")
Back to Journals » International Journal of Nanomedicine » Volume 11
Hyaluronan-conjugated liposomes encapsulating gemcitabine for breast cancer stem cells
Authors Han, Shin DH, Kim JS, Weon KY, Jang C, Kim J
Received 6 September 2015
Accepted for publication 9 December 2015
Published 5 April 2016 Volume 2016:11 Pages 1413—1425
DOI https://doi.org/10.2147/IJN.S95850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
Na-Kyung Han,1,* Dae Hwan Shin,1,* Jung Seok Kim,1 Kwon Yeon Weon,2 Chang-Young Jang,1 Jin-Seok Kim1
1Research Center for Cell Fate Control (RCCFC) and College of Pharmacy, Sookmyung Women’s University, Seoul, 2College of Pharmacy, Catholic University of Daegu, Gyeongbuk, Korea
*These authors contributed equally to this work
Abstract: Investigation of potential therapeutics for targeting breast cancer stem cells (BCSCs) is important because these cells are regarded as culprit of breast cancer relapse. Accomplishing this kind of strategy requires a specific drug-delivery system using the distinct features of liposomes. Studies on targeted liposomal delivery systems have indicated the conjugation of hyaluronan (HA), a primary ligand for CD44 surface markers, as an appropriate method for targeting BCSCs. For this study, enriched BCSCs were obtained by culturing MCF-7 breast cancer cells in nonadherent conditions. The enriched BCSCs were challenged with HA-conjugated liposomes encapsulating gemcitabine (2, 2-difluoro-2-deoxycytidine, GEM). In vitro study showed that the HA-conjugated liposomes significantly enhanced the cytotoxicity, anti-migration, and anti-colony formation abilities of GEM through targeting of CD44 expressed on BCSCs. In pharmacokinetic study, area under the drug concentration vs time curve (AUC) of the immunoliposomal GEM was 3.5 times higher than that of free GEM, indicating that the HA-conjugated liposomes enhanced the stability of GEM in the bloodstream and therefore prolonged its half-life time. The antitumor effect of the immunoliposomal GEM was 3.3 times higher than that of free GEM in a xenograft mouse model, probably reflecting the unique targeting of the CD44 receptor by HA and the increased cytotoxicity and stability through the liposomal formulation. Furthermore, marginal change in body weight demonstrated that the use of liposomes considerably reduced the systemic toxicity of GEM on normal healthy cells. Taken together, this study demonstrates that HA-conjugated liposomes encapsulating GEM show promise for the therapy of breast cancer in vitro and in a xenograft model by targeting the BCSCs.
Keywords: breast cancer stem cells, targeting, CD44 surface marker, EPR effect, drug delivery system
Introduction
Breast cancer is the most common cancer among women and one of the leading causes of cancer death worldwide.1 It is also considered the main cause of mortality and morbidity in women.2,3 Breast cancer presents as malignant tumors with invasion into normal healthy breast tissue and usually progresses or recurs after radiation therapy, indicating that the presence of a small fraction of breast cancer cells can cause regrowth of tumor cells.4 These cells are called breast cancer-initiating cells (BCICs) or breast cancer stem cells (BCSCs).5 Increasing data also indicate that most kinds of malignant solid cancers may include cancer stem cells (CSCs).5–9 Normal healthy stem cells have their own mechanisms that make them particularly resistant to anticancer drugs, such as enhanced multidrug resistance and increased expression of BCL-2 family proteins or producing proteins resistant to breast cancer drug.10–13 The increased expression of these proteins may enhance the resistance of BCSCs to current anticancer therapies.5 For this reason, an improved therapeutic strategy for targeting BCSCs is required to eliminate breast cancer. Generally, each type of CSC has its own cell surface markers.4 The population of BCSCs in breast cancer can be identified as CD44+/CD24−.5 Even though the functions of CD44 in BCSCs are not completely understood, recent studies indicate that advanced anticancer strategies to specifically eliminate BCSCs are needed to efficiently suppress malignant cancers and decrease the risk of recurrence.4,5 In this study, we suggest a drug-delivery system for targeting BCSCs using a CD44 marker and liposomes to suppress cancer migration and enhance the efficacy of breast cancer therapies.
Hyaluronan (HA), an extracellular matrix component, is an anionic high-molecular-weight glycosaminoglycan. HA can combine with several cell surface receptors including CD44.14,15 Some studies have reported that the use of HA as a ligand in a targeted delivery system is an effective strategy for cancer therapy.16–21 It is also well known that the use of polyethylene glycol (PEG) reduces immunogenic response and creates a hydrophilic barrier, which enables the delivery system to circulate in the body for an extended period.22–24 A previous study indicated that HA conjugated with polymer has PEG-like properties, creating a hydrophilic stealth shield and prolonging circulation time.25
Gemcitabine (2, 2-difluoro-2-deoxycytidine, GEM), a deoxycytidine analog, is known as an effective anticancer agent.26 It is effective against various types of cancers. In combination therapy, it can be used to treat ovarian cancer, breast cancer, and non-small-cell lung carcinoma.27 Recent studies suggest that the toxicity of GEM can be delivered through gap junctions. This phenomenon is known as the “bystander effect”, suggesting that anticancer therapy with GEM could be significantly enhanced in solid tumors that contain gap junctions.28,29 However, GEM must be administered frequently and at a very high dose due to its short half-life (32–94 minutes), resulting in cytotoxicity to healthy normal cells as well.30 Liposome-mediated targeted delivery can decrease the systemic toxicity of chemotherapeutics and overcome the resistance to anticancer agents, including GEM, thereby enhancing therapeutic effect.31–33 Even though the rapid degradation in the blood is one of the critical limitations of liposomes, this problem can be overcome by conjugating PEG or HA on the liposomes.22–25
The induction of apoptosis and suppression of cancer cell growth are significantly increased by liposomal delivery of GEM.34–36 Specific targeting of BCSCs can be accomplished by modifying liposomes with HA, a ligand for the CD44 surface marker, which is known to be overexpressed in BCSCs.
The goal of this study is to design an advanced targeted liposomal formulation against BCSCs including GEM as the “payload”. HA-conjugated liposomes were used to reduce the toxicity and improve the liposomal stability and cellular uptake of GEM into BCSCs. The enhancement of in vivo stability, pharmacokinetic properties, and the antitumor effect of the drug after intravenous injection can be achieved by using HA-conjugated liposomes as a drug-delivery vehicle. In this study, we suggest that HA-liposomal GEM (HA-L-GEM) may provide evidence for a new therapeutic strategy to eliminate malignant cancers, including breast cancer, by selectively targeting BCSCs.
Materials and methods
Materials
GEM HCl, known as Gemzar (Eli Lilly and Company, Indianapolis, IN, USA), was purchased from Shinwon Pharmacy Co. (Seoul, Korea). Cy5.5-labeled GEM was customized from GeneChem (Daejeon, Korea). Cholesterol, Hyaluronan (HA; molecular weight: 0.6–1.1 MDa), 1-ethyl-3-(3-dimethyl-aminopropyl-carbodiimide) (EDC), hexadecyltrimethylammonium bromide (CTAB), accutase, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), poly-2-hydroxyethyl methacrylate (polyHEMA), basic fibroblast growth factor (bFGF), human epidermal growth factor (hEGF), Sephadex G-75, insulin solution, crystal violet, dimethyl sulfoxide (DMSO), glutaraldehyde, bovine serum albumin (BSA), chloroform, boric acid, β-estradiol 17-cypionate, and methanol were purchased from Sigma-Aldrich (St Louis, MO, USA). Anti-CD24-fluorescein isothiocyanate (FITC) antibody, anti-CD44-phycoerythrin (PE) antibody, and isotype control antibodies were purchased from eBioscience (San Diego, CA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), Dulbecco’s phosphate-buffered saline (DPBS), trypsin-ethylenediaminetetraaceticacid (EDTA), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from WelGENE Inc. (Daegu, Korea). B-27 supplement and DMEM/F12 were purchased from CureBio (Seoul, Korea). l-α-Phosphatidylcholine from egg (EPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Bicinchoninic acid (BCA) protein assay kits were purchased from Bio-Rad (Hercules, CA, USA). All solvents and reagents were of analytical grade or of better quality.
Cell culture and identification of BCSCs
MCF-7 human breast cancer cells, purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea), were cultured as monolayers in DMEM containing 10% FBS. BCSCs were separated from MCF-7 cells as previously described.37,38 Briefly, MCF-7 cells were resuspended in stem cell-permissive medium including serum-free DMEM/F12 containing 5 μg/mL insulin, 0.01 μg/mL bFGF, 0.02 μg/mL hEGF, and 0.4% BSA and were seeded in polyHEMA-coated six-well plates at 40,000 cells/mL. Under this condition, the MCF-7 cancer cell population decreased while the MCF-7 CSC population increased and formed nonadherent sphere-shaped cell clusters, known as mammospheres.39 The mammospheres were harvested by centrifugation, then further placed at 10,000 cells/mL in the stem cell-permissive medium. After continuous culture in serum-free medium for 3 weeks, mammospheres were collected for identification of MCF-7 BCSC phenotype and purity. Briefly, the collected mammospheres were dissociated by accutase and washed in DPBS using gentle agitation. Immunostaining was performed after incubation with anti-CD24-FITC, anti-CD44-PE, or isotype controls for 30 minutes at 4°C in DPBS. The samples were then rinsed three times with DPBS and resuspended with 500 μL of DPBS. Flow cytometric analysis was carried out on a FACSCalibur™ flow cytometer (Becton Dickinson, San Jose, CA, USA) after removing the cell debris, dead cells, or doublets using forward/side scatter profiles.5
Tumor sphere-formation assay
The single cells of MCF-7 and BCSC obtained by enzymatic dissociation were plated in a 96-well plate with 100 μL of DMEM/F12 medium without serum. Every 3 days, 20 μL of medium was added to each well. The efficacy of clonogenesis was determined by calculating the percentage of wells including mammospheres that contained more than 50 cells on day 14. The spheres generated in each well were counted under a bright-field microscope.
Preparation of GEM formulations
Liposomes composed of EPC, DOPE, and cholesterol (3:1:1, molar ratio) were prepared by a freezing/thawing method.40 Briefly, 50 mmol of the lipid mixture was mixed with a methanol–chloroform (1:3, v/v) mixture in a round-bottomed flask, then the methanol–chloroform mixture was evaporated in a rotary evaporator (Laborota 4000; Heidolph Instruments, Milan, Italy) to make a thin film of lipid. The film of lipid was rehydrated with 1 mL of GEM solution and then subjected to nine cycles of freezing (liquid nitrogen) and thawing (65°C water bath), thereby accomplishing an increased encapsulation efficiency. Downsized unilamellar vesicles (ULVs) were obtained from multilamellar vesicles (MLVs) by extrusion through 450 and 200 nm cellulose membrane filters (Whatman Int, Ltd., Pittsburgh, PA, USA) five times each. Gel filtration using Sephadex G-75 (Sigma-Aldrich) was performed to remove unincorporated drug from ULVs.
EDC coupling for amide synthesis was used for conjugation of HA onto the surface of the liposomes.25,41 Briefly, 4 mg/mL of HA solution was activated with EDC for 2 hours at 37°C, followed by buffering with 0.1 M HCl to the final pH of 4. Then, the activated HA solution was mixed with liposomal GEM (L-GEM) (1:1 molar ratio of HA:liposome), followed by buffering with 0.1 M borated buffer to the final pH of 8.6, and incubated overnight at 37°C. The unreacted components were separated from the final product (HA-L-GEM) by Sephadex G-75 gel filtration.
Characterization of liposomal GEM
The zeta potential and size distribution of L-GEM and HA-L-GEM were measured by a dynamic laser light scattering system (NICOMP 380ZLS; Particle Sizing Systems Inc., Santa Barbara, CA, USA).
The encapsulation efficiency of drug into liposomes was measured using the Bligh and Dyer extraction method.42 Briefly, 250 μL methanol, 150 μL DPBS, and 1 mL chloroform were mixed with 100 μL of liposome sample. The mixture was centrifuged to separate into two layers: an upper aqueous phase including GEM and other hydrophilic materials and a lower organic phase including phospholipids and other hydrophobic materials. After removal of the organic phase, the aqueous phase was again mixed with fresh chloroform and centrifuged. These procedures were repeated three times to completely remove phospholipids in aqueous phase. Absorbance of the aqueous phase was measured at 268 nm. The encapsulation efficiency of GEM (%) and drug loading (%) were calculated using the following equation:
|
|
Determination of the amount of attached HA to liposome
The amount of phospholipids in the liposomes was determined by Bartlett’s phosphorus assay using KH2PO4 solutions as a standard.43 Briefly, 100 μL of phospholipid solution from each liposome sample obtained by the Bligh and Dyer extraction method and the standard KH2PO4 solutions were added to 400 μL of 10 N sulfuric acid in borosilicate glass tubes.42 The samples were heated at 165°C for 30 minutes. After cooling, 30 μL of hydrogen peroxide (30%) was mixed with each sample and the mixture was reheated at 165°C for 30 minutes. After further cooling, 4.6 mL of ammonium molybdate solution (22%, w/w) was mixed with each sample, followed by the addition of 200 μL of Fiske–Subbarow reagent. Then the samples were heated for 10 minutes. After cooling, the absorbance of each standard and sample solution was determined at 830 nm.
The amount of HA conjugated to liposome was quantified by the CTAB turbidimetric method using HA solutions as a standard.44,45 Briefly, 50 μL of HA samples extracted from each liposome solution by the Bligh and Dyer extraction method and the HA standard solutions were placed on a 96-well plate.42 The samples were mixed with 0.05 mL of 0.2 M sodium acetate buffer and incubated at 37°C for 10 minutes. Then CTAB solution was mixed with each sample and the absorbance of precipitation was determined at 570 nm.
Uptake of GEM by cells
Cy5.5-labeled GEM was used for observation of cellular uptake. BCSCs were incubated with DPBS, liposomes without drug (L-empty), HA-conjugated liposomes without drug (HA-L-empty), GEM in solution (free GEM), L-GEM, or HA-L-GEM. After 4 hours of incubation, cells were rinsed with DPBS and quantified by a flow cytometer. Fluorescent images of the cells were taken using a fluorescence microscope (IX71IX51; Olympus, Tokyo, Japan).
Inhibition of proliferation in MCF-7 cells and BCSCs
All studies including experiments related to human-derived cell lines were approved by the ethical review board of Sookmyung Women’s University. The anti-proliferation effect of various GEM formulations on MCF-7 cells and BCSCs was measured by an MTT assay. The MCF-7 cells and BCSCs were dissociated and plated in a 96-well plate at 2,500 cells/well density in 0.2 mL of medium containing serum to permit cells to attach to the plate. The media in each well was replaced with media containing DPBS, L-empty, HA-L-empty, free GEM (1 μM), L-GEM (1 μM), or HA-L-GEM (1 μM). After 48 hours of incubation, an MTT assay was performed. Briefly, MTT solution was mixed with each sample and incubated for 4 hours, after which the media was aspirated and DMSO was added to solubilize the formazan crystals. The plate was gently stirred and the absorbance of the samples was measured at 570 nm using an ELISA reader (EL 800; BioTek, Winooski, VT, USA). Cell growth (% of control) was calculated by the following equation:
|
where OD570(Sample) is the absorbance of the cells after treatment of the various GEM formulations, OD570(Control) is the absorbance of the cells after treatment of DPBS, and OD570(Original) is the absorbance of the cells at the time of sample treatment.
Clonogenic survival assay
A clonogenic assay was performed to evaluate the inhibitory ability of various GEM formulations on a single BCSC to grow into a colony.46 BCSCs were seeded in a six-well plate (2,000 cells/well) with serum-containing medium for cell adhesion. After 4 hours of incubation, medium containing DPBS (control), L-empty, HA-L-empty, free GEM (1 μM), L-GEM (1 μM), or HA-L-GEM (1 μM) was added to each well. After 14 days, media was aspirated and colonies were stained with crystal violet. The cells were rinsed gently with DPBS for three times. After drying, the colonies were observed and scored. The percentage of colony formation was calculated by the following equation:
|
In vitro anti-migration assay
To evaluate the migratory ability of the BCSCs, a wound-healing assay was performed.47 BCSCs were cultured in six-well plates with medium containing serum. After attachment of the cells on the bottom of the plates, an empty line was made in each plate by detaching a small fraction of the cells with a sterile pipette tip. The detached cells were gently removed from each well by washing with DPBS. The medium containing DPBS (control), L-empty, HA-L-empty, free GEM (1 μM), L-GEM (1 μM), or HA-L-GEM (1 μM) was then added to each well. Wound closure was quantified 24 hours after drug incubation. The cellular migratory rates were calculated by comparing the width of the wound before and after treatment.
In vitro protein adsorption assay
To evaluate the stability of liposomes in the blood, the amount of proteins adsorbed onto the liposomes was measured. When liposomes are unstable and aggregated, the amount of proteins adsorbed onto them is increased.48,49 Briefly, L-empty, L-GEM, HA-L-empty, or HA-L-GEM was mixed with 10% BSA (w/v) and incubated at 37°C. After 0.5, 3, 6, 24, and 48 hours of incubation, unadsorbed BSA was removed by centrifugation. BSA–liposome complexes were rinsed with DPBS for three times. The amount of adsorbed proteins was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
Pharmacokinetic study
All the animal studies were approved by the Institutional Animal Care and Use Committee of Sookmyung Women’s University (SMU-IACUC), Korea. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. The pharmacokinetic profile of various formulation was measured using Cy5.5-labeled GEM. Institute for Cancer Research (ICR) mice (male, 6 weeks old, 25–30 g) were intravenously (iv) injected with free GEM, L-GEM, or HA-L-GEM at a dose of 0.45 mg/kg. Blood samples of 0.1 mL were collected from the retro-orbital sinus at 1, 5, 10, 15, 30, 60, 120, 240, and 480 minutes after injection. The blood samples were centrifuged, after which 40 μL of serum was collected and subjected to extraction process using 10% Triton-X and methanol. After extraction, the amount of drug in each sample was quantified by measuring the fluorescent intensity using a plate reader at λex (excitation wavelength) =685 nm and λem (emission wavelength) =705 nm.
In vivo antitumor study using BCSC-xenografted mice
To establish the BCSC-induced tumor model, female immunodeficient nude mice (Charles River Laboratory, Yokohama, Japan) were used as described elsewhere.50,51 β-Estradiol 17-cypionate was subcutaneously injected into each mouse at a dose of 3 mg/kg once a week as a supplement for BCSC tumor growth, because the growth of MCF-7 tumors depends on the estrogen concentration in the body. Each group of mice was treated with DPBS (control, n=3) or various formulations of GEM (free GEM, L-GEM, or HA-L-GEM, 25 mg/kg, n=6) twice a week for 3 weeks. The size of the tumors was measured using a vernier caliper (Mitutoyo Co., Kanagawa, Japan), and the animals were weighed twice weekly. Tumor volume was calculated using the following equation:
|
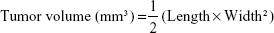
Statistical analysis
All the results except in vivo data are presented as the mean value ± standard deviation (SD). The parameters of the animal study are presented as the mean value ± standard error (SE). Statistical analysis was performed with a Student’s t-test. Statistical significance was assigned for P-value <0.05 (95% confidence level) or <0.01 (99% confidence level).
Results
Characterization of BCSCs
MCF-7 cells adhered to the surface of the culture dish and grew as monolayers, whereas BCSCs grew as nonadherent mammospheres (Figure 1A). For characterization of phenotypes for MCF-7 cells and BCSCs, both cells were mixed with anti-CD24-FITC and anti-CD44-PE. The population of BCSCs was identified as CD44+/CD24−.52 The CD44+/CD24− fraction of BCSCs and MCF-7 cells was 74.2% and 5.4%, respectively (Figure 1B). Another defining feature of BCSCs is their sphere-forming ability.39 Single-cell suspensions prepared from MCF-7 cells and BCSCs were incubated in stem-cell permissive medium and evaluated for their ability to form secondary spheres from single cells. Figure 1C and D shows that 71.3% of wells seeded with single cells from BCSCs exhibited secondary sphere formation, whereas only 11.9% of wells seeded with those from MCF-7 cells showed sphere formation. Thus, compared to MCF-7 cells, BCSCs separated from MCF-7 exhibited typical CSC-like features. Collectively, these results confirm the successful isolation of BCSCs from the MCF-7 cell population and verify their identity.
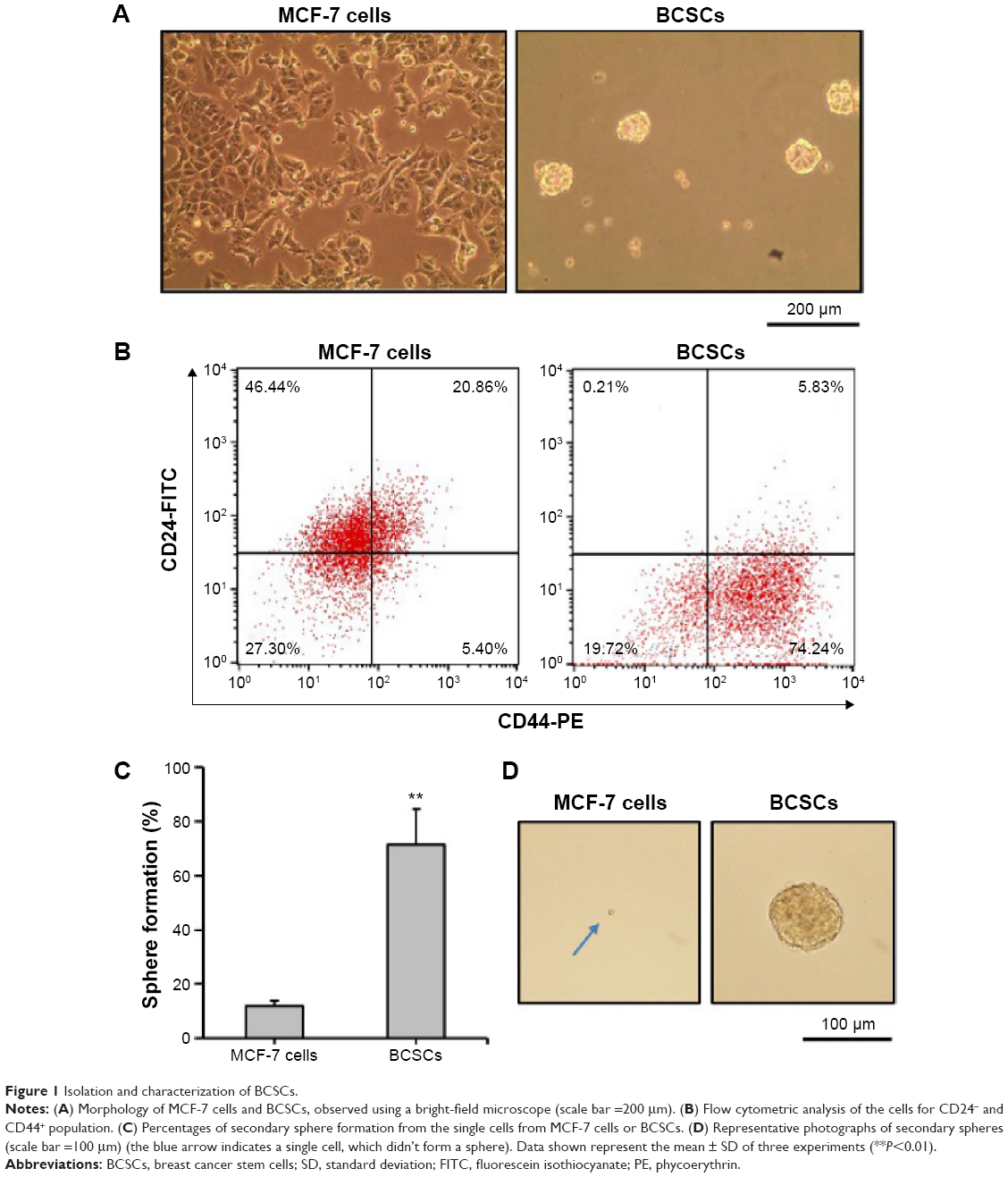 | Figure 1 Isolation and characterization of BCSCs. |
Characterization of GEM formulations
The mean diameters of L-GEM and HA-L-GEM were 177±15 and 212±15 nm, respectively. The zeta potentials were −3.49±0.73 mV and −19.0±3.9 mV for L-GEM and HA-L-GEM, respectively, due to anionic properties of HA. The encapsulation efficiencies of GEM were 14.6%±1.9% and 15.6%±2.5% for L-GEM and HA-L-GEM, respectively (Table 1). The amounts of liposome-conjugated HA and phospholipids in the liposomes were quantified by the Bradford assay and phosphorus assay, respectively, resulting in the ratio of HA:lipid to be 97.1±19.5 μg HA/μmol lipid.
 | Table 1 Mean diameter, zeta potential, encapsulation efficiency, and drug loading of L-GEM and HA-L-GEM |
Uptake of GEM by the cells
Various GEM formulations were tested for internalization into the BCSCs using Cy5.5-labeled GEM. As shown in Figure 2A, the cellular uptake of GEM at 4 hours after treatment was quite different for each of the various formulations. Free GEM showed little internalization by BCSCs, whereas L-GEM was internalized much more readily. Notably, GEM in HA-conjugated liposomes was internalized to a significantly greater extent than other formulations. Internalization percentages of GEM in BCSCs at 4 hours after treatment with DPBS (control), L-empty, HA-L-empty, free GEM, L-GEM, and HA-L-GEM, quantified by flow cytometry, were 1.93%, 2.00%, 1.85%, 20.3%, 28.9%, and 42.7%, respectively (Figure 2B). Specifically, the uptake of HA-L-GEM was two times higher than that of free GEM, probably reflecting the specific binding of HA to CD44 in BCSCs and resulting in increased uptake by the cells.
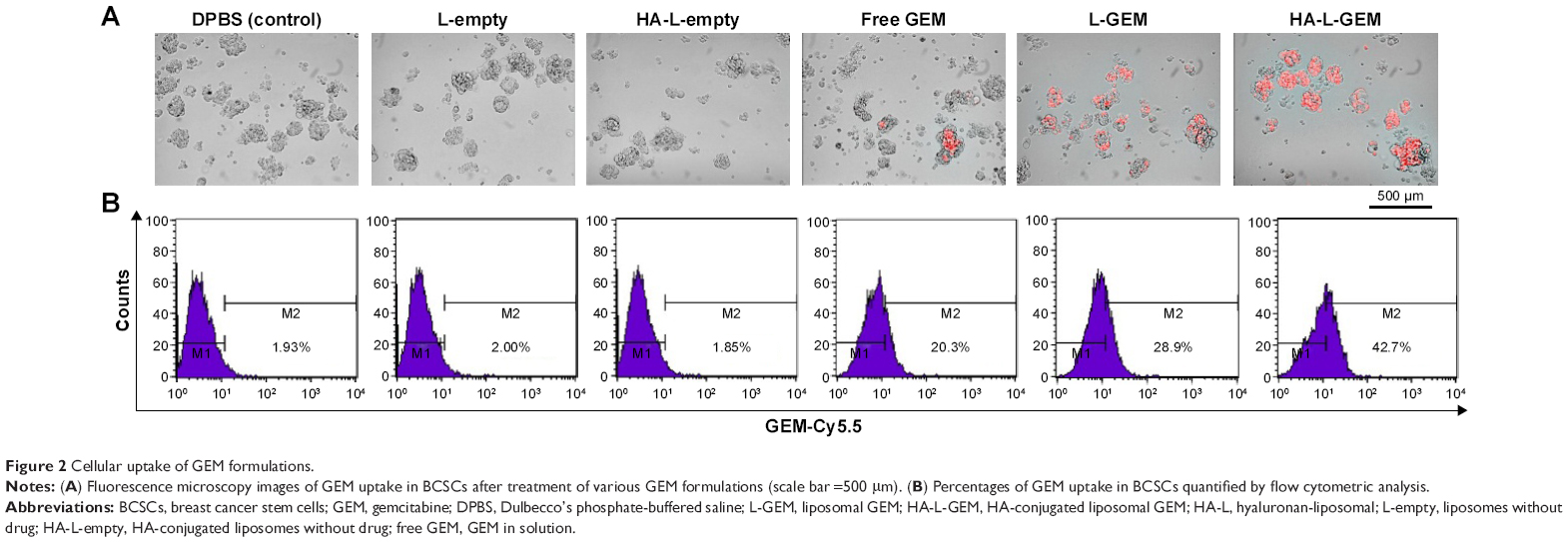 | Figure 2 Cellular uptake of GEM formulations. |
Inhibition of proliferation of MCF-7 cells and BCSCs
To evaluate the growth inhibition of the cells by GEM, MCF-7 cells or BCSCs were treated with various GEM formulations for 48 hours. As shown in Figure 3, the growth-inhibitory effect of L-empty and HA-L-empty on the cells was similar to the DPBS (control) group. On the other hand, the GEM group showed that BCSCs have enhanced chemo-resistance compared with MCF-7 cells. In MCF-7 cells, there was no evident difference between the free GEM (49.3%) and L-GEM (46.9%) groups, whereas a growth-inhibitory effect was enhanced when the cells were treated with HA-L-GEM (27.5%). The degree of growth inhibition on BCSCs was highest for HA-L-GEM, followed by L-GEM and free GEM. Also, the data from the HA-L-GEM group showed that targeting of the CD44 surface marker was more effective in BCSCs than in MCF-7 cells, presumably due to overexpression of CD44 in BCSCs.
 | Figure 3 Anti-proliferation effect of various formulations on MCF-7 cells and BCSCs. |
Clonogenic assay
Long-term cytotoxicity of various GEM formulations on BCSCs was measured by a clonogenic assay. Figure 4 shows that colony formation of the L-empty (94.4%) or HA-L-empty (91.5%) group was similar to that of the DPBS (control, 100%) group. On the other hand, colony formation was decreased when the cells were treated with free GEM (67.2%) and L-GEM (40.1%). Of all the formulations, HA-L-GEM was most effective, decreasing colony formation to 23.8%.
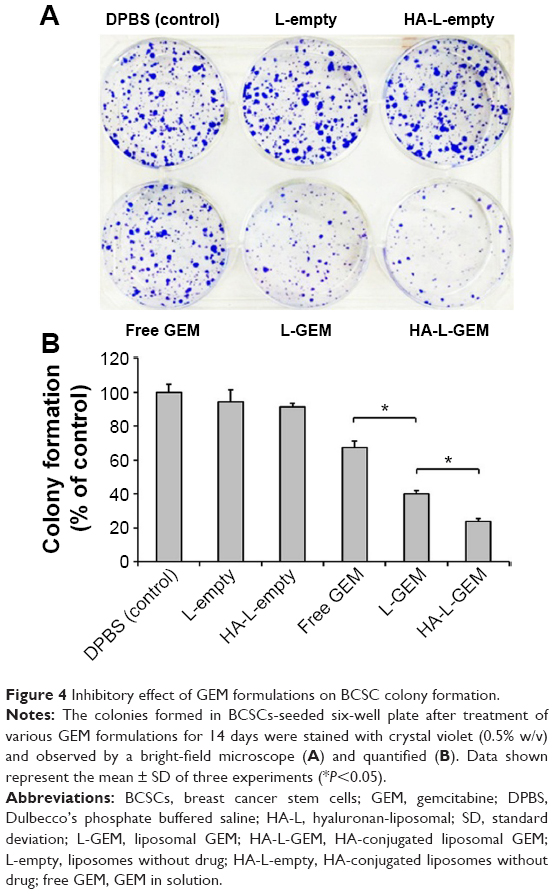 | Figure 4 Inhibitory effect of GEM formulations on BCSC colony formation. |
In vitro anti-migration assay
We investigated the effect of GEM formulations on tumor metastasis by evaluating the migratory capacity of BCSCs. As demonstrated in Figure 5, the migratory rates, calculated at 0 and 24 hours, show a considerable recovery of the empty area treated by DPBS (control, 100%), L-empty (92.3%), or HA-L-empty (92.1%). However, the cell migration rate was reduced by treatment of free GEM (72.1%) or L-GEM (50.7%). The greatest reduction occurred under treatment with HA-L-GEM (22.9%), indicating it is most effective at inhibiting migration among the cells tested. Taken together, these data suggest that HA-L-GEM is highly effective in attenuation of BCSC migration.
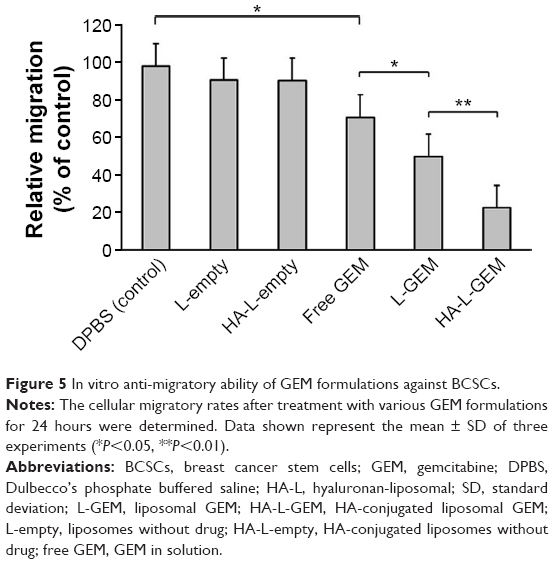 | Figure 5 In vitro anti-migratory ability of GEM formulations against BCSCs. |
In vitro serum protein adsorption assay
The stability of liposomes in the bloodstream is an important issue. One of the major factors causing the instability of liposomes is the presence of serum proteins. Serum proteins can aggregate with liposomes during circulation in the blood and break the lipid bilayer of liposomes, inducing unexpected drug leakage. Thus, the amount of proteins aggregated with liposomes was determined by BCA protein assay. As shown in Figure 6, the amount of proteins aggregated with non-HA-conjugated liposomes (L-empty and L-GEM) was significantly increased over the course of a 48-hour incubation period, whereas a very small amount of proteins was adsorbed onto the HA-conjugated liposomes (HA-L-empty and HA-L-GEM). Specifically, the amount of proteins aggregated with HA-conjugated liposomes was approximately two times less than that of nonconjugated liposomes (P<0.05). The absence of significant aggregation of proteins onto the HA-conjugated liposomes seems to reflect the fact that HA protects the liposome from interaction with serum proteins by forming a steric shield that can block the penetration of serum proteins into the liposomes.
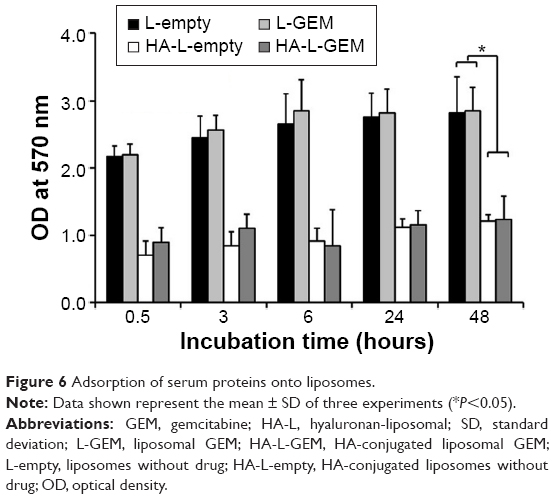 | Figure 6 Adsorption of serum proteins onto liposomes. |
Pharmacokinetic study
Pharmacokinetic behavior of GEM in various liposomal formulations was determined after iv injection into mice. As shown in Figure 7, free GEM quickly disappeared from the blood, whereas GEM from L-GEM and HA-L-GEM remained in the blood for much longer time. Based on a two-compartment model, pharmacokinetic parameters were calculated and described in Table 2.
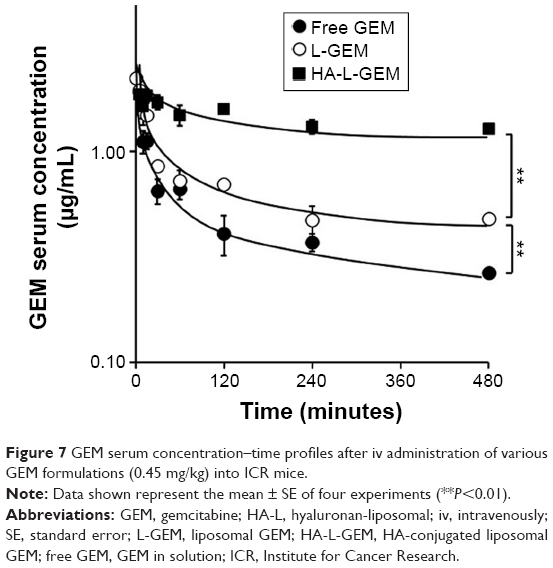 | Figure 7 GEM serum concentration–time profiles after iv administration of various GEM formulations (0.45 mg/kg) into ICR mice. |
 | Table 2 Pharmacokinetic parameters of GEM after iv injection of a variety of GEM formulations (0.45 mg/kg) |
With the HA-L-GEM formulation, both area under the curve (AUC) and half-life (t1/2α and t1/2β) increased, whereas total clearance time (CLt) decreased. Compared to the free GEM-treated group, the mean AUC values of the L-GEM- and HA-L-GEM-treated groups were elevated by approximately two and 3.5 times and the CLt decreased by approximately two and 4.5 times, respectively. These data indicated that using HA-conjugated liposomal formulation as a delivery carrier for GEM has an overall pharmacokinetic benefit.
Antitumor study in xenografted mice
The antitumor capacity of various GEM formulations was determined in BCSC-xenografted nude mice by measuring the tumor volumes after intravenous administration. The drug injections were given on days 0, 4, 7, 11, 14, and 18. Three weeks after the first injection, the mean tumor volume in the DPBS-treated (control) group was approximately five times bigger than the initial volume in the same group (256 vs 50 mm3), while that in the HA-L-GEM-treated group was only ~1.5 times bigger than the initial volume (76 vs 50 mm3), over a threefold reduction in tumor volume. The mean tumor volume in HA-L-GEM-treated group was smaller than that in free GEM or L-GEM-treated group. This result demonstrated that treatment with HA-L-GEM exerted an enhanced effect on the inhibition of BCSC-induced tumor growth (Figure 8A). Treatment with free GEM caused significant weight loss compared with DPBS control (~10%), suggesting a gross or systemic toxicity (Figure 8B), whereas neither L-GEM nor HA-L-GEM treatment caused severe weight loss. This result clearly suggests that the L-GEM formulations, both L-GEM and HA-L-GEM, are much less toxic than the free GEM.
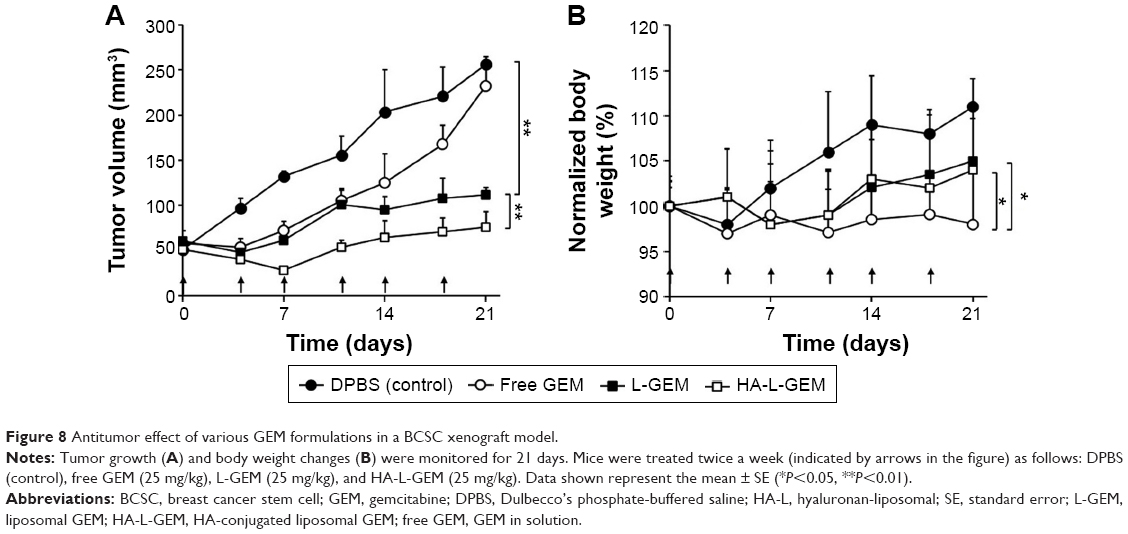 | Figure 8 Antitumor effect of various GEM formulations in a BCSC xenograft model. |
Discussion
Although its application for BCSC-targeted therapy shows a limited therapeutic efficacy, GEM has proven to be an effective drug against breast cancer in animal studies.53 To overcome the drawbacks and improve the inhibitory capacity of GEM against BCSC, a novel HA-conjugated GEM-encapsulated liposome is designed for targeting the CD44 receptor on BCSCs in this study.
To perform the targeting study, we isolated the BCSCs from the MCF-7 breast cancer cells using the CD44+/CD24− and confirmed their properties (Figure 1).37,38 The physicochemical characteristics of various types of liposomal formulations incorporating GEM were also evaluated by measuring the zeta potential and particle size. As proposed by Matsumura and Maeda, the enhanced permeability and retention effect of specific sized molecules, such as liposomes, is known to make these molecules accumulate in tumor sites at much higher concentrations than in nontumor sites.54 Liposomes with a diameter over 400 nm do not circulate well in the bloodstream owing to their rapid elimination by reticuloendothelial system (RES), while those with a size near 200 nm are known to stay in circulation for a longer period.55,56 The kinetics, stability, biodistribution, and interactions of liposome with targeted cells were affected by the surface charge on the shell. Liposomes with a negative charge rarely tend to aggregate and are comparatively much more stable in suspension during storage than those with a positive or neutral charge.57 Hence, we designed the liposomes with a suitable zeta potential (-20 to 0 mV) and particle diameter (~200 nm) for targeting BCSC (Table 1).
Metastasis is a key factor in the prognosis of breast cancer. CD44 serves not only as a marker of primary or parental BCSC but also as a marker of metastatic CSC.58 The migration and proliferation of tumor cells are important processes in metastasis.59 Metastasis of cancer is a complicated activity in which malignant cells such as CSCs split from the tumor, adhere to the degraded proteins of the nearby extracellular matrix, and move to different sites through the lymphatic fluid or the bloodstream.60 Therefore, cellular uptake, growth inhibition, clonogenic, and anti-migration (wound-healing) assays were performed to test the inhibitory effect of HA-L-GEM on these malignant CD44+ cells (BCSCs). HA, a targeting ligand, dramatically improved the liposomal delivery of GEM through the endocytosis of CD44, resulting in elevated GEM uptake into BCSCs (Figure 2). This finding is consistent with the data of MTT (short-term) and clonogenic (long-term) cytotoxicity assays, which collectively revealed that HA-L-GEM shows an improved growth-inhibitory effect on BCSCs (Figures 3 and 4). In addition, a wound-healing assay demonstrated that the migratory rate of BCSCs in HA-L-GEM-treated group was much smaller than that in other groups (Figure 5). This clearly shows that HA-L-GEM very effectively prevents the migration of BCSCs, suggesting the effective inhibition of metastasis in vivo. We also confirmed that stability of the HA-conjugated liposomes is much higher than that of non-HA-conjugated liposomes in the presence of plasma serum protein (Figure 6), due to the steric hindrance generated by the conjugation of HA and the resulting repulsive interactions between the HA and serum proteins. These results proved the advantage of HA-conjugated liposomes as an appropriate drug-delivery system for targeting BCSCs in vivo.
Based on these findings, we selected the HA-L-GEM for the pharmacokinetic and antitumor studies in vivo. To measure the effect of liposomal encapsulation and HA conjugation on the pharmacokinetics of HA-conjugated liposomes, we compared the pharmacokinetic properties of HA-L-GEM to those of free GEM and L-GEM. Indeed, HA-L-GEM showed prolonged circulation time owing to liposomal encapsulation and HA conjugation (Figure 7 and Table 2). Therefore, drugs were effectively protected by HA-conjugated liposomal encapsulation from RES uptake.
HA-L-GEM exerted significantly enhanced antitumor ability, inducing remission of BCSC-derived tumors in the xenografted nude mice (Figure 8A). Furthermore, minimal body weight loss of the animals proved that the HA attachment dramatically decreased the toxicity of GEM for normal healthy cells compared with the free GEM group (Figure 8B).
Based on these results, several sequential mechanisms can be suggested to explain this strong in vivo anti-BCSC effect of HA-L-GEM (Figure 9). After intravenous injection, GEM in the formulation is protected from RES uptake by steric shield created by HA-conjugated liposome, resulting in prolonged circulation in the blood. Because of the appropriate physicochemical properties of HA-L-GEM, enhanced permeability and retention effect helps to increase accumulation of the formulation in the tumor. The HA-L-GEM reaching the tumor site can selectively be internalized by BCSC due to the increased binding ability, which was proven in the uptake study in vitro. Bystander effect, one of the GEM-related anticancer effects,28,29 helps GEM in the BCSC to spread to the surrounding cancer cells through gap junctions, resulting in “multi-targeting” of BCSC and its surrounding cancer cells. Therefore, the HA-conjugated liposomal formulation incorporating GEM, described here, efficiently inhibited the growth of tumors by various anticancer mechanisms.
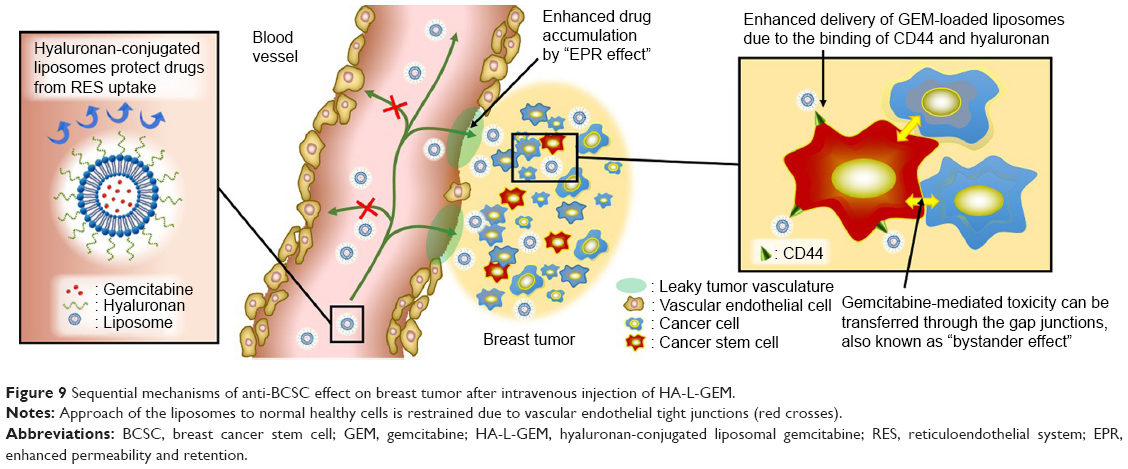 | Figure 9 Sequential mechanisms of anti-BCSC effect on breast tumor after intravenous injection of HA-L-GEM. |
Conclusion
We designed a new HA-conjugated liposomal formulation incorporating GEM for selectively targeting BCSCs and determined its anti-BCSC efficacy in vitro and in vivo. This delivery system enhanced the delivery of GEM to the BCSCs owing to the binding of HA to the CD44 surface marker, thus promoting effective regression of BCSCs with less toxicity for normal cells. On the basis of these findings, it is clearly suggested that this liposomal formulation has a high potential to be used as a promising delivery system for GEM treatment in breast cancer therapy.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (Ministry of Science, ICT and Future Planning [MSIP]) (no 2011-0030074) and the Basic Science Research Program through the NRF funded by the MSIP (2013R1A1A2062456 and 2014R1A2A2A01 004353). The authors would like to thank Ms Soobin Oh for her technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Warner E. Clinical practice. Breast-cancer screening. N Engl J Med. 2011;365(11):1025–1032. | ||
Vo AT, Millis RM. Epigenetics and breast cancers. Obstet Gynecol Int. 2012;2012:602720. | ||
Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66(4):1883–1890; discussion 1895–1896. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. | ||
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–110. | ||
Matsui W, Huff CA, Wang Q, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103(6):2332–2336. | ||
Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. | ||
Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89(7):1021–1031. | ||
Lagasse E, Weissman IL. bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med. 1994;179(3):1047–1052. | ||
Domen J, Gandy KL, Weissman IL. Systemic overexpression of BCL-2 in the hematopoietic system protects transgenic mice from the consequences of lethal irradiation. Blood. 1998;91(7):2272–2282. | ||
Zhou S, Schuetz JD, Bunting KD, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7(9):1028–1034. | ||
Turley EA, Noble PW, Bourguignon LY. Signaling properties of hyaluronan receptors. J Biol Chem. 2002;277(7):4589–4592. | ||
Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61(7):1303–1313. | ||
Coradini D, Pellizzaro C, Miglierini G, Daidone MG, Perbellini A. Hyaluronic acid as drug delivery for sodium butyrate: improvement of the anti-proliferative activity on a breast-cancer cell line. Int J Cancer. 1999;81(3):411–416. | ||
Luo Y, Prestwich GD. Synthesis and selective cytotoxicity of a hyaluronic acid-antitumor bioconjugate. Bioconjug Chem. 1999;10(5):755–763. | ||
Auzenne E, Ghosh SC, Khodadadian M, et al. Hyaluronic acid-paclitaxel: antitumor efficacy against CD44(+) human ovarian carcinoma xenografts. Neoplasia. 2007;9(6):479–486. | ||
Akima K, Ito H, Iwata Y, et al. Evaluation of antitumor activities of hyaluronate binding antitumor drugs: synthesis, characterization and antitumor activity. J Drug Target. 1996;4(1):1–8. | ||
Eliaz RE, Szoka FC Jr. Liposome-encapsulated doxorubicin targeted to CD44: a strategy to kill CD44-overexpressing tumor cells. Cancer Res. 2001;61(6):2592–2601. | ||
Lee H, Mok H, Lee S, Oh YK, Park TG. Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels. J Control Release. 2007;119(2):245–252. | ||
Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88(24):11460–11464. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145(3):178–181. | ||
Peer D, Margalit R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal Doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia. 2004;6(4):343–353. | ||
Carmichael J, Possinger K, Phillip P, et al. Advanced breast cancer: a phase II trial with gemcitabine. J Clin Oncol. 1995;13(11):2731–2736. | ||
Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17 Suppl 5:v7–v12. | ||
Pitts JD. Cancer gene therapy: a bystander effect using the gap junctional pathway. Mol Carcinog. 1994;11(3):127–130. | ||
Cottin S, Ghani K, de Campos-Lima PO, Caruso M. Gemcitabine intercellular diffusion mediated by gap junctions: new implications for cancer therapy. Mol Cancer. 2010;9:141. | ||
Grunewald R, Abbruzzese JL, Tarassoff P, Plunkett W. Saturation of 2′,2′-difluorodeoxycytidine 5′-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol. 1991;27(4):258–262. | ||
Choi M, Shin DH, Kim J. Repositioning of zoledronic acid for breast cancer using transferrin-conjugated liposome. J Pharm Investig. 2013;43:461–469. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Kim B, Shin DH, Kim HD, Kim JS. Antitumor effect of a newly synthesized celecoxib derivative encapsulated in liposome. J Pharm Investig. 2013;43(2):101–106. | ||
Cosco D, Bulotta A, Ventura M, et al. In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer. Cancer Chemother Pharmacol. 2009;64(5):1009–1020. | ||
Celia C, Malara N, Terracciano R, et al. Liposomal delivery improves the growth-inhibitory and apoptotic activity of low doses of gemcitabine in multiple myeloma cancer cells. Nanomedicine. 2008;4(2):155–166. | ||
Celia C, Calvagno MG, Paolino D, et al. Improved in vitro anti-tumoral activity, intracellular uptake and apoptotic induction of gemcitabine-loaded pegylated unilamellar liposomes. J Nanosci Nanotechnol. 2008;8(4):2102–2113. | ||
Zhang L, Yao HJ, Yu Y, et al. Mitochondrial targeting liposomes incorporating daunorubicin and quinacrine for treatment of relapsed breast cancer arising from cancer stem cells. Biomaterials. 2012;33(2):565–582. | ||
Ma X, Zhou J, Zhang CX, et al. Modulation of drug-resistant membrane and apoptosis proteins of breast cancer stem cells by targeting berberine liposomes. Biomaterials. 2013;34(18):4452–4465. | ||
Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–5511. | ||
Mayer LD, Hope MJ, Cullis PR, Janoff AS. Solute distributions and trapping efficiencies observed in freeze-thawed multilamellar vesicles. Biochim Biophys Acta. 1985;817(1):193–196. | ||
Peer D, Margalit R. Loading mitomycin C inside long circulating hyaluronan targeted nano-liposomes increases its antitumor activity in three mice tumor models. Int J Cancer. 2004;108(5):780–789. | ||
Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. | ||
Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234(3):466–468. | ||
Di Ferrante N. Turbidimetric measurement of acid mucopolysaccharides and hyaluronidase activity. J Biol Chem. 1956;220(1):303–306. | ||
Song J-M, Im J-H, Kang J-H, Kang D-J. A simple method for hyaluronic acid quantification in culture broth. Carbohydr Polym. 2009;78(3):633–634. | ||
Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–2319. | ||
Rodriguez LG, Wu X, Guan JL. Wound-healing assay. Methods Mol Biol. 2005;294:23–29. | ||
Kim CE, Lim SK, Kim JS. In vivo antitumor effect of cromolyn in PEGylated liposomes for pancreatic cancer. J Control Release. 2012;157(2):190–195. | ||
Shin DH, Xuan S, Kim WY, Bae GU, Kim JS. CD133 antibody-conjugated immunoliposomes encapsulating gemcitabine for targeting glioblastoma stem cells. J Mater Chem B. 2014;2(24):3707–3898. | ||
Varricchio L, Migliaccio A, Castoria G, et al. Inhibition of estradiol receptor/Src association and cell growth by an estradiol receptor alpha tyrosine-phosphorylated peptide. Mol Cancer Res. 2007;5(11):1213–1221. | ||
Yun BL, Cho N, Li M, et al. Intratumoral heterogeneity of breast cancer xenograft models: texture analysis of diffusion-weighted MR imaging. Korean J Radiol. 2014;15(5):591–604. | ||
Fillmore C, Kuperwasser C. Human breast cancer stem cell markers CD44 and CD24: enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007;9(3):303. | ||
Jones J, Takeda A, Tan SC, Cooper K, Loveman E, Clegg A. Gemcitabine for the treatment of metastatic breast cancer. Health Technol Assess. 2009;13 Suppl 2:1–7. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Maruyama K, Yuda T, Okamoto A, Kojima S, Suginaka A, Iwatsuru M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim Biophys Acta. 1992;1128(1):44–49. | ||
Liu D, Mori A, Huang L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim Biophys Acta. 1992;1104(1):95–101. | ||
Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. | ||
Liu H, Patel MR, Prescher JA, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A. 2010;107(42):18115–18120. | ||
Tang J, Zhang L, She X, et al. Inhibiting CD164 expression in colon cancer cell line HCT116 leads to reduced cancer cell proliferation, mobility, and metastasis in vitro and in vivo. Cancer Invest. 2012;30(5):380–389. | ||
Guo S, Xiao D, Liu H, Zheng X, Liu L, Liu S. Interfering with CXCR4 expression inhibits proliferation, adhesion and migration of breast cancer MDA-MB-231 cells. Oncol Lett. 2014;8(4):1557–1562. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.