outpatient charts reviewed for parent/patient report of hospitalization episodes and reasons for admission. We recorded the number and reasons for hospitalizations in the 2 years preceding and 2 years post-initiation of HU. We recorded and analyzed the laboratory results at baseline and the changes in laboratory results and dose of HU (mg/kg) at 1, 6, and 12 months of treatment. Data collected included Hb, hematocrit (Hct), MCV, white blood cell count (WBC), absolute neutrophil count (ANC), platelet count (PLT), absolute reticulocyte count, HbF%, lactate dehydrogenase (LDH), aspartate aminotransferase (AST), and total bilirubin.
Descriptive statistics were recorded and univariate analysis was performed using Student’s t-tests for continuous variables. Paired differences were compared at baseline and 12 months of treatment for laboratory data as well as the number of hospitalizations (overall and for pain). Wilcoxon-signed rank test was performed for paired difference for not normally distributed variables and Student’s t-test for paired mean for normally distributed variables. All patient data were analyzed using JMP10 software (SAS Institute Inc., Cary, NC, USA). Sample size calculations were completed using G*power data analysis (http://www.ats.ucla.edu/stat/gpower/indepsamps.htm).
Results
We identified 34 patients started on HU and 32 patients were evaluable based on consistent laboratory follow-up prior to and after starting HU. The mean age for starting HU in this cohort was 9.4±4.5 years without a difference in starting age by genotype (9.2 years in Hb SC vs 9.9 in Hb SB+ thalassemia). Thirty patients had matched data for pre- and post-HU hospitalizations; eleven patients were cared for in COA/UAB and 19 in UAB satellite clinics. Prior to starting HU, we identified no difference between the clinics (COA/UAB vs UAB satellite clinics) in the number of overall hospitalizations (1.8 vs 1.5 hospitalizations, P=0.46) and pain hospitalizations (1.2 vs 1.3 hospitalizations, P=0.47). As a cohort, these patients had significant reductions in total (1.6 vs 0.4 hospitalizations/year, P<0.001) and pain hospitalizations (1.5 vs 0.3 hospitalizations/year, P<0.001) after starting HU (Table 1). This benefit was also noted by genotype as patients with Hb SC demonstrated a reduction in total hospitalizations (1.8 vs 0.4 hospitalizations/year P<0.0001) and hospitalizations for pain (1.6 vs 0.3 hospitalizations/year P<0.001). Only seven patients with SB+ thalassemia had matched data which limits conclusions, although, in a larger sample, it could be expected to demonstrate marked reduction in overall hospitalizations (1.0 vs 0.1 hospitalizations/year, P=0.06) and pain hospitalizations (1.0 vs 0.07 hospitalizations/year, P=0.06) (Table 1).
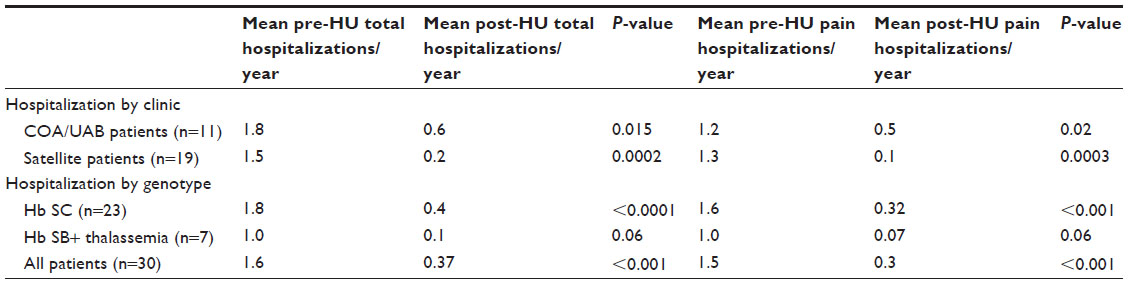 | Table 1 Hospitalizations per year prior to and after starting hydroxyurea (HU) |
Several laboratory markers were significantly altered for patients with Hb SC, Hb SB+ thalassemia, and the HU cohort as a whole. All groups with matched laboratory pairs had a statistical reduction in WBC (from 10 to 7.2×109/L, P=0.0003), ANC (from 5.0 to 3.2×109/L, P=0.007), and PLTs (from 344 to 282×109/L, P<0.0001) and an increase in MCV over 1 year (from 73.7 to 86.1 fL, P<0.001) (Table 2). HbF% was higher after 1 year in patients with Hb SC and the cohort as a whole but not statistically different in the small group of patients with Hb SB+ thalassemia (7 vs 9.5%, n=4, P=0.26) No differences were noted after 1 year in Hb, Hct, absolute reticulocyte count, LDH, bilirubin, or AST.
 | Table 2 Laboratory changes prior to and after starting hydroxyurea (HU) |
For the entire group, no significant difference was noted between mean starting dose and 1 year dose of HU (21.7±2.7 vs 22.3 ±3.3 mg/kg, P=0.3). At 1 year, four patients were receiving <20 mg/kg/day dosing and nine patients had been escalated to ≥25 mg/kg. The only significant association between dose and blood indices was that, at 1 year, patients receiving lower doses of HU also maintained a lower PLT (P=0.03). Clinically, an increased dose of HU at 1 year was significantly correlated with a larger decrease in number of total hospitalizations (P=0.03) and hospitalizations for pain over 2 years (P=0.04).
Discussion
The reduction in hospitalizations in this cohort provides clinical support for the use of HU in patients with Hb SC and SB+ thalassemia who suffer from vaso-occlusive crisis. Our data support the findings of one other cohort for patients with only Hb SC that demonstrated a decrease in hospitalizations for pain.13 The CHAMPS trial did not identify a difference in serious adverse events for patients with Hb SC, only 22 participants completed the full 44 weeks.7 Our cohort of 23 non-randomized patients with Hb SC followed for 2 years demonstrated a marked reduction in hospitalizations overall (almost two hospitalizations/year to 0.5/year) and for pain (1.6 pain hospitalizations/year to 0.4/year). Additionally, our data demonstrate that HU can increase MCV and HbF% as noted in prior studies of patients with Hb SC.7,13 Clinical researchers could consider designing a future randomized placebo vs HU trial with a primary objective to reduce hospitalizations for pain based on our results of decreasing from two hospitalizations/year to 0.5 hospitalizations/year. Assuming an alpha of 0.05, power of 0.8, and a 1:1 randomization with a standard deviation identified from this cohort of 2.35, 80 total participants would be needed to complete the study. Using a 50% reduction in hospitalizations for pain/year (two to one pain hospitalization) would require 170 participants to complete the study. These data also provide additional evidence that consistent hematologic changes occur when patients take HU, such as lower WBC, ANC, and PLTs and increase in HbF and MCV; yet HU again did not increase the Hb.13
The dosing strategy for HU in Hb SC and SB+ thalassemia is poorly understood. The only current randomized trial of HU vs placebo in young children with Hb SS and SB0 thalassemia used standard dosing of 20 mg/kg rather than dose titration based on a goal ANC and monitoring for hematologic toxicity.14,15 However, recent NIH clinical trials have incorporated dose titration of HU into trial design (Stroke with Transfusion Changing to Hydroxyurea, NCT 00122980; Transcranial Doppler with Transfusion Changing to Hydroxyurea, NCT 01425307; and Hydroxyurea to Prevent Brain Injury in Sickle Cell Disease, NCT 01389024). The mean starting dose of HU in this cohort was 21.7±2.4 mg/kg and the 1-year dose was 22.7±3.2 mg/kg. While no significant difference was noted in the dose, patients who were titrated to a higher dose experienced a significant decrease in total number of hospitalizations overall and fewer hospitalizations for pain than patients receiving lower doses of HU. This observed reduction in hospitalizations was not significantly associated with a lower ANC as noted in the multicenter study of HU.16 However, it may be of interest that our cohort of patients with Hb SC selected for multiple admissions had a higher mean WBC and ANC at baseline than patients enrolled in the CHAMPS study or other large cohort studies.1,7 Future randomized studies of HU aimed at reducing hospitalizations in this patient population should consider developing a plan for dose titration but understand that the potential benefit of titration may not be correlated with a decrease in ANC as noted in Hb SS patients.
Some patients in this cohort required dose reduction to <20 mg/kg. Patients requiring these lower doses were also identified as having significantly lower PLTs. Natural history studies have demonstrated that Hb SC patients develop progressively larger spleens which can contribute to “hypersplenism” as noted by mild thrombocytopenia and spleen dysfunction.17–19 In addition, HU may induce splenic regrowth in patients with Hb SC which may exacerbate this thrombocytopenia, limit dosing of HU, and increase risk of splenic complications.19–22 Patients with Hb SB+ thalassemia may also suffer from dysfunctional splenic function but this research is limited.23 This retrospective cohort did not demonstrate an association between splenomegaly noted on clinical exam with thrombocytopenia and HU dose. However, future studies of HU in this population should include a more rigorous method for monitoring splenic function and size. An additional concern for HU use in this population is the potential for promoting hyperviscosity in a population at risk for retinopathy.1,2,24 This cohort did not demonstrate a significant increase in Hb/Hct, potential surrogate markers of hyperviscosity. More rigorous methods of monitoring retinopathy should be evaluated in a future prospective study to evaluate the effect of HU on retinopathy in this population.
This study has some limitations worth noting. First, patients in this cohort included patients from the UAB satellite clinics as well as the COA/UAB Sickle Cell clinic in Birmingham. All patients who were admitted to COA/UAB underwent rigorous chart review of their hospitalization for this study. Patients who attend satellite clinics may be admitted to a local hospital and we rely on patient/parent report of hospitalizations as we may not receive official records from these outside hospitals. Therefore, we reported data in Table 1 for the confirmed patients from Birmingham clinic independently from patients who attended satellite clinics. While we did not identify a difference in hospitalization rate by clinic, the reliance on parent/patient report at UAB satellite clinics may be subject to recall bias. A second limitation of this study is that standard of care at our institution does not include strict measures for monitoring adherence. We hoped to minimize this limitation by evaluating hematologic changes at 1 year to exclude patients who admitted non-adherence and desired not to continue therapy within 1 year after HU initiation. Third, patients with Hb SC and SB+ thalassemia on HU are monitored in clinic more frequently than patients not on HU. Some reduction in hospitalization may be the result of closer follow-up with health care providers rather than medication effect.
Conclusion
This manuscript further identifies the hematologic changes that occur when patients with Hb SC and SB+ thalassemia are prescribed HU. In addition, these data support a clinical endpoint, reduction in hospitalizations for pain that could be used in a multicenter clinical trial. An obvious concern for conducting trials in Hb SC and SB+ thalassemia is that a recent NIH trial of HU in this population has already been closed for poor enrollment. Therefore, prior to conducting another clinical trial in this population, clinical trialists must develop a plan to identify barriers and facilitators to enrollment so that this future study can be designed to meet the parent’s and patient’s expectations for research as well as the scientific community’s.25 Scientific goals of an HU trial should include an overall goal to reduce hospitalizations but also aims to monitor splenic function and size as well as ocular manifestations. The morbidity and mortality associated with Hb SC and SB+ thalassemia and lack of any other proven therapy mandates the need for another clinical trial of HU in this population.
Acknowledgments
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under the award number UL1TR001417. The authors would like to acknowledge the assistance of Peng Li, PhD for providing biostatistical support through the UAB CCTS consultative services. The authors would like to thank the patients and parents in this study. Finally the authors would like to thank the Sickle Cell Nurse Practitioners (Kristen Osborn, Melissa Bagwell, Heather Carlton, Susan Dobbins, and Michelle Alleman) for providing excellent care to our patients.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflicts of interest to disclose.
References
Gualandro SF, Fonseca GH, Yokomizo IK, Gualandro DM, Suganuma LM. Cohort study of adult patients with haemoglobin SC disease: clinical characteristics and predictors of mortality. Br J Haematol. Epub 2015 Aug 10. | |
Serjeant GR, Ashcroft MT, Serjeant BE. The clinical features of haemoglobin SC disease in Jamaica. Br J Haematol. 1973;24(4):491–501. | |
Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. | |
Ballas SK, Lewis CN, Noone AM, Krasnow SH, Kamarulzaman E, Burka ER. Clinical, hematological, and biochemical features of Hb SC disease. Am J Hematol. 1982;13(1):37–51. | |
Lebensburger JD, Hilliard LM, Pair LE, Oster R, Howard TH, Cutter GR. Systematic review of interventional sickle cell trials registered in ClinicalTrials.gov. Clin Trials. Epub 2015 Jun 17. | |
Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. | |
Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol. 2011;152(6):771–776. | |
Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol. 1997;98(4):838–844. | |
Rogers ZR. Hydroxyurea therapy for diverse pediatric populations with sickle cell disease. Semin Hematol. 1997;34(3 Suppl 3):42–47. | |
Miller MK, Zimmerman SA, Schultz WH, Ware RE. Hydroxyurea therapy for pediatric patients with hemoglobin SC disease. J Pediatr Hematol Oncol. 2001;23(5):306–308. | |
Iyer R, Baliga R, Nagel RL, et al. Maximum urine concentrating ability in children with Hb SC disease: effects of hydroxyurea. Am J Hematol. 2000;64(1):47–52. | |
Loukopoulos D, Voskaridou E, Kalotychou V, Schina M, Loutradi A, Theodoropoulos I. Reduction of the clinical severity of sickle cell/beta-thalassemia with hydroxyurea: the experience of a single center in Greece. Blood Cells Mol Dis. 2000;26(5):453–466. | |
Yates AM, Dedeken L, Smeltzer MP, Lebensburger JD, Wang WC, Robitaille N. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer. 2013;60(2):323–325. | |
Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115(26):5300–5311. | |
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–1672. | |
Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol. 1997;34(3 Suppl 3):15–21. | |
Subbannan K, Ustun C, Natarajan K, et al. Acute splenic complications and implications of splenectomy in hemoglobin SC disease. Eur J Haematol. 2009;83(3):258–260. | |
Sills RH. Splenic function in children with hemoglobin SC disease and sickle beta-thalassemia. J Natl Med Assoc. 1983;75(10):991–994. | |
Aquino VM, Norvell JM, Buchanan GR. Acute splenic complications in children with sickle cell-hemoglobin C disease. J Pediatr. 1997; 130(6):961–965. | |
Huang Y, Ananthakrishnan T, Eid JE. Hydroxyurea-induced splenic regrowth in an adult patient with severe hemoglobin SC disease. Am J Hematol. 2003;74(2):125–126. | |
Koduri PR, Kovarik P. Acute splenic sequestration crisis in an adult with sickle beta-thalassemia. Ann Hematol. 2006;85(9):633–635. | |
Claster S, Vichinsky E. First report of reversal of organ dysfunction in sickle cell anemia by the use of hydroxyurea: splenic regeneration. Blood. 1996;88(6):1951–1953. | |
Barrios NJ, Kirkpatrick DV, Lohman D, McMullen CC, Wilson W, Humbert JR. Spleen function in children with sickle B+ thalassemia. J Natl Med Assoc. 1991;83(9):819–822. | |
Condon PI, Serjeant GR. Ocular findings in hemoglobin SC disease in Jamaica. Am J Ophthalmol. 1972;74(5):921–931. | |
Lebensburger JD, Sidonio RF, Debaun MR, Safford MM, Howard TH, Scarinci IC. Exploring barriers and facilitators to clinical trial enrollment in the context of sickle cell anemia and hydroxyurea. Pediatr Blood Cancer. 2013;60(8):1333–1337. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.