Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Hydroxychloroquine Ameliorates Hematuria in Children with X-Linked Alport Syndrome: Retrospective Case Series Study
Authors Sun L, Kuang XY, Zhang J, Huang WY ![]()
Received 25 October 2022
Accepted for publication 17 February 2023
Published 25 February 2023 Volume 2023:16 Pages 145—151
DOI https://doi.org/10.2147/PGPM.S394290
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Lei Sun, Xin-Yu Kuang, Jing Zhang, Wen-Yan Huang
Department of Nephrology and Rheumatology, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
Correspondence: Wen-Yan Huang, Email [email protected]
Purpose: As a rare collagen type IV hereditary kidney disease, X-linked Alport syndrome (XLAS) is the most common form of Alport syndrome, the prevalence of which is estimated at 1:10,000 of the population, four times higher than the prevalence rate of autosomal recessive Alport syndrome. To describe a series of eight XLAS children with persistent hematuria and proteinuria and the clinical outcomes after hydroxychloroquine (HCQ) treatment to assess its efficacy as early intervention.
Patients and Methods: The study retrospectively analysed 8 patients with persistent hematuria and proteinuria at different onset ages who were diagnosed with XLAS and been treated with HCQ. The urinary erythrocyte count, urinary albuminn were measured. Descriptive statistics were used to estimate the patients’ responses to HCQ treatment after one month, three months, and six months.
Results: After the first month, the three months, and the six months of HCQ treatment, the urinary erythrocyte counts of four, seven, and eight children were significantly reduced; the decreasing proteinuria was found in two, four, and five children. Only one child with increasing proteinuria was found after 1-month HCQ treatment. This proteinuria was maintained after 3-month HCQ treatment but decreased to minor after 6-month HCQ treatment.
Conclusion: We present the first potential efficacy of HCQ treatment in XLAS with hematuria and persistent proteinuria. It suggested that HCQ could be an effective treatment to ameliorate hematuria and proteinuria.
Keywords: X-linked Alport syndrome, hematuria, proteinuria, hydroxychloroquine, therapy
Introduction
A triad of symptoms including hemorrhagic nephritis, deafness, and ocular changes, first reported by Arthur C. Alport in 1927, was named Alport syndrome.1 As a rare hereditary disease, its incidence was reported 1/5000–1/50,000.2 Among the three genetic forms of Alport syndrome (X-linked form, autosomal recessive form, and autosomal dominant form), X-linked Alport syndrome (XLAS) is the most common affecting 80%-85% of Alport syndrome patients. XLAS is a rare hereditary nephritis caused by mutations in the COL4A5 gene encoding the type IV collagen α5 chain.3 These mutations result in the reduction of α5 chain in glomerular basement membranes.4 The prevalence of XLAS is estimated at around 1:10,000 of the population, a much higher prevalence rate than autosomal recessive and autosomal dominant forms.2
The mutations of COL4A5 gene cause the dysfunction of the glomerular filter; consequently, XLAS begins with isolated microscopic hematuria and progresses to moderate albuminuria and severe proteinuria gradually. Patients were eventually progress to renal failure as their illness progresses. The kidney failure risks for males with XLAS can be as high as 50%, 90%, and nearly 100% by age 25, 40, and 60.5
To date, the treatment of XLAS and the delay in progression to kidney failure has been full of challenges. Although interventions such as angiotensin-converting-enzyme (ACE) inhibitors, AT1-receptor antagonists, and conventional renin-angiotensin-aldosterone system (RAAS) inhibitor could delay the onset of proteinuria and kidney failure, there are no curative therapies for AS.6–9 In addition, the feasibility of gene therapy is uncertain.10 Exploration of new therapies for this common cause of kidney failure in children is a priority.
Hydroxychloroquine (HCQ), a well-known immunomodulator approved for treating systemic lupus erythematosus and rheumatoid arthritis,3 may play an important role in XLAS treatment. The treatment using HCQ in the patients with XLAS was inspired by a 17-year-old female who has 7-year history continued to present macroscopic hematuria and proteinuria (Patient No.1). As it happened, she had biopsies demonstrating glomerular IgA deposits (++), for which she was receiving HCQ. Surprisingly, therapy with HCQ produced a dramatic response to the patient’s renal status. After the first month of treatment, the macroscopic hematuria disappeared and stabilized after 6-month treatment. Meanwhile, the proteinuria characterized by the urinary albumin characterization index diminished from one plus protein (+ P) to negative (–) after 3-month treatment with HCQ.
Recently, HCQ has been studied in immunoglobulin A nephropathy (IgAN).11 A randomized controlled trial found that HCQ with optimized RAAS inhibition significantly reduced proteinuria in patients with IgAN over six months without adverse events.12 Similar to IgAN, inflammatory and fibrosis also play an important role in AS progression. HCQ may exert a therapeutic effect through its anti-inflammatory and immunomodulatory effects.This study describes a series of eight XLAS children with persistent hematuria and proteinuria at different onset ages and the clinical outcomes after they were admitted to the HCQ treatment at different treatment ages. The efficacy of HCQ as an early treatment intervention for XLAS disease was explored.
Materials and Methods
The study retrospectively analyzed 8 patients with persistent hematuria and proteinuria at different onset ages who were diagnosed with XLAS and been treated with HCQ. The inclusion criteria were listed the following: the children aged 0–18 years that confirmed XLAS; the patient have a persistent hematuria and proteinuria condition and were received HCQ treatment. Exclusion criteria: the duration of HCQ treatment was less than 6 months.
After obtaining approval from the ethics committee of Shanghai Children’s Hospital (No. 2019R063-E02) and written informed parental consent, all eight XLAS children with hematuria and proteinuria (five males and three females) were treated with HCQ from January 2019 to June 2021. At the time of research enrollment, the children had followed up and received Benazepril/Valsartan over 6 months. Demographics and clinical information including sex, the disease onset age, inclusion age for starting HCQ treatment, age for the onset of microalbuminuria, age for the progression from microalbuminuria to proteinuria, the family history, and hearing and ocular abnormality were retrospectively collected. Renal pathology data were also acquired in patients. For children with persistent hematuria and proteinuria of unknown etiology, a renal puncture biopsy was recommended. Given the nature of COL4A5 gene mutations in XLAS patients and the strong correlation between the age at kidney failure and COL4A5 genotype in males with XLAS,9 data on gene mutations were collected for the assistance of disease diagnosis and genotype-phenotype correlation analysis.
Diagnoses of XLAS were confirmed by the combination of pathologists’ interpretations and consultation with pediatric nephrologists in Shanghai Children’s Hospital. Alport syndrome was first suspected when there was persistent glomerular hematuria. The likelihood increased with a family history of Alport syndrome or renal failure; or when the characteristic clinical features (hearing loss, lenticonus, or retinopathy) were present. The diagnosis of XLAS was confirmed if the glomerular basement membrane (GBM) was lacked of the collagen IV a5 chains, if this chain was absent from skin, or a pathogenic mutation in COL4A5 was identified. All the exons and exon-intron boundaries were analyzed using next-generation sequencing in six patients. There were five missense mutations and one deletion mutation in gene COL4A5 (Table 1). Their renal function was normal. All were previously healthy, and HCQ medications had not been used.
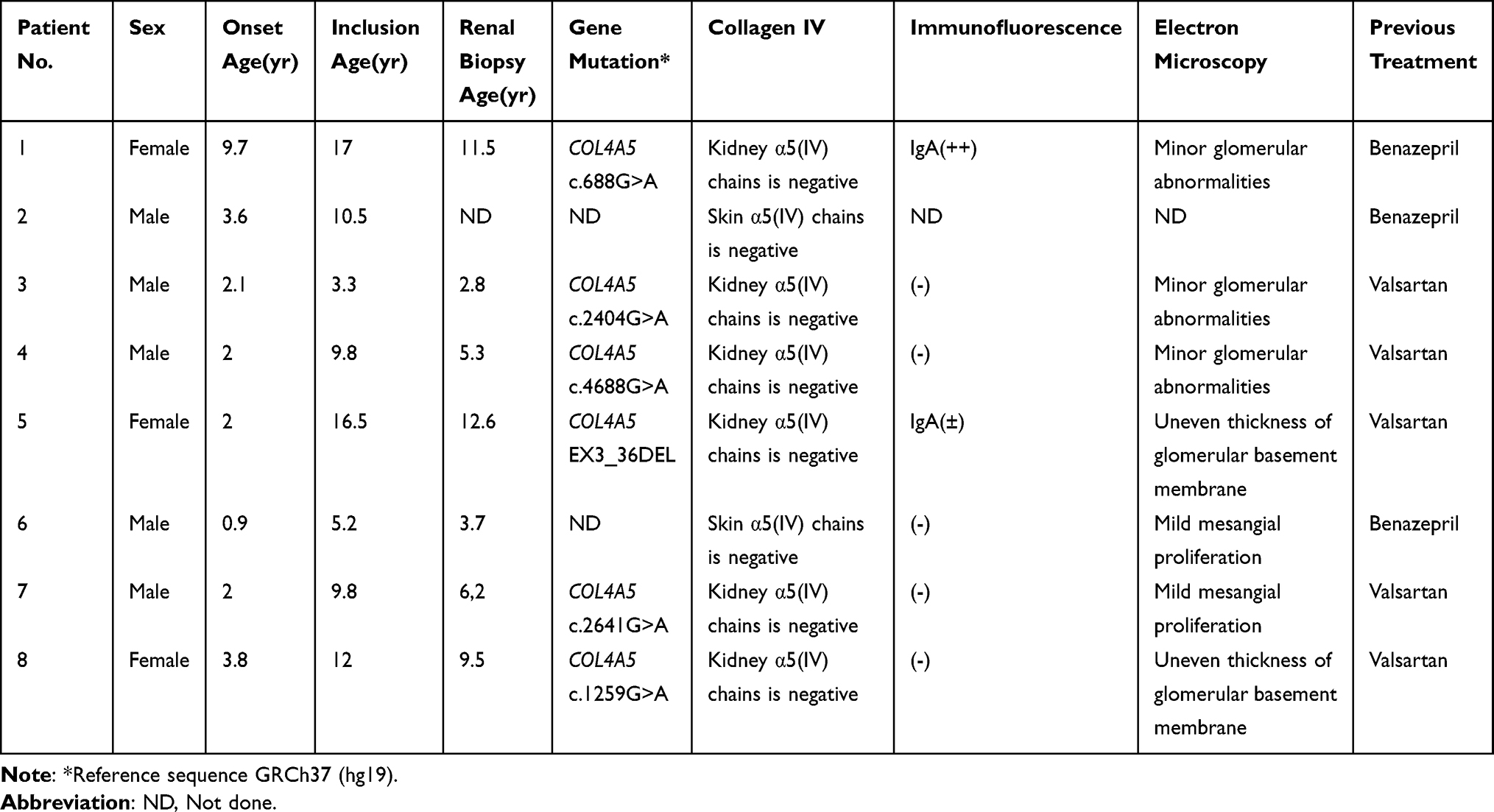 |
Table 1 Characteristics of the Eight Patients |
All the patients with XLAS were treated with HCQ orally at a dose of 6.5mg per kilogram twice a day for at least six months. Patients No.1, No.2, and No.6 were previously treated by Benazepril and the other patients were previously treated by Valsartan. During treatment with HCQ, all patients received Benazepril (2–4mg/m2. d). The dosage of Benazepril remains unchanged during 6-month HCQ treatment. Variation in hematuria was recorded by collecting the urinary erythrocyte count of first morning urine after the treatments. Proteinuria was tested by urinary albumin characterization of first morning urine. We recommend that patients come to our outpatient for first morning urine testing every 1 month. All the patients avoided urine testing during infection condition.
The severity of hematuria was graded into three categories according to the urinary erythrocyte count (> 100/High power Field (HP), 50–100/HP, < 50/HP). The severity of proteinuria was graded into six categories based on urinary albumin characterization: -(negative, less than 10 mg per dL), ± (trace, 10 to 20 mg per dL), + P (30 mg per dL), ++P(100 mg per dL), +++ P (300 mg per dL), ++++ P (1000 mg per dL). The intervention results at one month, three months, and six months during the HCQ treatment were retrospectively obtained. The liver and kidney function and ophthalmic examination during the HCQ treatment were obtained for monitoring adverse effects following HCQ therapy. Descriptive statistics were used to characterize the variations of the hematuria and proteinuria with the elapsed time after HCQ treatment. The severity of hematuria and proteinuria in the eight patients were evaluated by Chi-square test.
Results
Study Participants
The clinical characteristics and pedigree data of eight cases we included were summarized in Table 1 and Supplementary Table 1. Patients’ onset ages ranged from 0.9 to 9.7 years, and their inclusion ages ranged from 3.3 to 17. In all cases, the renal condition was manifested as different degrees of proteinuria and hematuria. Patients No.1 and No.3 showed macroscopic hematuria with minor glomerular abnormalities, and the other six patients showed microscopic hematuria. Apart from Patient No.2, all other patients had glomerular abnormalities or mesangial proliferation.
Patient Outcome
After the first month of HCQ treatment, the macroscopic hematuria of both patients (Patient No.1, No.3) was recovered. Also, the urinary erythrocyte counts of four patients (Patient No.1, No.2, No.3, No.4) were all reduced (Table 2) and maintained at 6 months follow-up. The urinary erythrocyte counts of the other four patients (Patient No.5, No.6, No.7, No.8) were then generally reduced at 3 months follow-up and continued to decrease at 6 months follow-up (Table 2). Patients with decreasing urinary erythrocyte count increased with the elapsed time after HCQ treatment from 4 to 8, as shown in Figure 1. This result is also illustrated in Figure 2. The severity of hematuria in the eight patients significantly decreased with the elapsed treatment time (p<0.05). Meanwhile, Figure 1 also shows that the number of patients with decreased proteinuria increased with the elapsed time after HCQ treatment from 2 to 5, though there is one patient with increasing proteinuria.
 |
Table 2 The Responses of the Eight Patients to HCQ |
 |
Figure 1 The varied number of patients with the elapsed time after HCQ treatment. |
 |
Figure 2 The hematuria changes of the eight patients to HCQ therapy (p<0.05). The severity of hematuria was categorised into three stages according to the urinary erythrocyte count (> 100/HP, 50–100/HP, < 50/HP). All patients maintained < 50/HP after the 6-month treatment of HCQ. |
All patients’ biochemical indicators of liver and kidney function were normal during the 24 weeks HCQ treatment. There was no significant difference in the change of eGFR. The patients’ ophthalmic examination and vision were normal after HCQ therapy. None of the patients had severe adverse events during treatment.
Discussion
In this series of eight XLAS children with different degrees of proteinuria and hematuria, the hematuria of the eight XLAS patients was significantly relieved following HCQ treatment. The proteinuria of the five XLAS patients was decreased after the 6-month HCQ treatment, other three patients had stable protein amount. There were limitations in the accuracy of urine protein characterization which may affect the statistics of the proteinuria level. In addition, insufficient sample size can also affect the statistical significance.
At present, no cure has been identified available for XLAS.13,14 RAAS inhibitor therapy only slows down the development of clinical symptoms.9 Now, many new drugs have been used in clinical studies to treat Alport syndrome (AS). Bardoxolone Methyl and lademirsen are the most concerned new drugs in recent years.13 Bardoxolone Methyl, an Nrf2 activator, which used as an antioxidant inflammation modulator in the CARDINAL Trial (NCT03019185).15 The completed Phase 2 clinical trial showed that Bardoxolone Methyl could increase the estimated glomerular filtration rate (eGFR) in patients with AS.16 Bardoxolone may be attractive for AS patients with declining GFR. Nevertheless, Bardoxolone Methyl increased the level of albuminuria, which is strongly positively associated with an increased risk of progression to kidney failure.16 For young AS patients with normal GFR, Bardoxolone Methyl seems undesirable.17 Lademirsen (SAR339375), which was used in the HERA trial (NCT02855268), can modify the microRNA-21 expression in the kidney. The microRNA-21 has been participated in regulating inflammatory responses and fibrotic. Since the decline of GFR was closely associated with increased interstitial fibrosis and tubular atrophy in AS patients. Anti-fibrotic and anti-inflammatory therapies such as lademirsen may benefit younger AS patients.18,19
Similarly, HCQ has anti-inflammatory and immunomodulatory effects. Several studies have demonstrated that HCQ reduces the production of cytokines, including TNF-α, IL-6, IFN-α.20,21 It is possible that the inhibition of TNF-α, IL-6, and IFN-α expression alleviates the inflammation response resulting from mesangial cell activation. The mesangial cell inflammatory response may contribute to subsequent podocyte and tubular injury, and HCQ could relieve the injury by inhibiting the inflammatory response. However, its exact role in linking inflammation and XLAS has remained unknown. Further research is required with a large number of cases to explore the linkage between inflammation and XLAS.
A wide range of side effects such as cardiovascular, dermatological, digestive, hematological, metabolic, ophthalmologic were reported to be associated with HCQ use.22 The occurrence of side effects was related to the duration and dosage of HCQ use. The daily dose associated with the best compromise between efficacy and safety is important. In our research, low doses for short-term use were safe to children apply.
Conclusion
In this case series, we investigated the efficacy and safety of combining HCQ and RAAS inhibition in XLAS with hematuria and persistent proteinuria for eight patients. Overall, HCQ may be a potential therapeutic option to ameliorate hematuria and proteinuria for XLAS. The XLAS patients also showed good tolerance and compliance with a combination of HCQ and RAAS inhibition therapy. The limitations of our study were that it was retrospective and the small sample size. Further studies with a high evidence-based medicine level were necessary to verify these observations. We have registered a randomized controlled trial at ClinicalTrials.gov (NCT04937907) to systematically explore the role of HCQ in treating children with XLAS.
Declaration
This study complies with the Declaration of Helsinki.
Consent to Publish
The participants have consented to the submission of the study to the journal.
Acknowledgments
All phases of this study were supported by the Shanghai municipal commission of science and technology (grant no. 19ZR1442300) and Shanghai Children’s Hospital (grant no. 2021YQ04, 2021YGZQ06). We are grateful for the support from the patients and their family members.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. 1927;1(3454):504–506. doi:10.1136/bmj.1.3454.504
2. Hertz JM, Thomassen M, Storey H, Flinter F. Clinical utility gene card for: Alport syndrome. Eur J Hum Genet. 2012;20:6. doi:10.1038/ejhg.2011.237
3. Hertz JM, Thomassen M, Storey H, Flinter F. Clinical utility gene card for: Alport syndrome - update 2014. Eur J Hum Genet. 2015;23:9. doi:10.1038/ejhg.2014.254
4. Wongtrakul P, Shayakul C, Parichatikanond P, et al. Immunohistochemical study for the diagnosis of Alport’s syndrome. J Med Assoc Thai. 2006;89(Suppl 5):S171–81.
5. Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649–657. doi:10.1681/asn.V114649
6. Grodecki KM, Gains MJ, Baumal R, et al. Treatment of X-linked hereditary nephritis in Samoyed dogs with angiotensin converting enzyme (ACE) inhibitor. J Comp Pathol. 1997;117(3):209–225. doi:10.1016/s0021-9975(97)80016-3
7. Gross O, Beirowski B, Koepke ML, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. doi:10.1046/j.1523-1755.2003.00779.x
8. Gross O, Licht C, Anders HJ, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81(5):494–501. doi:10.1038/ki.2011.407
9. Gross O, Tönshoff B, Weber LT, et al. A multicenter, randomized, placebo-controlled, double-blind Phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020;97(6):1275–1286. doi:10.1016/j.kint.2019.12.015
10. Daga S, Donati F, Capitani K, et al. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur J Hum Genet. 2020;28(4):480–490. doi:10.1038/s41431-019-0537-8
11. Yang YZ, Liu LJ, Shi SF, et al. Effects of Hydroxychloroquine on Proteinuria in Immunoglobulin A Nephropathy. Am J Nephrol. 2018;47(3):145–152. doi:10.1159/000487330
12. Liu LJ, Yang YZ, Shi SF, et al. Effects of Hydroxychloroquine on Proteinuria in IgA Nephropathy: a Randomized Controlled Trial. Am J Kidney Dis. 2019;74(1):15–22. doi:10.1053/j.ajkd.2019.01.026
13. Kashtan CE. Alport Syndrome: achieving Early Diagnosis and Treatment. Am J Kidney Dis. 2021;77(2):272–279. doi:10.1053/j.ajkd.2020.03.026
14. Kruegel J, Rubel D, Gross O. Alport syndrome--insights from basic and clinical research. Nat Rev Nephrol. 2013;9(3):170–178. doi:10.1038/nrneph.2012.259
15. Chertow GM, Appel GB, Andreoli S, et al. Study Design and Baseline Characteristics of the CARDINAL Trial: a Phase 3 Study of Bardoxolone Methyl in Patients with Alport Syndrome. Am J Nephrol. 2021;52(3):180–189. doi:10.1159/000513777
16. Baigent C, Lennon R. Should We Increase GFR with Bardoxolone in Alport Syndrome? J Am Soc Nephrol. 2018;29(2):357–359. doi:10.1681/asn.2017101062
17. de Zeeuw D, Akizawa T, Audhya P, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013;369(26):2492–2503. doi:10.1056/NEJMoa1306033
18. Gomez IG, MacKenna DA, Johnson BG, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125(1):141–156. doi:10.1172/jci75852
19. Guo J, Song W, Boulanger J, et al. Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome. Hum Gene Ther. 2019;30(7):865–881. doi:10.1089/hum.2018.205
20. van den Borne BE, Dijkmans BA, de Rooij HH, le Cessie S, Verweij CL. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol. 1997;24(1):55–60.
21. Willis R, Seif AM, McGwin G et al. Effect of hydroxychloroquine treatment on pro-inflammatory cytokines and disease activity in SLE patients: data from LUMINA (LXXV), a multiethnic US cohort. Lupus. 2012;21(8):830–835. doi:10.1177/0961203312437270
22. Fiehn C, Ness T, Weseloh C, et al. Safety management in treatment with antimalarials in rheumatology. Interdisciplinary recommendations on the basis of a systematic literature review. Z Rheumatol. 2021;80(Suppl1):1–9. doi:10.1007/s00393-020-00785-4
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.