Back to Journals » Drug Design, Development and Therapy » Volume 18
Hydroxy-Safflower Yellow A Mitigates Vascular Remodeling in Rat Pulmonary Arterial Hypertension
Authors Ji XY, Lei CJ, Kong S, Li HF, Pan SY, Chen YJ, Zhao FR, Zhu TT
Received 9 October 2023
Accepted for publication 13 February 2024
Published 20 February 2024 Volume 2024:18 Pages 475—491
DOI https://doi.org/10.2147/DDDT.S439686
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Xiang-Yu Ji,1– 3 Cheng-Jing Lei,2,3 Shuang Kong,2,3 Han-Fei Li,2,3 Si-Yu Pan,2,3 Yu-Jing Chen,2,3 Fan-Rong Zhao,2,3 Tian-Tian Zhu1– 3
1Department of Pharmacy, the First Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan, People’s Republic of China; 2College of Pharmacy, Xinxiang Medical University, Xinxiang, Henan, People’s Republic of China; 3Henan International Joint Laboratory of Cardiovascular Remodeling and Drug Intervention, Xinxiang, Henan, People’s Republic of China
Correspondence: Tian-Tian Zhu, College of Pharmacy, Xinxiang Medical University, Xinxiang, Henan, 453003, People’s Republic of China, Tel +86 15225990135, Email [email protected]
Purpose: The underlying causes of pulmonary arterial hypertension (PAH) often remain obscure. Addressing PAH with effective treatments presents a formidable challenge. Studies have shown that Hydroxysafflor yellow A (HSYA) has a potential role in PAH, While the mechanism underlies its protective role is still unclear. The study was conducted to investigate the potential mechanisms of the protective effects of HSYA.
Methods: Using databases such as PharmMapper and GeneCards, we identified active components of HSYA and associated PAH targets, pinpointed intersecting genes, and constructed a protein-protein interaction (PPI) network. Core targets were singled out using Cytoscape for the development of a model illustrating drug-component-target-disease interactions. Intersection targets underwent analysis for Gene Ontology (GO) functions and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment. Selected components were then modeled for target interaction using Autodock and Pymol. In vivo validation in a monocrotaline-induced PAH (MCT-PAH) animal model was utilized to substantiate the predictions made by network pharmacology.
Results: We associated HSYA with 113 targets, and PAH with 1737 targets, identifying 34 mutual targets for treatment by HSYA. HSYA predominantly affects 9 core targets. Molecular docking unveiled hydrogen bond interactions between HSYA and several PAH-related proteins such as ANXA5, EGFR, SRC, PPARG, PGR, and ESR1.
Conclusion: Utilizing network pharmacology and molecular docking approaches, we investigated potential targets and relevant human disease pathways implicating HSYA in PAH therapy, such as the chemical carcinogenesis receptor activation pathway and the cancer pathway. Our findings were corroborated by the efficacious use of HSYA in an MCT-induced rat PAH model, confirming its therapeutic potential.
Keywords: network pharmacology, molecular docking, hydroxy-safflower yellow A, pulmonary arterial hypertension
Introduction
Pulmonary arterial hypertension (PAH) is a chronic and progressive disorder marked by high pressures in the pulmonary arteries, which can lead to serious health consequences and decreased patient survival.1 The 6th World Symposium on Pulmonary Hypertension and subsequent updates to the 2022 guidelines have revised the hemodynamic definition of PAH to enhance diagnosis and treatment modalities. These guidelines now specify that normal pulmonary vascular resistance (PVR) should not exceed 2 Wood Units (WU), and differentiate pre-capillary PAH as having a PVR greater than 2 WU. A mean pulmonary artery pressure over 20 mm Hg coupled with a PVR above 2 WU are the updated criteria for diagnosing PAH.2 Patients diagnosed with PAH exhibit sustained pulmonary artery pressure, which contributes to detrimental structural alterations and a rise in resistance within the pulmonary vascular system—a phenomenon known as pulmonary vascular remodeling.3,4 As a result, the right ventricle faces a heightened workload that, without appropriate adaptation of the pulmonary arteries, triggers an escalation in PVR and ultimately progresses toward heart failure.5,6 The complexity of PAH’s pathophysiology, often exacerbated by its association with other health conditions, and a rising incidence and prevalence of idiopathic and hereditary forms—estimated at 5 to 25 cases per million individuals—present significant obstacles in clinical management.7 Although a study of 2967 US patients showed improved 1, 3, 5, and 7-year survival rates at 91%, 85%, 68%, and 49% respectively, PAH remains a grave concern with a heavy symptom burden and poor long-term prognosis, emphasizing the urgency for advanced research into the pathogenic mechanisms driving PAH.8,9 Despite progress in our understanding and treatment of this condition, current therapies fall short in halting the relentless progression of pulmonary vascular remodeling.
With an increasing understanding of the pathogenic mechanism of PAH and potential targets for drug treatment, many drugs are constantly emerging. Management primarily relies on pulmonary vasodilator drugs to lower blood pressure. However, the application of these medications is constricted due to their serious side effects. To give an example, the main adverse effects of endothelium receptor antagonists are hepatotoxicity, anemia, and edema,10 and adverse effects of phosphodiesterase inhibitors are mainly headaches, epistaxis, flushing, and dyspepsia;11 moreover, adverse effects of most prostanoids are pain or erythema at the site when given subcutaneously and their common side effects include headache, nausea, rash, and diarrhea.12 Meanwhile, approved treatments for PAH mainly target imbalances in pulmonary vasoactive pathways, which primarily enhance pulmonary vasodilation with weak effects on pulmonary vascular remodeling.13 Currently, Chinese herbal extracts are gradually becoming a research hotspot in recent years for their diverse bioactivities.
Safflower is a traditional Chinese herb that consists of the dried flowers of the Compositae plant safflower, which is native to Central Asia.14 At present, injectable Safflower extract is widely used for the treatment of cerebrovascular and cardiovascular diseases,15 and has been approved to treat angina pectoris by the State Food and Drug Administration since 2005.16 Hydroxysafflor yellow A (HSYA) is the main chemical component of safflower, which has been demonstrated to possess a number of pharmacological activities. HSYA plays an important role acting on cardiovascular and cerebrovascular disease17 for its anti-inflammatory and antioxidant properties18 and inhibitory effect of thrombin generation.19 A vitro study demonstrated that HYSA possesses vascular relaxation effects on rat pulmonary artery by activating the Kv channel in pulmonary artery smooth muscle cells (PASMCs),20 inhibition effect on vascular smooth muscle cells proliferation and migration through different signal pathway.21 Furthermore, the administration of HSYA phytosomes via intervaginal space injection decreased the deposition of collagen in the lungs of mice.22 Subsequent work further corroborated that HSYA protects against hypoxic-induced pulmonary hypertension by reversing the remodeling of the pulmonary artery through inhibiting the proliferation and hypertrophy of PASMCs,23 and attenuating the development of MCT-induced PAH by inhibiting vascular remodeling, decreasing vascular wall thickness and the degree of muscularization.24
Despite the existing literature highlighting the potential of HSYA as a therapeutic agent in ameliorating PAH, a thorough understanding of its modulatory mechanisms is currently absent. Network pharmacology stands at the forefront of biomedical research for unraveling the biological intricacies of diseases, facilitating the prediction of efficacious drug targets, and identifying critical biomarkers for a host of diseases and syndromes.25,26 This emergent discipline also enables the elucidation of the core mechanisms of action for small molecular entities by constructing comprehensive “compound-disease-gene-target” networks. Accordingly, our investigation is meticulously designed to systematically explore the primary targets and the corresponding mechanisms through which HSYA exerts its effects on PAH. Leveraging the combined strengths of network pharmacology and molecular docking, we aim to demystify the molecular underpinnings by which HSYA modulates PAH, with the postulations rigorously validated via experimental methodologies.
Materials and Methods
Screening of Active Ingredient Targets of HSYA
HSYA was retrieved from the Traditional Chinese Medicine Systems Pharmacology Database (TCMSP, https://old.tcmsp-e.com/tcmsp.php), and its corresponding SDF file was downloaded. Within the PharmMapper database (https://pubchem.ncbi.nlm.nih.gov/), the SDF file of HSYA was uploaded, and target files were retrieved with a constraint of a fit score above 0.6. This step yielded a prediction of 113 potential targets. The targets were further processed for their gene names using UniProt (http://www.uniprot.org/).
Identification of PAH-Associated Targets
The GeneCards database (http://www.genecards.org/)27 was queried for “pulmonary hypertension” to extract associated targets, setting a relevance score threshold of 5.2. A total of 1737 disease-related targets were retrieved.
Determination of Overlapping Targets Between HSYA and PAH
An intersection of HSYA and PAH targets was identified through the Venny 2.1.0 tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html),28 and a Venn diagram was generated to visualize the shared potential targets. The study protocol, including the use of human public domain data, was reviewed and approved by the Research Ethics Committee of Xinxiang Medical University.
Construction of PPI Network and Isolation of Core Targets
HSYA-PAH intersecting targets were input into the STRING database (https://cn.string-db.org/) for protein-protein interaction (PPI) analysis29. “Homo sapiens” was selected as the reference organism for PPI network construction, and results were exported in TSV format. This network data was imported into Cytoscape 3.9.1, where the “Network Analyzer” plugin helped identify core targets by setting the cut-off for degree centrality (DC), betweenness centrality (BC), and closeness centrality (CC) at values ≥ their respective mean values. This led to the identification of nine core targets, including Epidermal Growth Factor Receptor (EGFR), SRC proto-oncogene, non-receptor tyrosine kinase (SRC), Peroxisome Proliferator Activated Receptor-Gamma (PPARG), Endothelial Nitric Oxide Synthase (NOS3), Glutathione S-transferase P1 (GSTP1), Human Annexin A5 (ANXA5), Recombinant Human Progesterone Receptor (PGR), Estrogen Receptor 1 (ESR1), and Heat Shock Protein (HSP90AA1).
GO Functional and KEGG Pathway Enrichment Analysis
For functional insights, the core targets were input into the DAVID database (https://david.ncifcrf.gov/tools.jsp) for GO and KEGG pathway enrichment analysis. GO annotations span Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) categories. The top 20 BP, top 15 CC, and top 20 MF terms, along with KEGG pathways, were selected based on gene enrichment significance and then visualized as bubble charts using the online platform Bioinformatics (https://www.bioinformatics.com.cn/).
Molecular Docking Studies
To corroborate the predicted interactions, molecular docking was performed with nine key targets, including EGFR, FRC, and PPARG. HSYA’s 2D structure was sourced from PubChem and then converted to its 3D form using ChemBio3D Ultra (https://www.chemdraw.com.cn/xiazai.html). The protein structures were downloaded from the Protein Data Bank (https://www.rcsb.org/), followed by pre-processing of the ligand and receptor using AutoDock Tools (https://autodock.scripps.edu/).30,31 Molecular docking was completed with the aid of AutoDock Vina. Visualization of the docked complexes was accomplished using PyMOL (https://pymol.org/2/).
Animals and Treatments
Fifty 8-week-old male Sprague-Dawley rats were acquired from SPF (Beijing) Biotechnology Co., Ltd. (Identification No. SCXK Jing 2019–0010, License No. SCXK (Beijing) 2019–0010). All rats were sustained in an environmentally controlled facility featuring a temperature range of 22–26°C, 55±5% relative humidity, and a 12-hour light/dark cycle. Following a one-week acclimatization period, the rats were deemed suitable for experimental procedures. All procedures were executed in strict accordance with the National Institutes of Health guidelines for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978), and received approval from the Animal Care and Use Committee of Xinxiang Medical University (Approval No. XYLL-20230268, dated March 10, 2023).
Monocrotaline-Induced Pulmonary Hypertension Model
Monocrotaline (MCT) with ≥98% purity (Catalog No. C2401) was purchased from SIGMA. HSYA was obtained from Shanghai Yuanye Bio-Technology Co., LTD (Catalog No. S26799-1g, purity ≥ 90%). The rats were randomly divided into four groups: (i) control group (n = 10), (ii) HSYA group (n = 10), (iii) MCT group (n = 15), which received a single subcutaneous injection of 60 mg/kg MCT at the cervical region, and (iv) MCT+HSYA group (n = 15), which were administered daily oral doses of HSYA (220 mg/kg) subsequent to MCT injection for four weeks. Right ventricular function was evaluated using multiple echocardiographic parameters and invasive hemodynamic measurements. Under deep anesthesia with 0.1–0.2 mL of 3% PT-barbital per 100g body weight, we harvested the hearts and tibias of the rats prior to humane euthanasia. The weight of the right ventricle was measured relative to the body weight, tibial length, and the combined weight of the left ventricle plus the septum. Tissues collected for histopathological analysis were fixed in polyformaldehyde, with other samples flash-frozen and preserved at −80°C. The Xinxiang Medical University Animal Care and Use Committee’s standards were firmly adhered to for all animal-related procedures.
Echocardiographic Assessment
At the study’s culmination, the animals were lightly anesthetized with 2% ether and subjected to comprehensive echocardiographic analysis using the VEVO 2100 system with a 30 MHz probe (VisualSonics, Ontario, Canada). Stable echocardiographic acquisitions allowed for the precise quantification of pulmonary arterial time (PAT), pulmonary ejection time (PET), right ventricular wall thickness (RVWT), tricuspid annular plane systolic excursion (TAPSE) among other significant parameters.
Right Ventricular Systolic Pressure (RVSP) Measurement
With animals under deep anesthesia, the right external jugular vein was cannulated with a heparin-preloaded PE catheter connected to a pressure transducer (TaiMeng, Chengdu, China). The catheter was delicately inserted to gauge right ventricular systolic pressure (RVSP), with real-time pressure waveforms allowing accurate determination.
Tissue Sampling
After the RVSP evaluation, the animals were euthanized under deep anesthesia and their hearts excised. Right ventricular hypertrophy was quantified through weight ratios including right ventricular weight to the total heart weight (RV/(LV+S)) and right ventricular weight to tibial length (RV/TL). Lung tissue samples and pulmonary artery segments were also harvested, with some immediately frozen at −80°C and others fixed in 4% paraformaldehyde for histopathological examination.
Morphological Staining
Lung tissues were fixed for 16 hours in 4% paraformaldehyde, dehydrated sequentially in an alcohol gradient, and embedded in paraffin. Tissue sections (4 µm thick) underwent hematoxylin-eosin (HE) and Masson’s or Van Gieson’s (VG) staining. Examination of staining patterns was conducted with an Olympus light microscope (Olympus, Japan).
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
Total RNA was extracted from the lung tissues of MCT-treated rats using Trizol reagent (NCM Biotechnology, China). RNA concentration was normalized to 100 ng/μL and reverse-transcribed to cDNA, followed by PCR amplification with a specific kit (KR0501, Codonx Life Sciences) and 2×Taq PCR MasterMix (G3304, Servicebio). The PCR products were resolved by 1% agarose gel electrophoresis in TAE buffer, visualized, and documented with a gel imaging system. The data analysis employed Image J software, with results reported as ratios of target gene mRNA to GAPDH mRNA. The primer sequences for the genes are as follows (forward primer, F; reverse primer, R):
rattus-ANXA5-F: GAACTTACTCCTGGCTGT CGTGAAG
rattus- ANXA5-R: TCAGTCATCCTCCTCCACAG
rattus-SRC-F: ACCTCCCGCACGCAATTCAAC
rattus-SRC-R: CATCCACACCTCTCCGAAGCAAC
rattus-PGR-F: GCTACGGTGGAGGTGGAGGAG
rattus-PGR-R: GCGTGGTTCAGAGGCAGAATGG
rattus-EGFR-F: CACTACGCCGCCTGCTTCAAG
rattus-EGFR-R: TGGACAGTGGAGGTCAGACAGATG
rattus-PPARG-F: CTGCGGAAGCCCTTTGGTGAC
rattus-PPARG-R: GTGCTCTGTGACAATCTGCCTGAG
rattus-ESR1-F: ACTGTGCTGTGTGTAACGACTATGC
rattus-ESR1-R: CTGACGCTTGTGCTTCAACATTCTC
Statistical Analysis
The data are presented as mean ± standard Error of Mean (SEM). Group comparisons were conducted using unpaired T-tests (between two groups) or one-way ANOVA (where applicable), with SPSS software version 19.0 (Systat Software, San Jose, CA, USA) enlisted for statistical computations. A P-value of less than 0.05 was regarded as indicative of statistical significance.
Results
Network Analysis of PAH Compounds and Identification of Hub Genes
A comprehensive Venn diagram analysis conducted on the designated platform identified 34 intersecting genes between the known targets of HSYA and PAH, as depicted in Figure 1A. These 34 genes constituted the preliminary target pool for subsequent investigations. Utilizing the STRING database, PPI networks for these genes were constructed (see Figure 1B). Network and attribute files generated from the analysis were then imported into Cytoscape to construct a sophisticated drug-ingredient-target-disease network, demonstrated in Figure 1C. Here, core targets appear in blue, the disease is indicated by a red arrow, and active ingredients are represented with an orange triangle. For further refinement, the imported STRING TSV data was subjected to analysis using the Network Analyzer plugin within Cytoscape 3.9.1. Targets exhibiting DC, BC, and CC greater than or equal to the mean value were considered core, leading to the creation of a core target network map (see Figure 1D). The intensity of color correlates with the degree of target interconnectedness. Nine genes were ultimately designated as hub genes shared by HSYA and PAH, including EGFR, SRC, PPARG, NOS3, GSTP1, ANXA5, PGR, ESR1, and HSP90AA1.
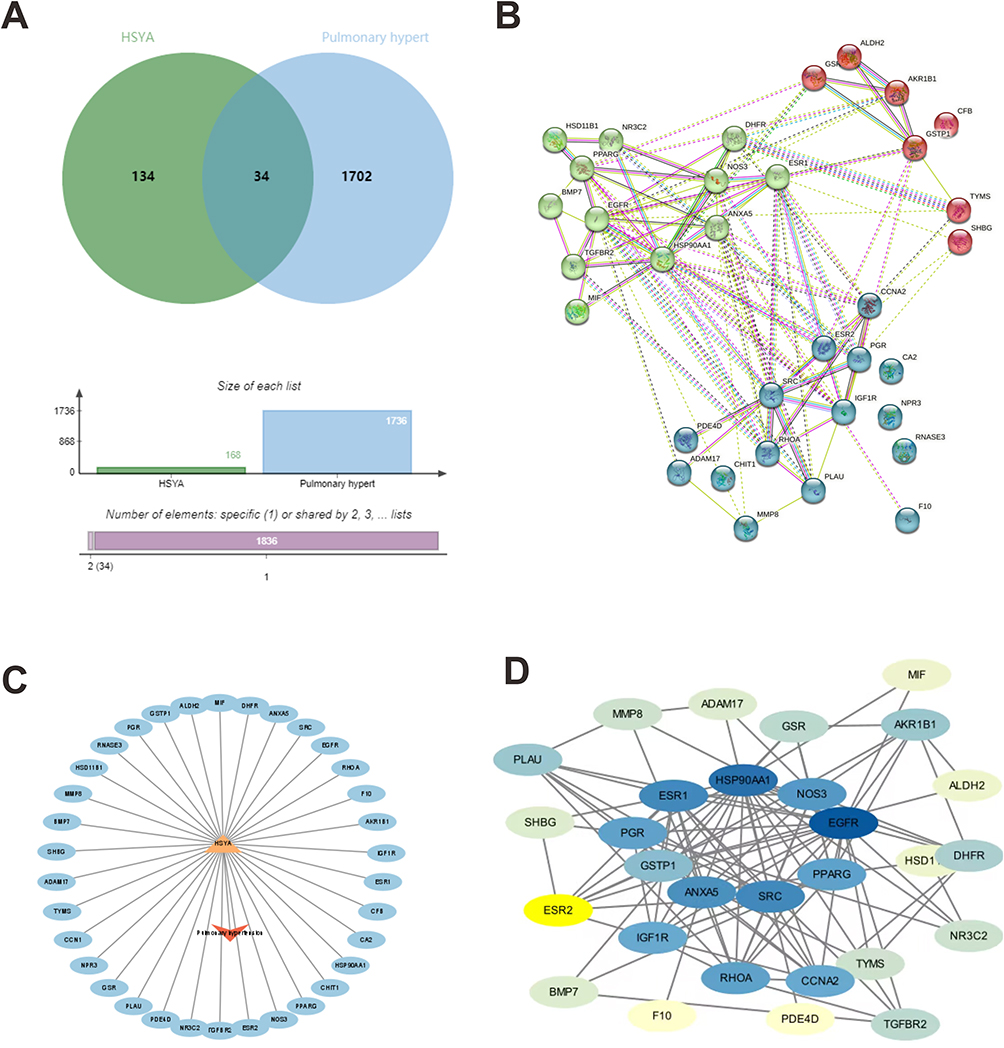 |
Figure 1 The “drug-ingredient-target” interaction network of HSYA on PAH treatment by pharmacologic analysis. (A) The intersection of the above drug targets and the disease targets; (B) PPI map generated from the STRING database among 34 genes; (C) drug-ingredient-target-disease network of HSYA; (D) PPI network processed by cytoscape software. The darker the color, the greater correlation of the targets. |
HSYA-PAH Enrichment Analysis
Elucidating the therapeutic mechanism of HSYA for PAH was achieved by subjecting the core targets to the DAVID database, from which GO data pertaining to BP, CC, and MF were extracted. Predominant GO categories were visualized in histograms (Figure 2), showcasing the top 20 BPs, 15 CCs, and 20 MFs, based on gene enrichment’s extent and statistical significance. In the GO-BP category, key areas of activity included positive regulation of protein kinase B signaling, negative regulation of apoptosis, and response to estrogen (Figure 2A). For GO-CC, core targets were principally localized to the cytoplasm, nucleus, nucleoplasm, and perinucleolar regions (Figure 2B), while in GO-MF, primary activities such as nitric oxide and enzyme regulatory functions, ATPase binding, among others were noted (Figure 2C).
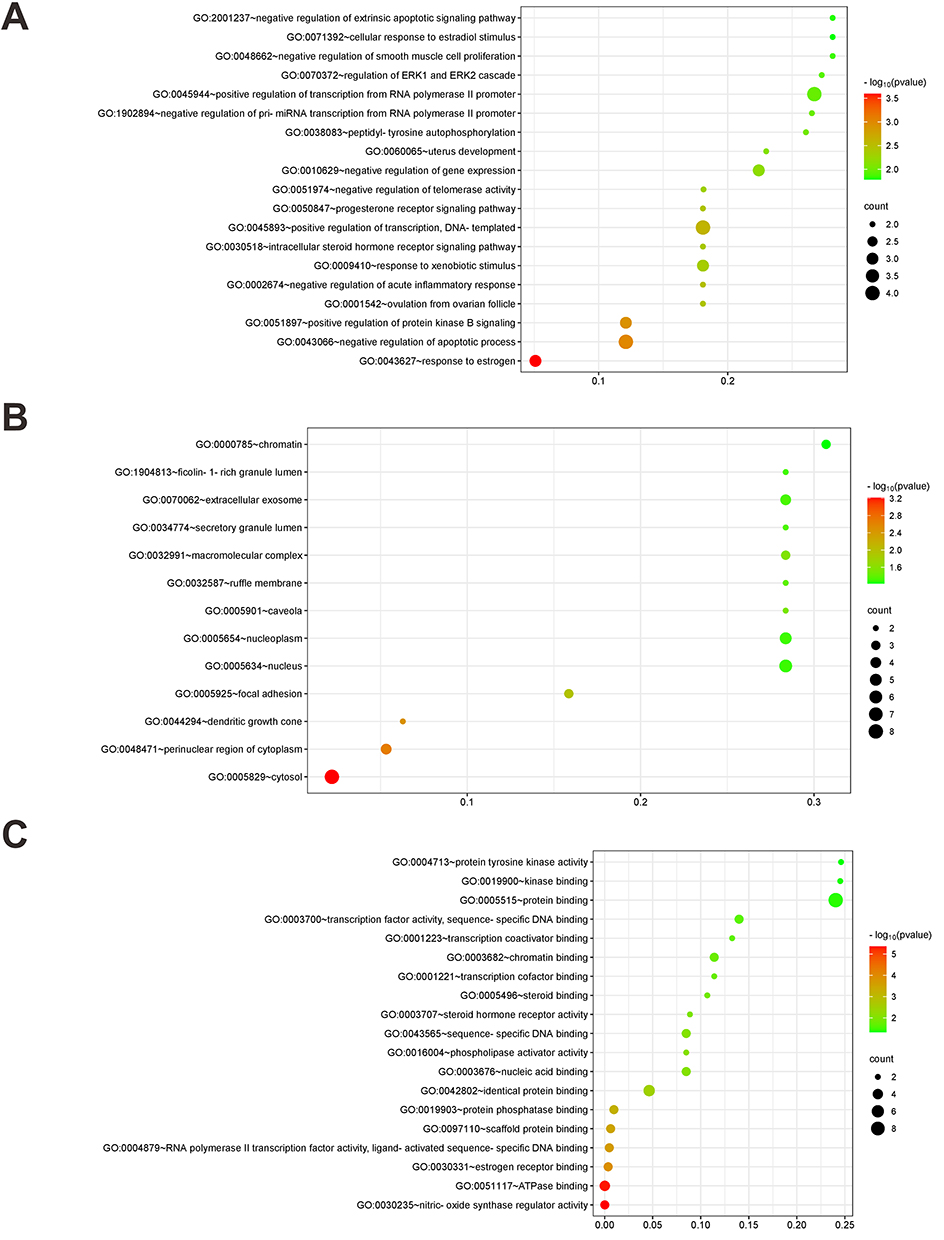 |
Figure 2 HSYA-PAH GO enrichment analysis. (A) The biological process (BP) of GO enrichment analysis; (B) The cell component (CC) of GO enrichment analysis; (C) The molecular function (MF) of GO enrichment analysis. The color represents the level of the P value, and the redder the color, the smaller the P value, the more significant the path. The size of the dot represents the number of genes. The larger the dot, the more genes are enriched to the pathway (The same is true of Figures 2 and 3 below). |
Pathway data garnered from the DAVID database facilitated the creation of a KEGG pathway classification summary diagram available on the Bioinformatics platform. The KEGG pathway enrichment encompassed 23 distinct pathways, predominantly involving human disease pathways such as the chemical carcinogenesis receptor activation and the cancer pathways (Figure 3).
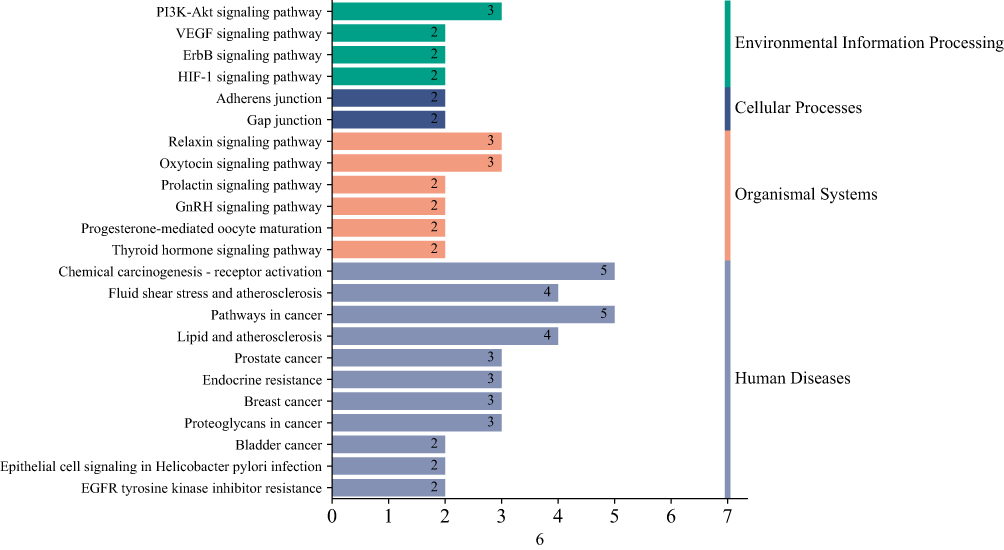 |
Figure 3 KEGG pathway enrichment analysis of key genes. The figure is a summary diagram of signal pathway classification, the different colors represent the different classification of signal pathways, and the numbers on the horizontal column represent the number of genes enriched in this pathway, namely the horizontal axis, and the vertical axis is the name of each pathway. |
Molecular Docking: Enhancing Specificity and Detailing Interaction Dynamics
In-depth molecular docking was conducted between HSYA and the nine essential core receptor proteins, revealing intricate details of their molecular interactions presented in Table 1. In the docking results, the minimum binding affinities of HSYA with ANXA5, SRC, PGR, EGFR, PPARG, and ESR1 were −7.8, −8.0, −8.4, −6.8, −7.1, and −7.0 kcal/mol, respectively. The binding energies, a pivotal indicator of ligand-receptor affinities, were considered robust at values below −5 kcal/mol. Higher absolute values of the binding energies implied more substantial hydrogen bond formations, suggesting a more stable ligand-receptor complex. Notably, HSYA demonstrated the most potent binding energy with the PGR, recorded at −8.4 kcal/mol, suggesting a significantly stable interaction.
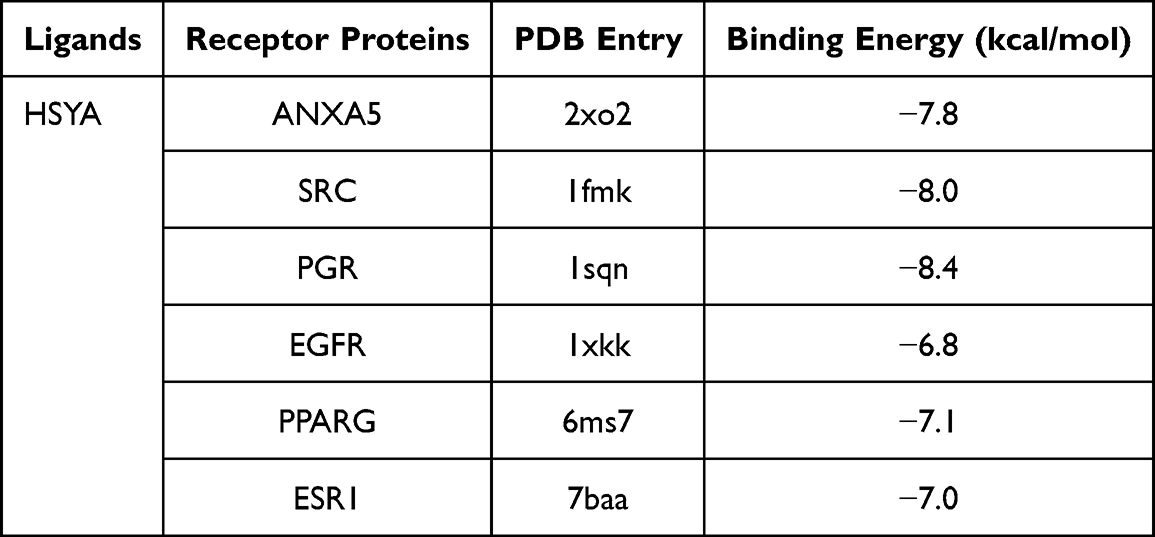 |
Table 1 The Molecular Docking of HSYA and the Proteins ANXA5, SRC, PGR, EGFR, PPARG, ESR1 |
Closer observation of the molecular docking outcomes through the PyMOL visualizations (Figure 4) highlighted multiple hydrogen bond formations as a commonality among the successful docking conformations. As shown in Figure 4A, HSYA combined with the GLN3 of ANXA5 in the form of hydrogen bonds. Similarly, in Figure 4B, HSYA interacted with the PHE150 of SRC via hydrogen bonds. Furthermore, as depicted in Figure 4C, HSYA combined with both the SER898 and PHE905 of PGR in the form of hydrogen bonds. Figure 4D demonstrated that HSYA engaged in hydrogen bonding with the residues of ARG776, GLN791, and LYS852 of SRC. Likewise, as illustrated in Figure 4E, HSYA established a hydrogen bond with the ARG234 of PPARG. Lastly, Figure 4F indicated that HSYA combined with the GLU20 and TYP48 of ESR1 in the form of hydrogen bonds. These visual mappings illuminated the spatial configurations and interaction sites, showing how the active components of HSYA formed hydrogen bonds at specific amino acid residues, further stabilizing the drug-target complex and suggesting high specificity of the interaction. This detailed molecular insight underscores the potential of HSYA in modulating key protein functions implicated in PAH, providing a strong impetus for ongoing therapeutic exploration and development.
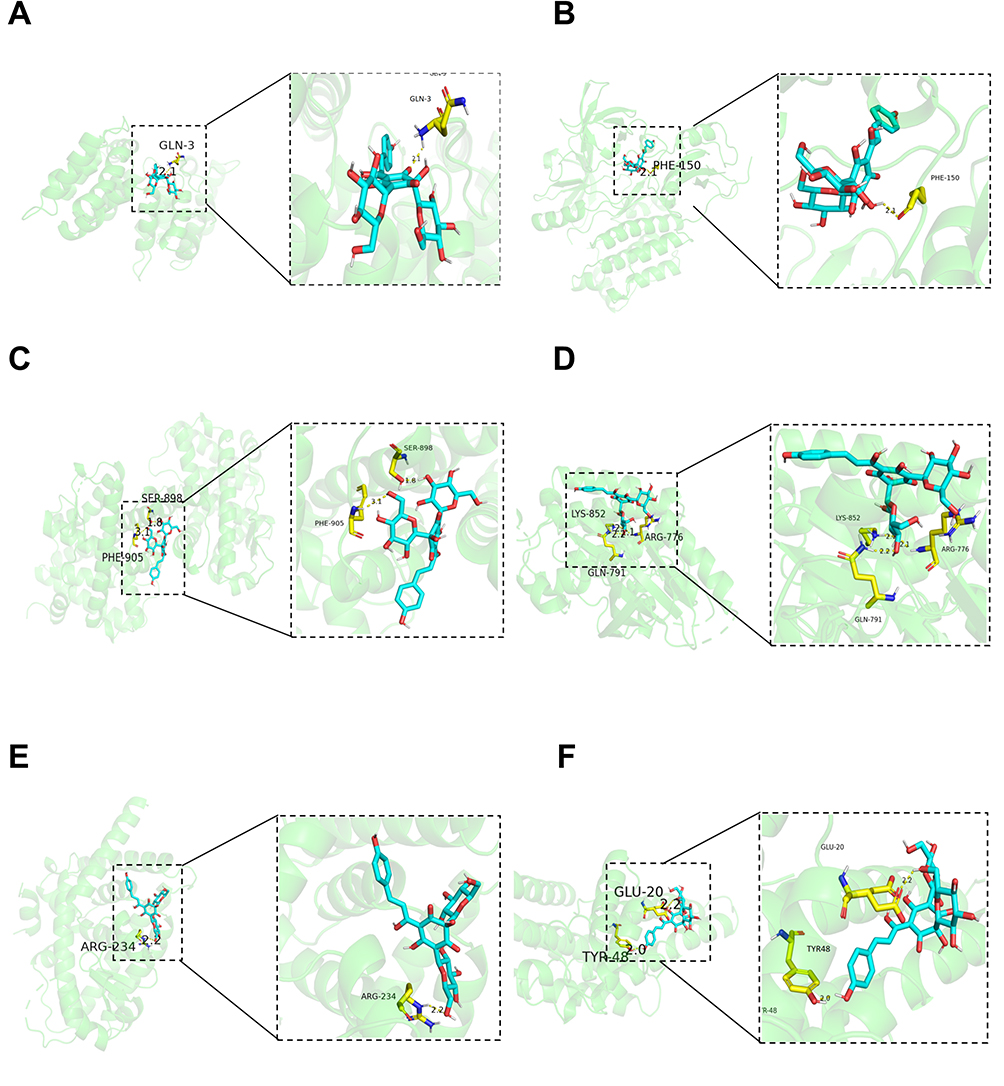 |
Figure 4 Molecular docking PYMOL image of key genes and HYSA. 3D molecular docking model of 6 key genes with HYSA. Gray, red, white, Orange, and purple stick represents an atom of C, O, H, P, and N, respectively. HYSA is represented as blue strip, and yellow strip represents amino acid residues having interaction with the key genes. A to F represents key genes such as ANXA5, SRC, PGR, EGFR, PPARG, ESR1. |
HSYA Attenuated MCT-Induced RVSP Increases and Improved Right Ventricular Function in PAH Rats
Four weeks post-administration of MCT, the pulmonary arterioles in rats developed significant thickening when compared with the normotensive control group, evidenced by a marked increase in wall thickness percentage (WT%) and wall area percentage (WA%), indicative of pulmonary arteriole remodeling. This remodeling resulted in increased vascular resistance and heightened strain on the right ventricle, contributing to right ventricular hypertrophy.
When compared to the control group, the body weight of rats in the MCT group was obviously lowered. However, the body weight of rats in the MCT+HSYA group was higher than that in the MCT group (Figure 5A), which indicates the beneficial role of HSYA on MCT-induced PAH. Furthermore, Rats in the MCT group developed a significant elevation of RVSP, RV/(LV + S), and RV/tibial length in comparison to those in the control group, indicating that the development of RV hypertrophy and PAH model was built successfully (Figure 5B and C). However, those variations were significantly attenuated by HYSA. Additionally, echocardiographic Doppler assessments (Figure 5E-J) showed pronounced abnormalities in the PAT, PET, PAT/PET, RVAW, and TAPSE in MCT-induced PH rats, similarly, these indices of right ventricular function in the MCT+HSYA rats closely resembled those of the control group, suggesting that HSYA can reduce the pathological progression of PAH.
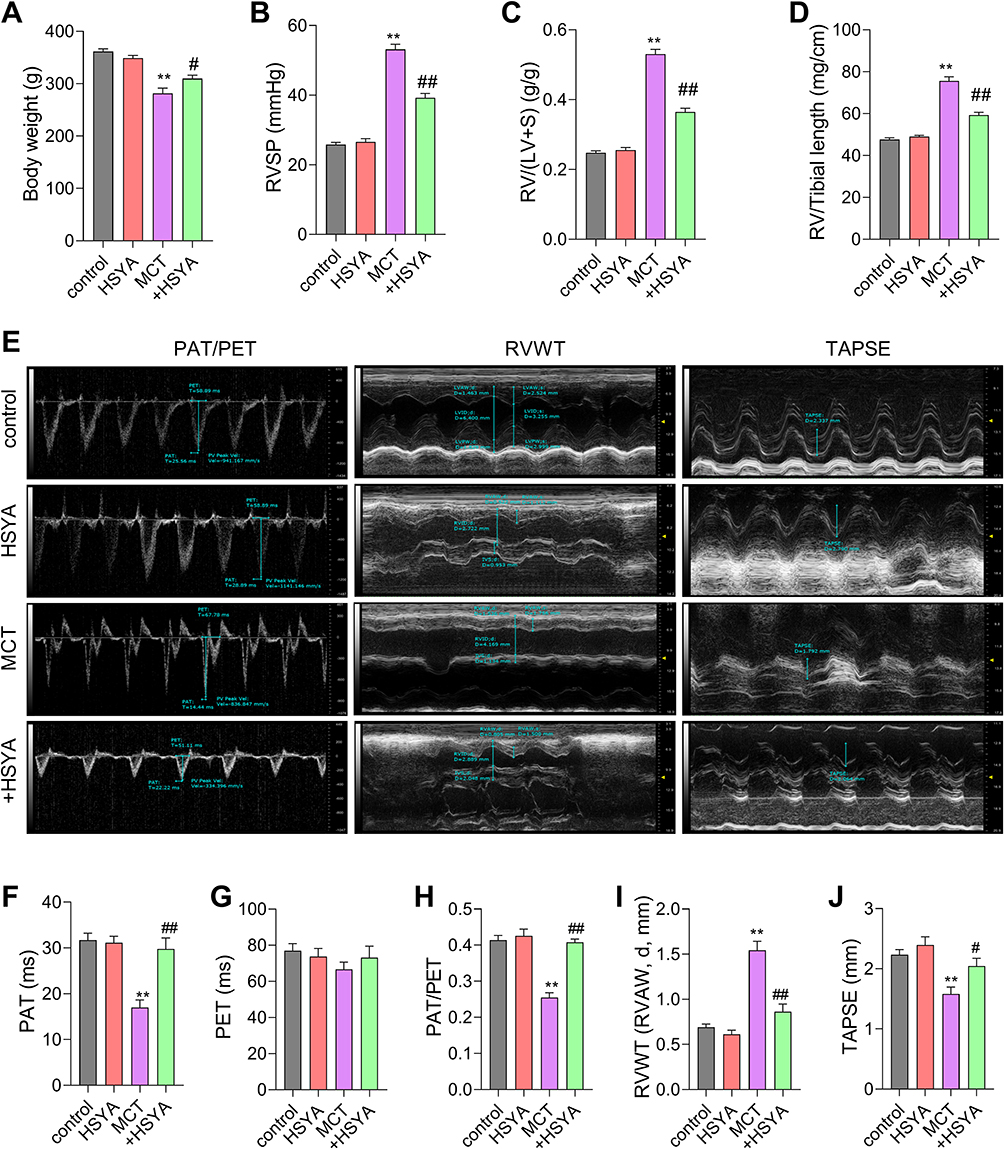 |
Figure 5 The effect of HSYA on RVSP and RV function in PAH rats. (A) The Body weight of rats; (B) Statistical Results of RVSP in rats; (C) The ratio of RV weight to left ventricular+ventricular septal weight (RV/(LV+S)) in rats; (D) The ratio of RV weight to tibial length (RV/TL) in rats; (E) Pulmonary artery blood flow acceleration time (red) (PAT), Pulmonary artery ejection time (white) (PET), Right Ventricular Wall Thickness (RVWT) and Tricuspid annular plane systolic excursion (brown) (TAPSE) in rats were measured by echocardiograms in Doppler model; (F-J) Statistical Results of PAT, PET, PAT/PET, RVWT and TAPSE in (E). All values were expressed as mean±SEM. n=10, **P<0.01 vs control, #P<0.05, ##P<0.01 vs MCT. |
HSYA Alleviated Pulmonary Vascular Remodeling and Collagen Deposition in MCT-Induced PAH Rats
Morphological evaluations through HE, Masson’s trichrome, and VG staining elucidated significant enhancements in pulmonary vascular wall thickness and perivascular collagen fibrosis in the MCT rat model relative to controls. In contrast, PAH rats treated with HSYA exhibited similar histopathological features to the control rats, indicating a therapeutic effect of HYSA (Figures 6A-F). Additionally, the analysis of right ventricular remodeling and collagen deposition (Figure 7) highlighted increased markers of hypertrophy and fibrosis in the MCT group when compared to the control group, however, this variation was attenuated significantly in the HSYA-treated PAH rats, signaling an improvement in cardiac pathology comparable to control (Figures 7A-F).
 |
Figure 6 The effect of HSYA on pulmonary vascular remodeling and collagen deposition in lung tissues of PAH rats. (A) Morphological analysis was performed by HE staining in lung tissue; (B) Quantification of the ratio of pulmonary artery wall thickness to vessel diameter in (A); (C) Masson staining in lung tissue; (D) Quantification of the fibrosis area in (C); (E) VG staining in lung tissue; (F) Quantification of the fibrosis area in (E). All values were expressed as mean±SEM. n=10, **P<0.01 vs control, ##P<0.01 vs MCT. |
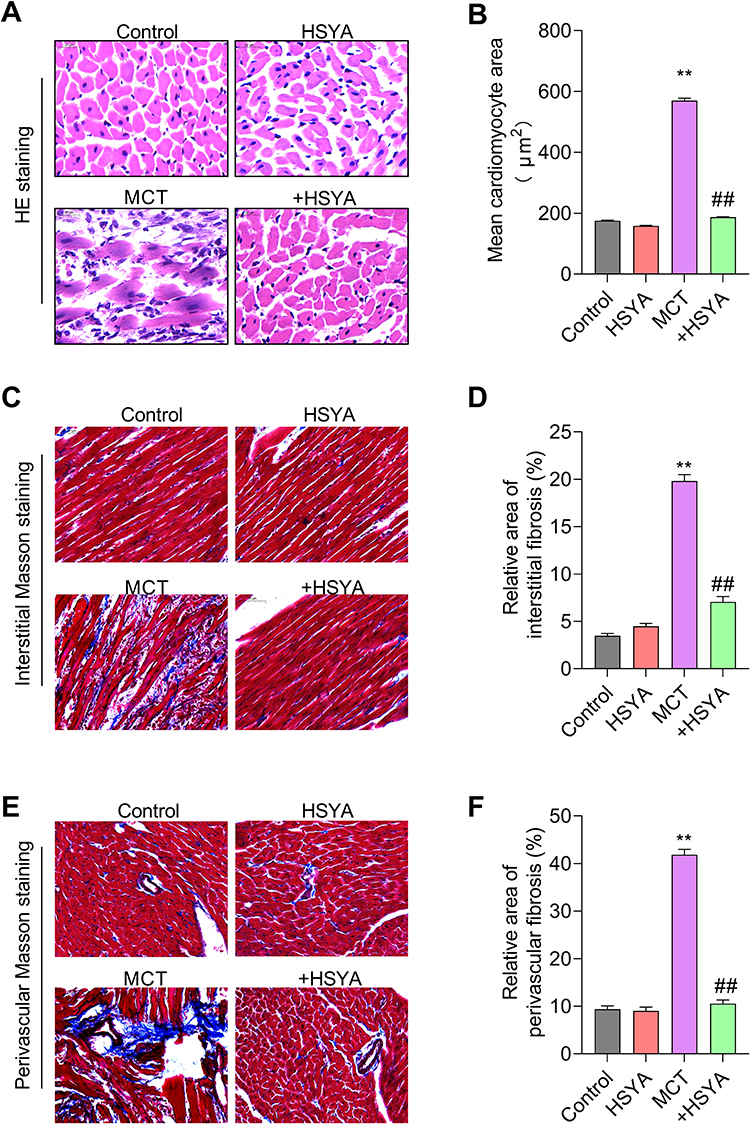 |
Figure 7 The effect of HSYA on right ventricular remodeling and collagen deposition in cardiac tissues of PAH rats. (A) HE staining in right ventricle; (B) Mean cardiomyocyte area in (A); (C) Interstitial Masson staining in right ventricle; (D) Relative area of interstitial fibrosis in (C); (E) Perivascular VG staining in right ventricle; (F) Relative area of perivascular fibrosis in (E). All values were expressed as mean±SEM. n=10, **P<0.01 vs control, ##P<0.01 vs MCT. |
HSYA Mitigated Right Ventricular Remodeling and Collagen Deposition in MCT-Induced PAH Rats
We next evaluate the efficacy of HSYA on right ventricular changes in MCT-induced PAH. As shown in Figure 7A and B, representative images of HE staining and quantitative analysis of the mean cardiomyocyte area showed that HYSA effectively attenuated MCT-induced myocardial hypertrophy. Similar changes were observed in the right ventricular fibrosis. As revealed by interstitial Masson staining (Figure 7C and D) and perivascular Masson staining (Figure 7E and F), a significant elevation of interstitial fibrosis and perivascular fibrosis were observed in the right ventricle of MCT-treated, which was markedly decreased by the treatment of HYSA. Thus, the above observations demonstrated that HYSA attenuates right ventricular remodeling and fibrosis in MCT-induced PAH rats.
HSYA Exerted Protect Role on PAH Through Regulating the Expression of ANXA5, SRC, PGR, EGFR, PPARG, and ESR
To further clarify the mechanism of HYSA on PAH, we detected hub genes expression patterns. As shown in Figure 8A and B, the expression of ANXA5, SRC, PGR, PPARG, and ESR was downregulated, while the expression of EGFR was upregulated in lung tissues. However, this change could be reversed by HYSA, suggesting the restorative capacity of HSYA. Importantly, there were no significant alterations in the expression of these genes between the HSYA-treated group and the control group, indicating a minimal effect of HYSA on gene expression in normal rats.
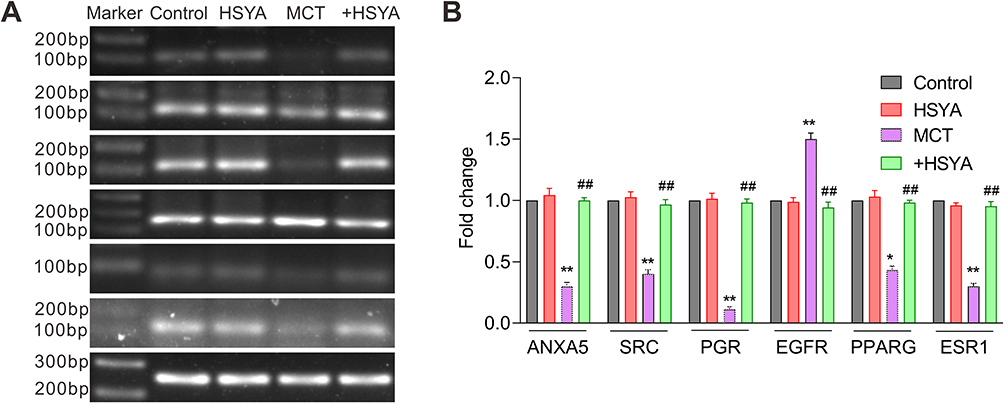 |
Figure 8 The effects of HSYA on genes expression in lung tissues of PAH rats. (A) Representative images of nucleic acid electrophore diagram via RT-PCR. (B) Quantitative analysis of (A). All values were expressed as mean±SEM. n=10, *P<0.05, **P<0.01 vs control, ##P<0.01 vs MCT. |
Discussion
Multiple studies have demonstrated that HSYA plays a vital protective role against hypoxia-induced pulmonary hypertension23 and MCT-PAH,24 effectively reversing the remodeling of the pulmonary artery and attenuating right ventricular hypertrophy. To delve deeper into the effects of HSYA on PAH, we conducted a network pharmacology analysis to uncover the underlying mechanisms. Our findings highlight several critical targets influenced by HSYA in PAH, including proteins such as ESR1, HSP90AA1, NOS3, EGFR, PPARG, SRC, ANXA5, GSTP1, and PGR. These proteins localize to cytosolic-nuclear interfaces, membranes, and the pericellular matrix,32 and are involved in key biological processes such as protein kinase B signaling regulation, the negative regulation of apoptotic processes, estrogen responses, and ATPase interactions. They are implicated in pathways that mirror cancer biology, such as carcinogenesis receptor activation, hinting at a therapeutic anti-PAH potential for HSYA.
KEGG pathway enrichment analysis further revealed that the central hub genes are distributed across 23 distinct pathways, mainly pertaining to human diseases, including receptor activation in chemical carcinogenesis and cancer-related pathways. This suggests functional parallels between vascular cells in PAH and neoplastic diseases.33 To elucidate the protective role of these hub targets in HSYA’s action against PAH, we performed molecular docking studies. The docking results indicated that 6 out of the 9 key targets, namely ESR1, EGFR, PPARG, SRC, ANXA5, and PGR, exhibit covalent binding to HSYA. A universally recognized concept in molecular docking is that the lower the binding energy, the greater the affinity of the compound for its target.34 Docking simulations suggest that ANXA5, SRC, PGR, EGFR, PPARG, and ESR1 have significant binding interactions with HSYA, particularly PGR, which showed the most stable binding.
To verify our computational predictions, we conducted extensive experimental studies. The results from animal models confirmed that the MCT-induced PAH successfully replicated the disease’s hallmarks, such as vascular remodeling, collagen accumulation in lung tissues, and right ventricular restructuring. In line with previous findings,24,35 HSYA exerted a protective effect, ameliorating vascular remodeling, reducing collagen deposition, and improving cardiac function. Our multidisciplinary investigation thus substantiates the therapeutic potential of HSYA in the management of PAH.
To further elucidate the role of pivotal hub genes in PAH, we assessed the expression of these genes in the lung tissues of rat models using RT-PCR. Intriguingly, all hub target genes, including ANXA5, SRC, PGR, EGFR, PPARG, and ESR1, exhibited abnormal expression in the lungs of rats with PAH. This phenomenon was notably reversed following HYSA treatment.
ANXA5, a widely expressed intracellular protein that binds specifically to phosphatidylserine, may impact cellular signaling, inflammation, and fibrosis processes.36,37 Its anti-inflammatory and anticoagulatory activities are well-documented.38 SRC, belonging to the SRC family kinases, plays a pivotal role in modulating signaling pathways in response to environmental changes at the cellular level.39 Previous research highlighted that Src phosphorylation at Tyr416 increased concomitant with pulmonary artery pressure in a rat model,40 and the Src kinase–EGFR–NADPH oxidase signaling axis is critical for pulmonary hypertension progression under chronic hypoxia.41 Notably, our findings showed a decrease in SRC expression in PAH rats, which contrasts with prior studies, prompting us to investigate Src phosphorylation in future research.
As for PGR, a nuclear receptor that modulates numerous target genes, clinical observations suggest a higher survival rate in women despite a greater prevalence of PAH as compared to men.42 Yet, animal studies indicated milder symptoms in females across hypoxia and MCT models43, and even the development of PAH in specific transgenic mouse strains.43,44 The indispensable role of PGR in the antiproliferative effects of progesterone on PASMCs has been previously established.45 Our study corroborates this notion, showing reduced PGR expression in PAH rats and how HYSA can counteract this change.
The EGFR, a part of the ERBB family of receptor tyrosine kinases, orchestrates numerous biological functions such as cell proliferation.46 Despite the controversial role of EGFR in PAH—with evidence linking it to chronic hypoxia-induced pulmonary vasoconstriction,47 antagonists of EGFR have been demonstrated to be effective in inhibiting vascular cell proliferation in both cell culture and various animal PAH models.48–50 Our results are consistent with the view that EGFR upregulation occurs in PAH and HYSA treatment can reverse this trend.
Moreover, PPARG deficiency has been implicated in the pathogenesis of PAH, and the therapeutic restoration of PPARG function is known to mitigate PH in preclinical models.51,52 The inhibitory effects of FGF21 on hypoxia-induced PASMC proliferation and migration, mediated by PPARG upregulation, further affirm PPARG’s central role in PAH.53,54
ESR1, or estrogen receptor, which regulates genes involved in growth, metabolism, reproduction, and other functions, also has a notably higher expression in PASMCs from female PAH patients than males.55 Remarkably, both ERα56 and ERβ57,58 are seen as exerting a protective effect against PH, potentially synergizing to ameliorate the condition.59,60 In line with these findings, ESR1 expression was down-regulated in PAH rats and subsequently normalized by HYSA treatment.
In a word, our study confirms that HSYA has a therapeutic impact on PAH by modulating the expression of ANXA5, SRC, PGR, EGFR, PPARG, and ESR1. However, the detailed mechanisms by which these genes modulate PAH have yet to be fully delineated and warrant more extensive future investigations.
Conclusion
This study harnessed the combined power of cutting-edge network pharmacology and precise molecular docking techniques to pinpoint crucial molecular targets and pathways implicated in pulmonary hypertension. Substantiation of these targets was achieved through meticulous experimental validation. Our results, particularly the upregulation of key genes such as ANXA5, SRC, PGR, PPARG, ESR1 and downregulation of EGFR following HSYA treatment in a PAH rat model, contribute meaningful insights that could revolutionize drug discovery efforts. They hold significant promise for forging fresh exploratory avenues and clinical interventions in the realm of pulmonary hypertension, potentially heralding a new dawn in therapeutic modalities.
Data Sharing Statement
The authors confirm that the data supporting the findings of this study are available within the article.
Acknowledgments
This project was supported by funding from the National Natural Science Foundation of China (81800051, 82300073), Key Scientific Research Projects of Henan Province (22A180010), Henan Province science and technology research project (212102310319), Key scientific research project of Henan University (22A180010), Research Foundation of Xinxiang Medical University (XYBSKYZZ202108), and the Provincial College Students Innovation and Entrepreneurship Training Program of Xinxiang Medical University (202310472017).
Author Contributions
The author played a substantial role in the review, whether in the conception, design, or all of these aspects, agreed on the journal to which the article has been submitted, reviewed and agreed on all versions of the article before submission, during revision, the final version accepted for publication, and any significant changes introduced at the proofing stage, and agreed to take responsibility and be accountable for the contents of the article.
Fan-Rong Zhao: substantially revised or critically reviewed the article; Xiang-Yu Ji: execution and written the article; Cheng-Jing Lei: execution, acquisition of data, analysis and interpretation; Shuang Kong, Han-Fei Li, and Si-Yu Pan: acquisition of data, analysis and interpretation; Yu-Jing Chen: substantially revised or critically reviewed the article; Tian-Tian Zhu: conception, study design.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barlow A, Barlow B, Koyfman A, et al. Pulmonary arterial hypertension in the emergency department: a focus on medication management. Am J Emerg Med. 2021;47:101–108. doi:10.1016/j.ajem.2021.03.072
2. Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61.
3. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53.
4. Zhao C, Le X, Li M, et al. Inhibition of Hsp110-STAT3 interaction in endothelial cells alleviates vascular remodeling in hypoxic pulmonary arterial Hypertension model. Respir Res. 2023;24(1):289. doi:10.1186/s12931-023-02600-5
5. Luna-López R, Ruiz Martín A, Escribano Subías P. Pulmonary arterial hypertension. Med Clin. 2022;158(12):622–629. doi:10.1016/j.medcli.2022.01.003
6. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017;367(3):643–649. doi:10.1007/s00441-016-2539-y
7. Hajra A, Safiriyu I, Balasubramanian P, et al. Recent Advances and Future Prospects of Treatment of Pulmonary Hypertension. Curr Probl Cardiol. 2023;48:101236. doi:10.1016/j.cpcardiol.2022.101236
8. Humbert M, Sitbon O, Guignabert C, et al. Treatment of pulmonary arterial hypertension: recent progress and a look to the future. Lancet Respir Med. 2023;11(9):804–819. doi:10.1016/S2213-2600(23)00264-3
9. Gurevich S, Prins KW. The evolving role of interventional cardiology in the treatment of pulmonary hypertension. Catheter Cardiovasc Interv. 2021;97(4):E446–e453. doi:10.1002/ccd.29146
10. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. doi:10.1056/NEJMoa012212
11. Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi:10.1056/NEJMoa050010
12. Tapson VF, Gomberg-Maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–688. doi:10.1378/chest.129.3.683
13. Auth R, Klinger JR. Emerging pharmacotherapies for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs. 2023;32(11):1025–1042. doi:10.1080/13543784.2023.2274439
14. Yan K, Wang X, Zhu H, et al. Safflower yellow improves insulin sensitivity in high-fat diet-induced obese mice by promoting peroxisome proliferator-activated receptor-γ2 expression in subcutaneous adipose tissue. J Diabetes Investig. 2020;11(6):1457–1469. doi:10.1111/jdi.13285
15. Kong D, Xia W, Zhang Z, et al. Safflower yellow injection combined with conventional therapy in treating unstable angina pectoris: a meta-analysis. J Tradit Chin Med. 2013;33(5):553–561. doi:10.1016/S0254-6272(14)60021-2
16. Zhang Q, Peng JH, Zhang XN. A clinical study of Safflower Yellow injection in treating coronary heart disease angina pectoris with Xin-blood stagnation syndrome. Chin J Integr Med. 2005;11:222–225. doi:10.1007/BF02836509
17. Sun X, Wei X, Qu S, et al. Hydroxysafflor Yellow A suppresses thrombin generation and inflammatory responses following focal cerebral ischemia-reperfusion in rats. Bioorg Med Chem Lett. 2010;20:4120–4124. doi:10.1016/j.bmcl.2010.05.076
18. Chen J, Pan M, Wang J, et al. Hydroxysafflor yellow A protects against colitis in mice by suppressing pyroptosis via inhibiting HK1/NLRP3/GSDMD and modulating gut microbiota. Toxicol Appl Pharmacol. 2023;467:116494. doi:10.1016/j.taap.2023.116494
19. Wu Y, Wang L, Jin M, Zang BX. Hydroxysafflor yellow A alleviates early inflammatory response of bleomycin-induced mice lung injury. Biol Pharm Bull. 2012;35:515–522. doi:10.1248/bpb.35.515
20. Bai Y, Lu P, Han C, et al. Hydroxysafflor yellow A (HSYA) from flowers of Carthamus tinctorius L. and its vasodilatation effects on pulmonary artery. Molecules. 2012;17:14918–14927. doi:10.3390/molecules171214918
21. Jingshan Z, Qianyu G, Shaohong L, et al. Hydroxysafflor yellow A inhibits rat vascular smooth muscle cells proliferation possibly via blocking signal transduction of MEK-ERK1/2. Zhonghua Xin Xue Guan Bing Za Zhi. 2015;43:728–731.
22. Li T, Han D, Li Z, et al. Hydroxysafflor Yellow A Phytosomes Administered via Intervaginal Space Injection Ameliorate Pulmonary Fibrosis in Mice. Pharmaceuticals;2022. 15. doi:10.3390/ph16010015
23. Li L, Dong P, Hou C, et al. Hydroxysafflor yellow A (HSYA) attenuates hypoxic pulmonary arterial remodelling and reverses right ventricular hypertrophy in rats. J Ethnopharmacol. 2016;186:224–233. doi:10.1016/j.jep.2016.04.004
24. Han X, Zhang Y, Zhou Z, et al. Hydroxysafflor yellow A improves established monocrotaline-induced pulmonary arterial hypertension in rats. J Int Med Res. 2016;44(3):569–584. doi:10.1177/0300060515597931
25. Zhang R, Zhu X, Bai H, Ning K. Network Pharmacology Databases for Traditional Chinese Medicine: review and Assessment. Front Pharmacol. 2019;10:123. doi:10.3389/fphar.2019.00123
26. Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–690. doi:10.1038/nchembio.118
27. Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards Suite: from Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics. 2016;54(1):1.30.1–1.30.33. doi:10.1002/cpbi.5
28. Oliveros JC (2007-2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. Available from: https://bioinfogp.cnb.csic.es/tools/venny/index.html.
29. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–d613. doi:10.1093/nar/gky1131
30. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model. 2021;61(8):3891–3898. doi:10.1021/acs.jcim.1c00203
31. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
32. Wang L, Zhu W, Sun R, et al. Network Pharmacology Prediction and Molecular Docking-Based Strategy to Discover the Potential Pharmacological Mechanism of Wen-Yu-Jin against Pulmonary Fibrosis in a Mouse Model. Evid Based Complement Alternat Med. 2022;2022:7753508. doi:10.1155/2022/7753508
33. Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. 2011;147(1):4–12. doi:10.1016/j.ijcard.2010.07.003
34. Kadioglu O, Chan A, Cong Ling Qiu A, et al. Artemisinin Derivatives Target Topoisomerase 1 and Cause DNA Damage in Silico and in Vitro. Front Pharmacol. 2017;8:711. doi:10.3389/fphar.2017.00711
35. Feng J, Guo J, Yan J, et al. Luhong Formula and Hydroxysafflor yellow A protect cardiomyocytes by inhibiting autophagy. Phytomedicine. 2023;110:154636. doi:10.1016/j.phymed.2022.154636
36. Yoshida S, Minematsu N, Chubachi S, et al. Annexin V decreases PS-mediated macrophage efferocytosis and deteriorates elastase-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L852–60. doi:10.1152/ajplung.00066.2012
37. Krahling S, Callahan MK, Williamson P, Schlegel RA. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999;6(2):183–189. doi:10.1038/sj.cdd.4400473
38. Luo C, Ji X, Fan J, et al. Annexin A5 promotes macrophage activation and contributes to pulmonary fibrosis induced by silica particles. Toxicol Ind Health. 2016;32(9):1628–1638. doi:10.1177/0748233715572744
39. De Kock L, Freson K. The (Patho)Biology of SRC Kinase in Platelets and Megakaryocytes. Medicina. 2020;56. doi:10.3390/medicina56020056
40. Liu P, Gu Y, Luo J, et al. Inhibition of Src activation reverses pulmonary vascular remodeling in experimental pulmonary arterial hypertension via Akt/mTOR/HIF-1 signaling pathway. Exp Cell Res. 2019;380(1):36–46. doi:10.1016/j.yexcr.2019.02.022
41. Norton CE, Sheak JR, Yan S, et al. Augmented Pulmonary Vasoconstrictor Reactivity after Chronic Hypoxia Requires Src Kinase and Epidermal Growth Factor Receptor Signaling. Am J Respir Cell Mol Biol. 2020;62(1):61–73. doi:10.1165/rcmb.2018-0106OC
42. Jacobs W, van de Veerdonk MC, Trip P, et al. The Right Ventricle Explains Sex Differences in Survival in Idiopathic Pulmonary Arterial Hypertension. Chest. 2014;145(6):1230–1236. doi:10.1378/chest.13-1291
43. Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307(1):L7–L26. doi:10.1152/ajplung.00337.2013
44. Morris H, Denver N, Gaw R, et al. Sex Differences in Pulmonary Hypertension. Clin Chest Med. 2021;42(1):217–228. doi:10.1016/j.ccm.2020.10.005
45. Hu WP, Xie L, Hao SY, et al. Protective effects of progesterone on pulmonary artery smooth muscle cells stimulated with Interleukin 6 via blocking the shuttling and transcriptional function of STAT3. Int Immunopharmacol. 2022;102:108379. doi:10.1016/j.intimp.2021.108379
46. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59:21–26. doi:10.1016/j.ijrobp.2003.11.041
47. Norton CE, Broughton BR, Jernigan NL, et al. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid Redox Signal. 2013;18:1777–1788. doi:10.1089/ars.2012.4836
48. Dahal BK, Cornitescu T, Tretyn A, et al. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2010;181(2):158–167. doi:10.1164/rccm.200811-1682OC
49. Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–431. doi:10.1161/CIRCULATIONAHA.105.540542
50. Toby IT, Chicoine LG, Cui H, et al. Hypoxia-induced proliferation of human pulmonary microvascular endothelial cells depends on epidermal growth factor receptor tyrosine kinase activation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L600–6. doi:10.1152/ajplung.00122.2009
51. Tseng V, Sutliff RL, Hart CM. Redox Biology of Peroxisome Proliferator-Activated Receptor-γ in Pulmonary Hypertension. Antioxid Redox Signal. 2019;31(12):874–897. doi:10.1089/ars.2018.7695
52. Khandekar MJ, Banks AS, Laznik-Bogoslavski D, et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc Natl Acad Sci U S A. 2018;115(3):561–566. doi:10.1073/pnas.1717776115
53. Cai G, Liu J, Wang M, et al. Mutual promotion of FGF21 and PPARγ attenuates hypoxia-induced pulmonary hypertension. Exp Biol Med. 2019;244(3):252–261. doi:10.1177/1535370219828692
54. Liu J, Cai G, Li M, et al. Fibroblast growth factor 21 attenuates hypoxia-induced pulmonary hypertension by upregulating PPARγ expression and suppressing inflammatory cytokine levels. Biochem Biophys Res Commun. 2018;504(2):478–484. doi:10.1016/j.bbrc.2018.09.004
55. Wright AF, Ewart MA, Mair K, et al. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res. 2015;106:206–216. doi:10.1093/cvr/cvv106
56. Frump AL, Goss KN, Vayl A, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308(9):L873–90. doi:10.1152/ajplung.00006.2015
57. Matori H, Umar S, Nadadur RD, et al. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension. 2012;60(2):425–430. doi:10.1161/HYPERTENSIONAHA.112.191445
58. Umar S, Partow-Navid R, Ruffenach G, et al. Severe pulmonary hypertension in aging female apolipoprotein E-deficient mice is rescued by estrogen replacement therapy. Biol Sex Differ. 2017;8:9. doi:10.1186/s13293-017-0129-7
59. Lahm T, Albrecht M, Fisher AJ, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980. doi:10.1164/rccm.201107-1293OC
60. Lahm T, Crisostomo PR, Markel TA, et al. Selective estrogen receptor-alpha and estrogen receptor-beta agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1486–93. doi:10.1152/ajpregu.90667.2008
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.