Back to Journals » International Journal of Nanomedicine » Volume 17
Hybrid Membrane Nanovaccines Combined with Immune Checkpoint Blockade to Enhance Cancer Immunotherapy
Authors Zhao P ![]() , Xu Y, Ji W, Li L, Qiu L, Zhou S, Qian Z, Zhang H
, Xu Y, Ji W, Li L, Qiu L, Zhou S, Qian Z, Zhang H
Received 2 November 2021
Accepted for publication 22 December 2021
Published 7 January 2022 Volume 2022:17 Pages 73—89
DOI https://doi.org/10.2147/IJN.S346044
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yan Shen
Peiqi Zhao,1,* Yuanlin Xu,2,* Wei Ji,3 Lanfang Li,1 Lihua Qiu,1 Shiyong Zhou,1 Zhengzi Qian,1 Huilai Zhang1
1Department of Lymphoma, Tianjin’s Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, 300060, People’s Republic of China; 2Department of Lymphatic Comprehensive Internal Medicine, Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, Henan, 450001, People’s Republic of China; 3Public Laboratory, Tianjin’s Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, 300060, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Peiqi Zhao; Huilai Zhang
Department of Lymphoma, Tianjin’s Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, 300060, People’s Republic of China
Tel +86-22-23340123
Email [email protected]; [email protected]
Purpose: Cancer vaccines are a promising therapeutic approach in cancer immunotherapy and can inhibit tumor growth and prevent tumor recurrence and metastasis by activating a sustained antitumor immunoprotective effect. However, the therapeutic effect of cancer vaccines is severely weakened by the low immunogenicity of cancer antigens and the immunosuppressive microenvironment in tumor tissues.
Methods: Here, we report a novel hybrid membrane nanovaccine, composed of mesoporous silica nanoparticle as a delivery carrier, hybrid cell membranes obtained from dendritic cells and cancer cells, and R837 as an immune adjuvant (R837@HM-NPs). We investigated the anti-tumor, tumor recurrence and metastasis prevention abilities of R837@HM-NPs and their mechanisms of action through a series of in vivo and ex vivo experiments.
Results: R837@HM-NPs not only provide effective antigenic stimulation but are also a durable supply of the immune adjuvant R837. In addition, R837@HM-NPs promote antigen endocytosis into dendritic cells via various receptor-mediated pathways. Compared with HM-NPs or R837@HM-NPs, R837@HM-NPs in combination with an immune checkpoint blockade showed stronger antitumor immune responses in inhibiting tumor growth, thus eliminating established tumors, and rejecting re-challenged tumors by regulating the immunosuppressive microenvironment and immunological memory effect.
Conclusion: These findings suggest that the hybrid membrane nanovaccine in combination with immune checkpoint blockade is a powerful strategy to enhance antitumor immunotherapy without concerns of systemic toxicity.
Keywords: hybrid membrane, immune checkpoint blockade, immunotherapy, mesoporous silica nanoparticle, nanovaccine
Graphical Abstract:

Introduction
Cancer immunotherapy can control and eradicate cancer cells by restarting and maintaining the cancer-immunity cycle and restoring the body’s normal antitumor immune response.1,2 By intervening in various parts of the cancer-immunity cycle (release of cancer cell antigens, cancer antigen presentation, priming and activation, trafficking of T cells to tumors, infiltration of T cells into tumors, recognition of cancer cells by T cells, and killing of cancer cells), researchers have developed a variety of cancer immunotherapy approaches. Among these, anti-programmed cell death-1 (αPD-1) and anti-programmed cell death ligand-1 (αPD-L1) antibodies have shown significant antitumor activity in a variety of tumor types.3,4 However, only a fraction of patients can achieve long-term clinical benefit from the treatment for numerous reasons, including the suppressive tumor microenvironment, cancer cell heterogeneity, and drug side effects.5,6 In addition, this immunotherapy strategy requires continuous use, which imposes a significant financial burden on patients.
Cancer vaccines have now become a promising therapeutic approach in cancer immunotherapy. Cancer vaccines mainly kill cancer cells by introducing tumor antigens into the patients, enhancing immunogenicity, activating the patient’s own immune system, and inducing cellular and humoral immune responses, and prevent tumor metastasis and recurrence by generating a sustained immune response.7,8 However, weak immunogenicity of tumor antigens has limited the development of cancer vaccine technology.9 Thus, improving the immunogenicity of tumor antigens is key to therapeutic cancer vaccines. Cancer cell membrane vesicles have recently been proposed as a new vaccine platform for cancer immunotherapy.10,11 However, downregulation of tumor antigen expression on cancer cell membranes weakens the immune system, thus hindering the success of this biological approach.12 Researchers have fed dendritic cells (DCs) with antigen-encoding DNA or mRNA to compel DCs to express tumor antigen on the cell surface,13 thus providing artificially programmed antigen-presenting cells (APCs) as cell-typed vaccines. In addition, a novel cancer vaccine derived from the fusion of DCs with cancer cells, can provide strong expression of the entire tumor antigen complexes and the co-stimulatory molecules, and this leads to a strong antitumor immune response.14
The addition of immune adjuvants is another effective strategy to enhance the immunogenicity of tumor antigens. Toll-like receptor (TLR) has a key role in natural and adaptive immunity; thus, TLR agonists have been extensively studied as potential adjuvants for cancer vaccines.15–17 TLR agonists produce immunostimulatory effects by inducing co-stimulatory molecules on DCs and inflammatory cytokines that favor the development of T helper cell type 1 (Th1) immune responses.18 Of these, TLR7 agonist R837 has been approved for the treatment of external genital warts caused by human papillomavirus infection, and CpG oligodeoxynucleotides have been widely used for cancer treatment.19 Currently, various nano-drug delivery systems have proven to be effective in the co-delivery of antigens and adjuvants due to their versatility and flexibility. They have achieved an active role in enhancing immune responses.20–22 Mesoporous silica nanoparticle (MSN) is widely used as a promising carrier for antigen and drug delivery due to its good biocompatibility and high carrying capacity.23,24
Scheme 1 shows our design for a novel hybrid membrane nanovaccine, that is composed of MSN as a delivery carrier and a hybrid cell membrane obtained from DCs and cancer cells, and R837 serves as an immune adjuvant (R837@HM-NPs). R837@HM-NPs have several novelties. First, R837@HM-NPs can be efficiently delivered to secondary lymphoid tissues such as lymph nodes and APCs, because nanovaccines of medium size can be retained in lymph nodes for a considerable period of time.25 Second, R837@HM-NPs can improve the pharmacokinetic properties of the drug payload by encapsulating adjuvants, thus further enhancing the immunomodulatory effects. Third, R837@HM-NPs derived from DCs and cancer cells are not only capable of direct T-cell immune activation via APCs, but also carry tumor antigens that can be recognized by DCs, thus inducing a DC-mediated T-cell immune activation. The combination of these two pathways provides a potent antitumor immune response. Finally, and most importantly, the vaccine platform is flexible, thus allowing loading of different antigens and adjuvants for accommodating diverse functions and treating different types of cancers. We hypothesize that our prepared R837@HM-NPs can induce DC maturation and stimulate T cell production (Scheme 1). After being combined with αPD-1, R837@HM-NPs can achieve remarkable synergistic responses in inhibiting tumor growth, eliminating established tumors, as well as rejecting re-challenged tumors by regulating the immunosuppressive microenvironment, promoting the antitumor immune response and immunological memory effects.
 |
Scheme 1 Schematic illustration of hybrid membrane derived nanovaccine for enhancing cancer immunotherapy. |
Materials and Methods
Materials
Roswell Park Memorial Institute (RPMI) 1640 medium and fetal bovine serum (FBS) were purchased from Gibco Life Technologies (USA). The antibodies of CD3, CD4, CD8, CD11b, CD11c, CD44, CD45, CD62L, CD80, CD86, CD206, Foxp3, F4/80 and the enzyme-linked immunosorbent assay (ELISA) test kits of IL-6, IL-12p40, IFN-γ and TNF-α were acquired from Sigma-Aldrich (USA). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was purchased from Thermo Fisher Scientific (USA). TUNEL Apoptosis Detection Kit were obtained from Vazyme (China). Interleukin-4 (IL-4) were purchased from Beyotime Institute of Biotechnology (China). Granulocyte-macrophage colony-stimulating factor (GM-CSF) were purchased from BioLegend, Inc (USA). R837 (imiquimod) was obtained from InvivoGen (France). αPD-1 was obtained from Bio X Cell (USA). All reagents were directly used without purification unless specified mentioned.
4T1 (triple negative breast cancer cell line), HBL-100 (human mammary epithelial cell line) and B16F10 (murine melanoma cell line) cells were bought from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in a RPMI 1640 medium containing 10% FBS, penicillin (100 UI/mL), and streptomycin (100 UI/mL) with 5% CO2 at 37 °C. Bone marrow-derived dendritic cells (BMDCs) were isolated from the tibia and femur of the BALB/c mice according to an established method.14
BALB/c mice at 6 weeks of age were obtained from SiPeiFu Biotechnology Co., Ltd (Beijing, China). All procedures were performed under sterile conditions. Details about the treatment schedule are shown in the figure legends. All animal experiments were approved by the Animal Ethical and Welfare Committee of Tianjin Medical University Cancer Institute and Hospital, and all animal studies were conducted in accordance with the guidelines of this committee.
Method of Fusing of BMDCs and 4T1 Cells
The method of fusing bone marrow-derived dendritic cells (BMDCs) and 4T1 cells has been described in previous research.14 Briefly, 4T1 cells were inactivated by pretreatment with 20% ethanol for 15 min in an ice bath. BMDCs were then mixed with inactive 4T1 cells in a 2:1 ratio and centrifuged at 500 g. Next, serum-free RPMI 1640 was slowly dripped into the above mixture. The solution was centrifuged and resuspended in the RPMI 1640 containing 10% fetal bovine serum (FBS) and interleukin-4 (IL-4, 10 ng/mL). Hybrid cells were cultured at 37°C in a humid atmosphere and the medium was changed every other day.
Preparation of R837@HM-NPs
To achieve hybrid membrane encapsulation, R837@MSN was first mixed with hybrid membrane at a weight ratio of 2:1 and then resuspended in phosphate buffered saline (PBS). The mixed solution was sonicated in a cold-water bath until the solution became clear. We then used an Avanti mini-extruder to extrude 11 times and centrifuged (2000 rpm × 5 min) to remove the excess hybrid membrane vesicles. Finally, the R837@HM-NPs was obtained.
Cellular Uptake of HM-NPs in BMDCs
To observe whether HM-NPs could promote cellular uptake of antigens by BMDCs, HM-NPs were incubated with BMDCs for 4 h at 37 °C, followed by three rounds of rinsing with cold PBS to remove the extracellular HM-NPs. BMDCs were then labeled with 4, 6-diamidino-2-phenylindole (DAPI) and observed under confocal laser scanning microscope (CLSM).
Endocytic Mechanism of R837@HM-NPs in BMDCs
To further explore the pathway of R837@HM-NPs into BMDCs, we pretreated these cells with several cellular uptake inhibitors and blocked the corresponding endocytic pathway.26,27 BMDCs were treated with filipin (15 μg/mL), amiloride (10 μg/mL), chlorpromazine (10 μg/mL), monensin (10 μg/mL), methyl-β-cyclodextrin (10 μg/mL), and ethylisopropylamiloride (20 μg/mL) at 37 °C for 2 h. The pretreated cells then were co-incubated with R837@HM-NPs for 4 h and untreated cells were used as a negative control. The cells were then trypsinized, centrifuged, and lysed by ultrasonication. Finally, R837 was extracted from the cells and assayed by reverse phase high performance liquid chromatography.
Effect of R837@HM-NPs on BMDCs in vitro
To verify whether R837@HM-NPs could promote BMDC activation and antigen cross-presentation, immature BMDCs were co-cultured with R837@HM-NPs in a 5% CO2 cell incubator at 37°C for 4 h. BMDCs were then stained with fluorescently labeled antibodies against CD11c, CD80, and CD86. Events were collected and plotted by flow cytometry. Meanwhile, cytokines such as interleukin-6 (IL-6), interleukin-12p40 (IL-12p40) and tumor necrosis factor-α (TNF-α) released from culture supernatants were quantified by ELISA according to the manufacturer’s protocols.
In vitro Antitumor Ability of R837@HM-NP-Activated Immune Cells
To obtain single cell suspensions of spleen, the mouse spleen was first removed and placed in a culture dish containing pre-cooled RPMI 1640 culture medium, and excess connective tissues and fat were removed. The spleen was then gently ground to a celiac fluid, followed by filtration through a 70-µm nylon membrane to obtain single cell suspensions of spleen. Red blood cell lysis buffer was then added, suspended for 10 min, and then centrifuged (800 g, 5 min). The cells were then washed twice with PBS and resuspended with 5 mL of 1640 culture medium.
To further evaluate the activation levels of T lymphocytes, BMDCs were pre-treated with PBS, R837, R837@MSN, HM-NPs and R837@HM-NPs for 24 h. T lymphocytes were then added to BMDCs at a ratio of 5 (BMDCs to T cells) and co-cultured for 24 h. The T lymphocytes were washed three times with PBS, stained with CD3, CD4, and CD8 antibodies; and then counted by flow cytometry. The killing ability of R837@HM-NP-activated immune cells to HBL100, 4T1 and B16F10 (the ratio of activated splenic lymphocytes to target cells was 5:1) was measured via a lactate dehydrogenase microplate Assay.
Prophylactic Effect of R837@HM-NPs
To test the preventive effect of R837@HM-NPs on tumors, BALB/c mice were first intravenously inoculated with R837@HM-NPs in the left groin three times every four days. One week after the last R837@HM-NPs injection, 4T1 cells (5×106) were subcutaneously injected into the left flank of the mice. The tumor volume (0.5 × length × width2) and survival time of mice were recorded. Cytokines in blood, including IL-6, IL-12p40, and TNF-α were detected by ELISA. Activated T cells were isolated from the tumors, stained with CD3, CD4 and CD8, and then analyzed by flow cytometry.
Therapeutic Effect of R837@HM-NPs in Combination with αPD-1
To evaluate the therapeutic effect of R837@HM-NPs on established tumors, 4T1 cells (5×106) were injected subcutaneously into the left flank of BALB/c mice. Seven days later, the tumor-bearing mice were randomly divided into five groups (n = 6 per group), including PBS, HM-NPs, αPD-1, R837@HM-NPs and R837@HM-NPs+αPD-1. The treatment drugs were injected intravenously every four days for a total of three injections (10 mg/kg for αPD-1 monotherapy or combination treatment groups). The tumor volume and body weight of the mice were recorded every two days during the treatment period.
Tumor tissues and major organs were removed, dehydrated, embedded, sectioned for 3 μm, and stained with hematoxylin and eosin (H&E) for histological analysis. The apoptosis of tumor cells was detected by TUNEL assays. Activated T cells were isolated from the tumors, stained with CD3, CD4 and CD8, and then analyzed by CLSM and flow cytometry. Regulatory T-cells (Treg, CD4+ Foxp3+), M1 phenotype macrophages (CD80+CD11b+F4/80+) and M2 phenotype macrophages (CD206+CD11b+F4/80+) in the tumors were also analyzed by flow cytometry. Cytokines in serum, including IL-6, IL-12p40, and TNF-α were detected by ELISA. The whole blood was collected and analyzed by blood biochemistry analyzer (MNCHIP, Tianjin) and fully automated hematology analyzer (MC-6200VET) through the end of the experiments.
RNA Sequencing and Bioinformatic Analysis
The methods of RNA sequencing and bioinformatic analysis refer to previous literature.28 RNA samples were extracted from the tumor tissues treated as described above and RNA library preparation used a VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2. RNA-seq was then performed on an Illumina HiSeq X ten PE150. The degree of infiltration at different immune cell types in tumor tissues treated as described above was then evaluated using a gene set enrichment analysis (GSEA).29 The data were analyzed by R software (version 4.0.5) using GSVA, LIMA and GSEABase packages provided by Bioconductor. The biological functions of immune-related genes were mainly derived from gene ontology enrichment analysis based on differential gene expression profiles.
Immunological Memory Effect of R837@HM-NPs in Combination with αPD-1
To evaluate the immunological memory effect of R837@HM-NPs in combination with αPD-1, we need to establish the tumor-bearing mice model by subcutaneously injecting 4T1 cells (5×106) into the left flank of mice. After 1 week, we eliminated the left tumor with the following treatment (surgery, αPD-1, R837@HM-NPs and R837@HM-NPs+αPD-1). After 25 days, 4T1 cells (2×106) were injected subcutaneously into the right flank of the mice. The tumor volume of the right flank and survival time of mice were also recorded from the day of right tumor inoculation. The expression levels of effector memory T cells (TEM, CD3+CD8+CD44+CD62L−) and central memory T cells (TCM, CD3+CD8+CD44+CD62L+) in the spleen were detected by flow cytometry at day 35. Interferon-γ (IFN-γ) in serum was also detected by ELISA.
Statistical Analysis
Unless otherwise stated, all experiments were repeated three times. Experimental results were presented as mean ± standard deviation. Statistical differences between groups were calculated using the Tukey’s test. *P<0.05 means statistically different and **P < 0.01 means significantly different.
Results and Discussion
Construction and Characterization of R837@HM-NPs
The most critical step in the preparation of R837@HM-NPs is the synthesis of hybrid membrane. In this experiment, we chose 4T1 cells as the model cancer cell line. Hybrid membranes were synthesized using DCs and 4T1 cells according to the previous method.23 To express “eat-me” signals on the surface of hybrid membrane, we first treated 4T1 cells with ethanol.30 Ethanol-treated 4T1 cells were then mixed with DCs in PBS at a ratio of 1:2. The entire process continued for 2 min at 37°C, and the cells were then collected and washed with RPMI 1640 medium and cultured continuously for six days under normal conditions, thus allowing the expression of peptide-loaded major histocompatibility complex (pMHC), co-stimulatory molecules, and lymph node homing receptors on the hybrid membrane.31 The characteristics of the hybrid membrane depend on its membrane proteins, and thus we used Western blotting analysis to measure protein markers on the hybrid membrane. The results showed that the cell membrane-specific markers pan cadherins and Na+/K+-ATPase were well preserved. In contrast, the Lamin B1 (nuclear envelope marker), calnexin (endoplasmic reticulum membrane marker) and HSP 60 (mitochondrial marker) did not appear on the hybrid membrane (Figure S1).
The hybrid membrane was next extruded with the MSN according to a reported procedure.26 In addition to the antigen and delivery system, adjuvants were another key element in the nanovaccine delivery system. R837 is an important adjuvant that interacts with tumor-associated antigens to produce tumor-specific immune responses and is involved in the regulation of the immune response. R837 was thus loaded into the MSN during the synthesis process.
Figure 1 and Table 1 show that coating a hybrid membrane to the MSN surface led to a subtle increase in the hydrodynamic diameter of MSN. The surface zeta potential of R837@HM-NPs increased from −17.5 to −15.6 mV, similar to that of hybrid membrane -derived vesicles. This also shows that the MSN had been successfully wrapped in a hybrid membrane. Key physicochemical parameters that affect lymphatic uptake have been identified, including size, charge, and hydrophobicity. Nanoparticles with an average diameter of less than 100 nm can be diffused and transported through the tissue interstitium, across the weak cell-cell junctions of the lymphatic endothelium, and subsequently drained into the lymph nodes. Moreover, the negative surface charge of R837@HM-NPs provides an additional advantage for lymph node targeting. Thus, the electrostatic repulsion of the negatively charged interstitial matrix allows the R837@HM-NPs to move faster and to accumulate more efficiently in the draining lymph nodes.32,33
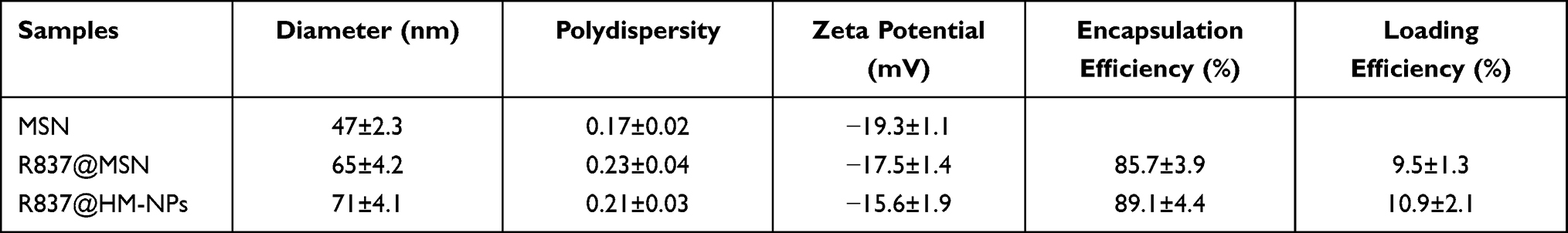 |
Table 1 Physicochemical Characterization of R837@HM-NPs |
 |
Figure 1 Physicochemical characterization of R837@HM-NPs. Size distributions (A–C) from dynamic light scattering analysis of MSN, R837@MSN and R837@HM-NPs. (D) Evidence for the coverage of hybrid membrane coated on the surface of MSN by confocal microscopy. (E) Size changes of R837@HM-NPs over seven successive days in PBS. (F) Zeta potential changes of R837@HM-NPs in seven successive days in PBS. (G) In vitro release profiles of R837 from R837@MSN and R837@HM-NPs in PBS (at pH 5.0 and pH 7.4). (H) Cell viability of BMDCs incubated with PBS, MSN and HM-NPs at different concentrations measured at 48 h. |
To verify whether the hybrid membrane was wrapped around the surface of MSN, we next labeled the hybrid membrane with DID dye. Figure 1D shows that a generally uniform red shell is observed on the surface of the MSN, indicating that the MSN was successfully covered by the hybrid membrane. The diameter of R837@HM-NPs in the PBS and RPMI 1640 medium containing 10% FBS remained stable for 7 days, indicating good stability in the physiological state. This in turn lays the foundation for in vivo biological application (Figures 1E and S2).14,34
The R837 content in the supernatant was measured to monitor the release kinetics of R837 from R837@MSN and R837@HM-NPs in PBS at pH 5.0 and 7.4. R837 had a burst release of 68% from R837@MSN during the first day in PBS at pH 7.4 followed by a slower and more sustained release, which was faster than the release behavior of R837 from R837@HM-NPs (42%) in the first day. These data indicate that the hybrid membrane can prevent premature drug release. Under the same conditions, the release rate at pH 5.0 was significantly higher than that at pH 7.4 (Figure 1G).
The safety of nanovaccine vector systems is a key issue that must be considered before biological applications. To evaluate the in vitro safety of MSN and HM-NPs, BMDCs and 4T1 cells were co-cultured with different concentrations of MSN and HM-NPs for 48 h, and their cell viability was assayed by MTT. As shown in Figures 1H and S3, the survival rates of both cells were not significantly different compared with the control group even at a concentration of 300 μg/mL, indicating that MSN and HM-NPs have good in vitro biocompatibility.
Cellular Uptake of HM-NPs
After the vaccine is injected into the body, the antigen is first processed by APCs (including macrophages and DCs) to activate the innate immune system.7 Hence, the cellular uptake function of nanovaccines is a critical prerequisite to evaluate nanovaccine effectiveness. To evaluate the uptake of HM-NPs by BMDCs, BMDCs were co-incubated with calcein-loaded MSN and HM-NPs and examined by CLSM and flow cytometry, respectively. Figure 2 shows that the cellular uptake efficiency in the HM-NPs group was significantly higher than that in the MSN group, which was consistent with the flow cytometry results. The enhanced cellular uptake of HM-NPs containing antigen and adjuvant by APCs facilitated a stronger immune response.
 |
Figure 2 Cellular uptake and endocytic mechanism of HM-NPs in BMDCs. (A) CLSM images of BMDCs after 4 h incubation with calcein-loaded MSN and HM-NPs. (B) Cellular uptake of MSN and HM-NPs in BMDCs by flow cytometric analyses after 4 h of incubation. Control indicates the autofluorescence of untreated cells. (C) Cellular uptake efficiency of R837@HM-NPs in BMDCs in the presence of different endocytosis inhibitors. |
DCs can achieve nanoparticle endocytosis through multiple mechanisms, including macropinocytosis, as well as clathrin and caveolae-dependent endocytosis.35 To elucidate the endocytic pathway of HM-NPs involved in BMDCs in detail, cellular uptake analysis was performed in the presence of various endocytosis inhibitors such as filipin, amiloride, chlorpromazine, monensin, methyl-β-cyclodextrin and ethylisopropylamiloride (Figure 2C). The results showed that filipin, amiloride, chlorpromazine, methyl-β-cyclodextrin and ethylisopropylamiloride showed significant inhibitory effects on the cellular internalization of HM-NPs. Amiloride and ethylisopropylamiloride were inhibitors of macropinocytosis and can block macropinocytosis by lowering submembrane pH and blocking Rac 1 and Cdc 42 signaling.36 In addition, filipin, chlorpromazine and methyl-β-cyclodextrin reduced HM-NP internalization by 30.81%, 43.12% and 49.24%, respectively, indicating that caveolae and clathrin-mediated endocytosis play an important role in HM-NP internalization in addition to macropinocytosis.
Activation of BMDCs in vitro
DCs are a typical APC and can deliver vaccine information to T cells and later play an important role in cancer immunotherapy.37 Maturation of DCs is often accompanied by antigen presentation and subsequent initiation of immune response, thus upregulation of co-stimulatory molecules (CD80, CD86) on the cell surface is often considered to be a marker of DC maturation.38,39 Upregulation of CD80 and CD86 was greatest in BMDCs in the R837@HM-NPs group as a result of the DC maturation versus treatment of R837 and R837@MSN (Figure 3A and B).
 |
Figure 3 The immune stimulatory effect of R837@HM-NPs on BMDCs in vitro. (A) Representative flow cytometry data to show BMDCs maturation induced by different treatments in vitro. (B) Representative statistical data to show BMDCs maturation induced by different treatments in vitro. ELISA results of IL-6 (C), IL-12p40 (D), and TNF-α (E) secretion from BMDCs after R837@HM-NPs treatment. (F) Flow cytometric analyses of the expression of CD4 and CD8, the markers for T cell activation, after in vitro incubation of T cells with PBS, R837, R837@MSN, HM-NPs, and R837@HM-NPs for 24 h. *P < 0.05, **P < 0.01. |
The immune-related cytokines secreted by BMDCs are another important indicator of DC maturation and were detected here using ELISA. Consistent with the BMDC maturation results, the R837@HM-NPs treatment induced the BMDCs to secrete more IL-6, IL-12p40 and TNF-ɑ (Figure 3C-E). Both IL-12 and TNF-ɑ are cytokines necessary for stimulating T cell proliferation and inducing tumor-specific CTL responses.40,41 Our data suggest that R837@HM-NPs activate BMDC maturation more effectively than free R837 and R837@MSN.
DCs can recognize antigens and then process them into antigen peptides in the form of pMHC when transferred to nearby draining lymph nodes, where pMHCs are presented to T cell receptors that activate T cells and kill cancer cells.42 Next, R837@HM-NPs-treated BMDCs were first co-cultured with T cells and their induction of T-cell activation was examined by flow cytometry. The results showed that the percentage of CD3+ CD8+ CTLs was higher in the R837@HM-NPs-treated BMDCs group than the other control groups (Figures 3F and S4). Activated T cells by R837@HM-NPs-treated DCs were next cultured with HBL-100, 4T1, and B16F10 cells at a number ratio of 5:1 (T cells versus HBL-100, 4T1, or B16F10 cells) to detect the killing ability of T cells on cancer cells. The results showed that the cell-killing effect of activated T cells on 4T1 cells was significantly stronger than the other cells; minimal cell toxicity was observed in HBL-100 and B16F10 groups (Figure S5), indicating that R837@HM-NPs have strong immunological specificity.
R837@HM-NPs as a Vaccine for Tumor Prevention
The tumor-preventive effect of the nanovaccine was next evaluated by a series of in vivo experiments. First, BALB/c mice were immunized with different formulations of vaccines and were challenged to study antitumor immunity. Then 4T1 cells were inoculated subcutaneously 7 days after the last immunization (Figure 4A). Mice in the R837@HM-NPs group had significantly smaller tumor volumes (Figure 4B and C) at all time points and significantly longer survival times than all other groups (Figure 4D). Consistent with the in vitro results (Figure 3), vaccination with R837@HM-NPs induced the highest expression of CD80 and CD86 in DCs at the inguinal lymph nodes of treated mice compared with that induced by R837 alone or R837@MSN treatment (Figures 4F and S6). Figure 4E shows that the R837@HM-NPs group had the highest expression of inflammatory factors (IL-6, IL-12p40 and TNF-α), confirming that it induces a stronger immune response. Next, the T cell responses induced by R837@HM-NPs in vivo were also investigated (Figures 4F and S7). The results showed that R837 induced 27.99% of CD8+ T cells, while the HM-NPs treatment led to a significant increase in the frequency of CD8+ T cells 40.14%. These data suggest that HM-NPs enhanced the response of systemic T cells in vivo. In addition, R837@HM-NPs were more effective than single HM-NPs, perhaps because of the synergistic activation of TLR and the hybrid membranes.
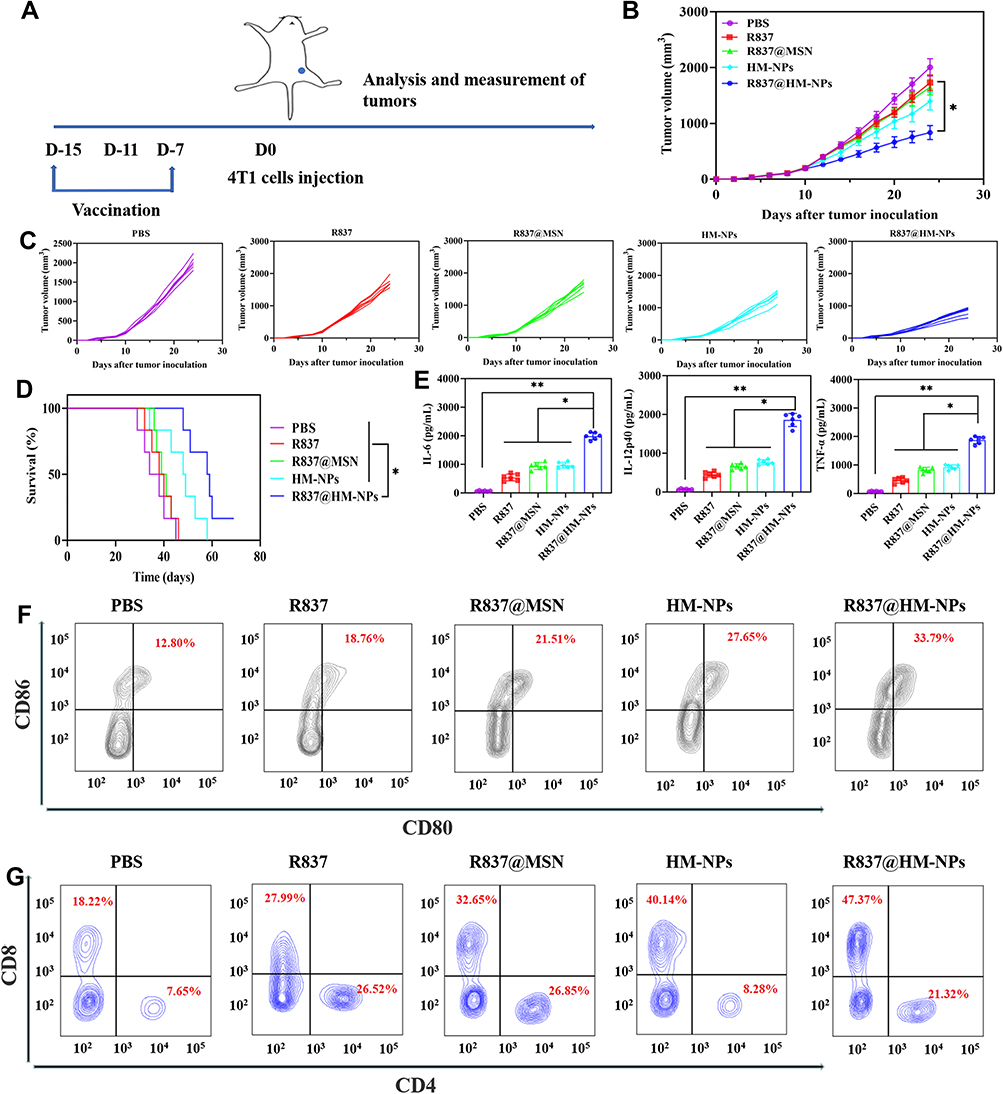 |
Figure 4 The preventive efficacy of R837@HM-NPs nanovaccine for tumor occurrence in vivo. (A) Schemes of tumor prevention experiment design. The average tumor growth curves (B) and individual tumor growth curves (C) for different groups of mice as indicated (n = 6). (D) Survival curves as a function of treatment type. (E) Cytokine concentrations including IL-6, IL-12p40 and TNF-α in serum from treated mice. (F) Representative flow cytometry data to show DCs maturation in lymph nodes induced by different treatments in vivo. (G) Flow cytometric analysis of CD4+ and CD8+ T cells in tumors, gated on CD3+ T cells. *P < 0.05, **P < 0.01. |
Therapeutic Effect of R837@HM-NPs in Combination with αPD-1 on Tumors
To evaluate the antitumor effect of R837@HM-NPs, we successfully established a 4T1 tumor model by subcutaneously injecting 4T1 cells into the left flank of each mouse. The flow chart of the in vivo experiments is shown in Figure 5A. At 24 d after injection of 4T1 cells, the tumor volume in PBS-treated mice was 2191 mm3, while a significant tumor growth inhibition was observed in R837@HM-NPs-treated mice with an average tumor volume of 646 mm3. Only injection of R837 slightly inhibited the tumor growth. It was particularly encouraging that the best treatment effect was observed in the R837@HM-NPs combined with αPD-1 group (Figures 5B, C and E and S8). In addition, the results of H&E staining and TUNEL assays in tumor tissues showed that R837@HM-NPs in combination with αPD-1 effectively promoted necrosis and apoptosis of tumor cells versus all other groups (Figure 5F and G). This is consistent with studies in recent years whereby cancer vaccines combined with immune checkpoint blockade can improve antitumor efficacy.43,44
 |
Figure 5 In vivo therapeutic effect of R837@HM-NPs nanovaccine in combination with αPD-1. (A) Schematic illustration of combining R837@HM-NPs-based vaccination with αPD-1 therapy to inhibit established 4T1 tumors. The average tumor growth curves (B) and individual tumor growth curves (E) for different groups of mice as indicated (n = 6). (C) Weights of tumors from 4T1-bearing mice receiving different treatments. (D) Body weight profiles of tumor-bearing mice during treatment. Representative H&E (F) and TUNEL (G) images of tumor tissues in different treatment groups at the end of the experiments. Cell nuclei were stained blue, and TUNEL was stained green. Scale bar, 100 μm (HE); 20 μm (TUNEL). (H) Representative immunofluorescence images of tumors showing CD8+ (green) and CD4+ (red) infiltration in the tumor tissues for all the treatment groups. Scale bar, 20 μm. *P < 0.05, **P < 0.01. |
To understand the mechanisms of how R837@HM-NPs in combination with αPD-1 act to enhance the antitumor immune response, we next analyzed the status of T cells and tumor microenvironment in this scenario in detail. First, immunofluorescence staining and flow cytometric analysis (Figures 5H and 6A and B) showed that the infiltrating CD8+ T cells were significantly higher in mice in the R837@HM-NPs+αPD-1 group than in all other groups, indicating the strongest T cell-mediated immune response in the combined treatment group. We next examined the status of Tregs in each group of mice. Tregs play a suppressive role in antitumor immunity by inhibiting CTL responses through multiple cellular and molecular mechanisms.45 The proportion of Tregs (Figure 6C and D) was reduced and the CD8+/Tregs ratio (Figure 6I) was significantly higher in the R837@HM-NPs+ αPD-1 group compared with the PBS group and the R837@MSN group. Similar results have been reported in other studies: R837 prevented the expansion of Tregs.2 In addition, R837@HM-NPs combined with αPD-1 induced a switch from anti-inflammatory to pro-inflammatory macrophages during treatment (Figure 6E-H). These findings suggest that R837@HM-NPs in combination with αPD-1 can favorably regulate the tumor microenvironment by decreasing Tregs, expanding the CD8+/Treg ratio, and promoting macrophage polarization to M1 phenotype. These steps lead to remodeling of the immune microenvironment of tumors making them sensitive to αPD-1 treatment. The secretion of cytokines is also important for stimulating antitumor immunity. Cytokines such as IL-6, IL-12p40 and TNF-α (Figure 6J-L) were detected in serum by ELISA. The highest levels of inflammatory factors in the R837@HM-NPs+αPD-1 group could be seen, further indicating that a combination of R837@HM-NPs with αPD-1 could induce a stronger immune response.
 |
Figure 6 The immune activation mechanism of R837@HM-NPs in combination with αPD-1 on inhibition of 4T1 tumor growth. Representative flow cytometry data of tumor infiltrating CD4+CD8+T cells (A), CD4+Foxp3+T cells (C), CD80+CD11b+F4/80+ cells (E), and CD206+CD11b+F4/80+ cells (G) for different groups of mice after various treatments. Representative statistical data of tumor infiltrating CD4+CD8+T cells (B), CD4+Foxp3+T cells (D), CD80+CD11b+F4/80+ cells (F), and CD206+CD11b+F4/80+ cells (H) for different groups of mice after various treatments. (I) Flow cytometric analysis assessing the relative abundance of CD8+/Treg for different groups of mice after various treatments. Cytokine levels of IL-6 (J), IL-12p40 (K), and TNF-α (L) in serum from tumor-bearing mice isolated at 48 h after the last injection. (M) Heat map analysis of ex tumors after receiving different treatments as indicated through transcriptome sequencing (RNA-seq). (N) Gene ontology biological process analysis of ex tumors after receiving different treatments as indicated. *P < 0.05, **P < 0.01. |
The number and activation status of tumor-infiltrating immune cells are closely related to the effects of immunotherapy.46 There was a strong immune response against the tumor when the number of infiltrating lymphocytes in tumor tissues was significantly increased.28,46,47 Therefore, we conducted a preliminary exploration of the antitumor molecular mechanisms in each treatment group. Figure 6M and N show a heat map analysis obtained from tumor transcriptome data: The combination treatment of R837@HM-NPs with αPD-1 resulted in a good accumulation of immune effector cells including B cells, granulocytes, macrophages, memory CD4+ T cells, NK cells, NK/T cells and T cells. The results were further confirmed by gene ontology biological process analysis, and the combination treatment group against 4T1 tumors exhibited a strong immune response.
Biosafety is an important prerequisite for clinical applications of nanovaccines. Therefore, we evaluated toxicity in vivo. Body weight was normal in all groups, indicating that no toxicity occurred during treatment with the various agents (Figure 5D). There were no obvious pathological changes in major organs such as heart, liver, lung, spleen, and kidney (Figure 7A). In addition, the biochemical indexes and blood count of the mice remained in the normal range, indicating that R837@HM-NPs combined with αPD-1 therapy is safe (Figure 7B).
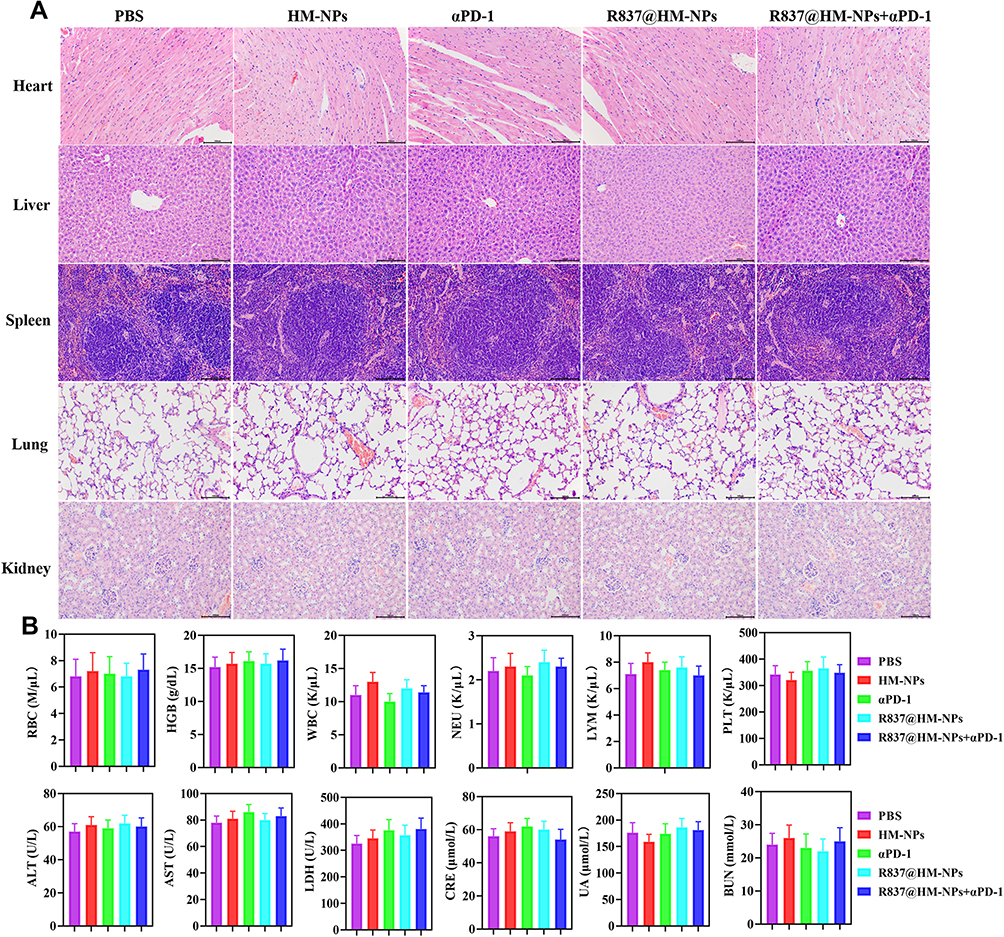 |
Figure 7 Biocompatibility analysis of R837@HM-NPs in combination with αPD-1. (A) Representative histological examinations of H&E staining organs from mice under different treatments. Scale bars, 100 µm. (B) Biosafety analysis in the aspect of blood routine, liver function and renal function (n = 6). |
Immune Memory Effect R837@HM-NPs in Combination with αPD-1 on Rechallenged Tumors
The organism undergoes immunization with tumor-specific antigens to produce the immune memory effect and provide long-term immune protection against tumor recurrence.48 Thus, we investigated the efficacy of hybrid membrane nanovaccine in the prevention of tumor recurrence through immune memory effects. BALB/c mice were inoculated subcutaneously into the left flank of the mice with 5×106 4T1 cells on day 0 and then immunized with surgery, αPD-1, R837@HM-NPs and R837@HM-NPs+αPD-1. A second batch of 4T1 tumor cells was then inoculated subcutaneously into the right flank of the mice on day 25 (Figure 8A). The results showed that R837@HM-NPs+αPD-1 realized the best tumor therapeutic benefit and significantly prolonged the median survival time of mice (Figures 8B and C and S9).
 |
Figure 8 The long-term immunological memory effects of R837@HM-NPs nanovaccine in combination with αPD-1 therapy against the rechallenged 4T1 tumor model. (A) Schematic illustration of R837@HM-NPs nanovaccine in combination with αPD-1 therapy against the rechallenged 4T1 tumor model. (B) Tumor growth curves of the right tumor for different groups of mice (n = 6). (C) Survival curve of mice treated with various formulations after inoculation of the right tumor. Representative flow cytometry data (D) and statistical data (E and F) of TEM and TCM in the spleen analyzed by flow cytometry (gated on CD3+CD8+ T cells) on day 35. (G) Concentration of IFN-γ in serum from tumor-bearing mice isolated on day 35.*P < 0.05, **P < 0.01. |
To further investigate the immunological memory effect induced by the combined immunotherapy, spleen was next collected from mice in each treatment group on day 35. Memory CD8+T cells were classified into TEM and TCM subsets based on the expression pattern of CD62L and CD44.49 The results showed that the percentage of TEM and TCM in mice was significantly higher in the combination group of R837@HM-NPs+ αPD-1 than in other control groups (Figure 8D-F). In addition, TEM could produce protective immune responses such as cytokines (eg, IFN-γ) after a second encounter with the same pathogen. As shown in Figure 8G, higher concentrations of IFN-γ were obtained in the combination treatment group. The results indicated that a combination of R837@HM-NPs and αPD-1 produced the strongest immune memory response and prevented tumor recurrence.
Conclusions
In summary, we developed a novel and efficacious vaccine nanoplatform composed of hybrid membranes obtained from DCs and cancer cells for antigen delivery and homogenous targeting, MSN as a nanocarrier, and R837 as an immune adjuvant. R837@HM-NPs can effectively stimulate DC maturation, promote antigen presentation, and induce DCs to secrete large amounts of pro-inflammatory cytokines. In vivo experiments confirmed that a combination of R837@HM-NPs and αPD-1 therapy not only effectively inhibited tumor growth and prolonged the survival time of tumor-bearing mice by promoting CD8+ T cell-mediated immune response and modulating the tumor immunosuppressive microenvironment, but also suppressed tumor recurrence. This synergistic treatment approach can safely and effectively trigger a stronger immune response, thus offering broad prospects for clinical applications.
Acknowledgments
The authors gratefully acknowledge National Natural Science Foundation of China (Grant 81402575). We thank LetPub (www.letpub.com) for its linguistic assistance and scientific consultation during the preparation of this manuscript.
Disclosure
The authors declare no financial or non-financial conflicts of interest for this work.
References
1. Finck A, Gill SI, June CH. Cancer immunotherapy comes of age and looks for maturity. Nat Commun. 2020;11(1):3325. doi:10.1038/s41467-020-17140-5
2. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21(6):360–378. doi:10.1038/s41568-021-00346-0
3. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–2501. doi:10.1056/NEJMc1713444
4. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–150. doi:10.1038/s41568-019-0116-x
5. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi:10.1126/science.aaa8172
6. Park JA, Cheung NV. Limitations and opportunities for immune checkpoint inhibitors in pediatric malignancies. Cancer Treat Rev. 2017;58:22–33. doi:10.1016/j.ctrv.2017.05.006
7. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–222. doi:10.1038/nrc.2016.154
8. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18(3):168–182. doi:10.1038/nri.2017.131
9. Qiu F, Becker KW, Knight FC, et al. Poly(propylacrylic acid)-peptide nanoplexes as a platform for enhancing the immunogenicity of neoantigen cancer vaccines. Biomaterials. 2018;182:82–91. doi:10.1016/j.biomaterials.2018.07.052
10. Kroll AV, Fang RH, Jiang Y, et al. Nanoparticulate delivery of cancer cell membrane elicits multiantigenic antitumor immunity. Adv Mater. 2017;29:47. doi:10.1002/adma.201703969
11. Ochyl LJ, Bazzill JD, Park C, et al. PEGylated tumor cell membrane vesicles as a new vaccine platform for cancer immunotherapy. Biomaterials. 2018;182:157–166. doi:10.1016/j.biomaterials.2018.08.016
12. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–1036. doi:10.1038/ni.3518
13. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi:10.1038/nri2173
14. Liu WL, Zou MZ, Liu T, et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat Commun. 2019;10(1):3199. doi:10.1038/s41467-019-11157-1
15. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–680. doi:10.1038/90609
16. Fleer A, Krediet TG. Innate immunity: toll-like receptors and some more. A brief history, basic organization and relevance for the human newborn. Neonatology. 2007;92(3):145–157. doi:10.1159/000102054
17. Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc Natl Acad Sci U S A. 2014;111(34):12294–12299. doi:10.1073/pnas.1400478111
18. Dowling JK, Mansell A. Toll-like receptors: the Swiss army knife of immunity and vaccine development. Clin Transl Immunol. 2016;5(5):e85. doi:10.1038/cti.2016.22
19. Vacchelli E, Galluzzi L, Eggermont A, et al. Trial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1(6):894–907. doi:10.4161/onci.20931
20. Hu CM, Zhang L, Aryal S, et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. doi:10.1073/pnas.1106634108
21. Zhang C, Long L, Xiong Y, et al. Facile engineering of indomethacin-induced paclitaxel nanocrystal aggregates as carrier-free nanomedicine with improved synergetic antitumor activity. ACS Appl Mater Interfaces. 2019;11(10):9872–9883. doi:10.1021/acsami.8b22336
22. Rajput MKS, Kesharwani SS, Kumar S, et al. Dendritic cell-targeted nanovaccine delivery system prepared with an immune-active polymer. ACS Appl Mater Interfaces. 2018;10(33):27589–27602. doi:10.1021/acsami.8b02019
23. Zhao P, Qiu L, Zhou S, et al. Cancer cell membrane camouflaged mesoporous silica nanoparticles combined with immune checkpoint blockade for regulating tumor microenvironment and enhancing antitumor therapy. Int J Nanomedicine. 2021;16:2107–2121. doi:10.2147/IJN.S295565
24. Zhao Q, Sun X, Wu B, et al. Construction of homologous cancer cell membrane camouflage in a nano-drug delivery system for the treatment of lymphoma. J Nanobiotechnology. 2021;19(1):8. doi:10.1186/s12951-020-00738-8
25. Ni Q, Zhang F, Liu Y, et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci Adv. 2020;6(12):eaaw6071. doi:10.1126/sciadv.aaw6071
26. Zhao P, Li L, Zhou S, et al. TPGS functionalized mesoporous silica nanoparticles for anticancer drug delivery to overcome multidrug resistance. Mater Sci Eng C Mater Biol Appl. 2018;84:108–117. doi:10.1016/j.msec.2017.11.040
27. Liu D, Liu J, Ma B, et al. A simple self-adjuvanting biomimetic nanovaccine self-assembled with the conjugate of phospholipids and nucleotides can induce a strong cancer immunotherapeutic effect. Biomater Sci. 2021;9(1):84–92. doi:10.1039/d0bm01333a
28. Zhang D, Lin Z, Wu M, et al. Cytosolic delivery of thiolated neoantigen nano-vaccine combined with immune checkpoint blockade to boost anti-cancer T cell immunity. Adv Sci (Weinh). 2021;8(6):2003504. doi:10.1002/advs.202003504
29. Hilton HG, Rubinstein ND, Janki P, et al. Single-cell transcriptomics of the naked mole-rat reveals unexpected features of mammalian immunity. PLoS Biol. 2019;17(11):e3000528. doi:10.1371/journal.pbio.3000528
30. Song W, Kuang J, Li CX, et al. Enhanced immunotherapy based on photodynamic therapy for both primary and lung metastasis tumor eradication. ACS Nano. 2018;12(2):1978–1989. doi:10.1021/acsnano.7b09112
31. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38(8):577–593. doi:10.1016/j.it.2017.05.006
32. Rao DA, Forrest ML, Alani AW, Kwon GS, Robinson JR. Biodegradable PLGA based nanoparticles for sustained regional lymphatic drug delivery. J Pharm Sci. 2010;99(4):2018–2031. doi:10.1002/jps.21970
33. Liu H, Irvine DJ. Guiding principles in the design of molecular bioconjugates for vaccine applications. Bioconjug Chem. 2015;26(5):791–801. doi:10.1021/acs.bioconjchem.5b00103
34. Lv Y, Liu M, Zhang Y, et al. Cancer cell membrane-biomimetic nanoprobes with two-photon excitation and near-infrared emission for intravital tumor fluorescence imaging. ACS Nano. 2018;12(2):1350–1358. doi:10.1021/acsnano.7b07716
35. Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi:10.1146/annurev.immunol.22.012703.104538
36. Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188(4):547–563. doi:10.1083/jcb.200908086
37. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5(8):617–628. doi:10.1038/nri1670
38. Noh YW, Jang YS, Ahn KJ, Lim YT, Chung BH. Simultaneous in vivo tracking of dendritic cells and priming of an antigen-specific immune response. Biomaterials. 2011;32(26):6254–6263. doi:10.1016/j.biomaterials.2011.05.013
39. Yang R, Xu J, Xu L, et al. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano. 2018;12(6):5121–5129. doi:10.1021/acsnano.7b09041
40. Huang L, Mellor AL. Metabolic control of tumour progression and antitumour immunity. Curr Opin Oncol. 2014;26(1):92–99. doi:10.1097/CCO.0000000000000035
41. Liu Q, Chen X, Jia J, et al. pH-Responsive poly(D,L-lactic-co-glycolic acid) nanoparticles with rapid antigen release behavior promote immune response. ACS Nano. 2015;9(5):4925–4938. doi:10.1021/nn5066793
42. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35(2):814–824. doi:10.1016/j.biomaterials.2013.10.003
43. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489–496. doi:10.1038/nmat4822
44. Chao Y, Xu L, Liang C, et al. Combined local immunostimulatory radioisotope therapy and systemic immune checkpoint blockade imparts potent antitumour responses. Nat Biomed Eng. 2018;2(8):611–621. doi:10.1038/s41551-018-0262-6
45. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. doi:10.1038/nri1806
46. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi:10.1038/ni.2703
47. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–821. doi:10.1038/s41423-020-0488-6
48. Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi:10.1111/j.0105-2896.2006.00391.x
49. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi:10.1038/44385
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.