Back to Archived Journals » International Journal of High Throughput Screening » Volume 5
Human pluripotent stem cells as tools for high-throughput and high-content screening in drug discovery
Authors Allison T, Powles-Glover N, Biga V, Andrews P, Barbaric I
Received 15 November 2014
Accepted for publication 21 January 2015
Published 11 March 2015 Volume 2015:5 Pages 1—13
DOI https://doi.org/10.2147/IJHTS.S58113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Wayne Wahls
Thomas F Allison,1 Nicola S Powles-Glover,2 Veronica Biga,3 Peter W Andrews,1 Ivana Barbaric1
1Centre for Stem Cell Biology, Department of Biomedical Science, University of Sheffield, Sheffield, 2AstraZeneca, Alderley Edge, 3Centre for Signal Processing and Complex Systems, Department of Automatic Control and Systems Engineering, University of Sheffield, Sheffield, UK
Abstract: A significant bottleneck in drug discovery is the lack of suitable models for sensitive, reliable, and rapid assessment of lead molecules in preclinical stages of drug discovery. Human pluripotent stem cells (hPSCs) derived either from early human blastocysts (human embryonic stem cells) or by reprogramming somatic cells to a pluripotent state (human-induced pluripotent stem cells) can be propagated extensively in vitro while retaining the ability to differentiate into any specialized cell type within the body. In this review, we discuss how these unique features of hPSCs could offer a way of producing relevant in vitro models amenable to high-throughput testing for drug discovery. We summarize recent progress in inducing differentiation of hPSCs to specific cell types, and describe the ongoing efforts in applying hPSCs and their differentiated derivatives in disease modeling, drug discovery, and developmental toxicology. Moreover, we review the applications of high-content imaging assays in detecting the changes in the phenotype of hPSCs and their differentiated progeny. Finally, we highlight challenges that need to be overcome in order for the application of hPSC technology to fully benefit drug discovery.
Keywords: human pluripotent stem cells, drug discovery, high-content assays
Introduction
Drug discovery is an expensive and lengthy process, hampered by high attrition rates (∼90%) of new drug candidates.1 Two key factors contributing to attrition are lack of efficacy and safety concerns.2 Current preclinical programs for drug safety include both in vivo and in vitro tests, with later preclinical stages relying mainly on animal data prior to progression into man. Older retrospective reviews estimated that preclinical tests failed to detect adverse drug reactions in humans in as many as 30% of cases.3 A more recent review still showed that unacceptable safety is one of the most important reasons for failure, accounting for more than half of all project closures, and that the majority of these failures occurred before clinical testing.4 This review also highlights the importance of having a solid understanding of the drug target biology and the disease indication for a successful outcome of a drug project.4
As previously indicated, in vitro models are widely utilized in preclinical development, and these are often humanized models. A particular advantage of in vitro assays is the opportunity for a high-throughput approach, which would enable filtering out unsuitable compounds at early stages of drug development. However, most of the in vitro studies use either primary cells or transformed cell lines, and both cell sources have significant drawbacks. Primary human lines can be difficult to source, and their tendency to senesce rapidly in culture contributes to reduced robustness of in vitro assays due to batch-to-batch variability of cells.5 Development of transformed cell lines has alleviated the issue of senescence. However, transformed cell lines often harbor large genomic aberrations, and consequently, they can significantly differ from the cell type or tissue of origin. Thus, there is a significant and urgent need for improved assays that can reliably predict human response to drugs.
The derivation of human pluripotent stem cells (hPSCs) has opened up potential new and powerful avenues to tackle the persistent issues in drug development. In this review, we summarize the main features that hPSCs posses that make them amenable to high-throughput assays for testing drug safety and efficacy. Furthermore, we describe the ongoing efforts in applying hPSC-based assays to drug discovery and delineating the mechanisms of action of new drug candidates. Finally, we discuss tools and methods that need to be improved to ensure that hPSC-based assays fulfill the potential of transforming the landscape of drug discovery.
Characteristics of hPSCs and their potential for applications in drug discovery
hPSCs include human embryonic stem cells (hESCs) derived from early human blastocysts6 and human-induced pluripotent stem cells (hiPSCs) created by reprogramming somatic cell types to a pluripotent state.7,8 Several important characteristics distinguish hPSCs from either primary or immortalized cell lines. hPSCs are karyotypically normal cells that can grow in culture extensively due to their unlimited self-renewing capacity.6 Because of their ability to differentiate into any cell type in vitro, hPSCs can provide unlimited supply of differentiated cell types.9 This unique dual ability to self-renew and to differentiate makes hPSCs an ideal source of cells for drug discovery applications, whereby undifferentiated cells could be expanded and directed to differentiate into a cell type of interest (Figure 1). For such an approach to be advantageous over the existing in vitro models, the expansion and maintenance of large numbers of undifferentiated cells and their subsequent differentiation to desired cell types must be robust, reliable, and efficient. However, efficient mass culture of hPSCs has been hampered by a tendency of these cells to undergo apoptosis, particularly when plated as single cells or at a low plating density.10,11 The molecular mechanism underpinning this pronounced susceptibility to apoptosis involves activation of the Rho-associated coiled-coil-containing kinase pathway, which in turn causes actomyosin hyperactivation of dissociated cells.11,12 More recently, hPSCs were also shown to have constitutively activated Bax, a proapoptotic protein that controls activation of caspases.13 The priming for cell death in early embryonic cells may have evolved as a protection mechanism against genetic damage in the developing embryo.14,15 However, in the context of in vitro culture, poor viability of cells creates conditions for culture adaptation, whereby genetic variants harboring mutations that allow cell growth under suboptimal conditions may expand and overtake the culture.16–19 Differences in the behavior of culture-adapted cells compared to their wild-type counterparts, including resistance to apoptosis20,21 and reduced tendency to differentiate,22,23 could be detrimental if variant cells were to be used in applications such as drug screening and toxicology. Hence, the issue of culture adaptation brought sharply into focus the need for optimizing culture conditions for hPSC expansion and maintenance.
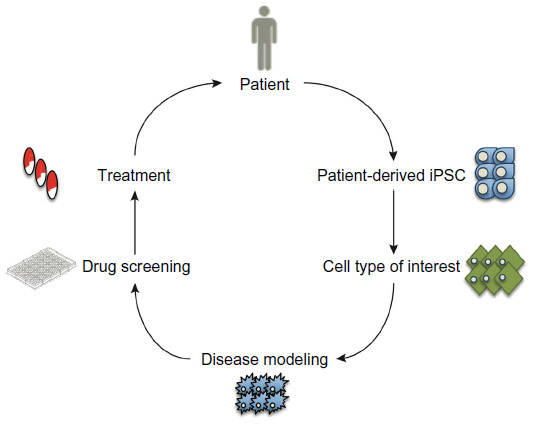 | Figure 1 A model for drug discovery using patient-derived hiPSCs. |
The first hPSC lines were derived and maintained in two-dimensional culture system on feeder layers of mitotically inactivated mouse embryonic fibroblasts (MEFs) in a medium supplemented with fetal bovine serum.6 Factors contained within the serum and secreted by MEFs support the self-renewal of hPSCs. However, such ill-defined culture conditions are highly undesirable for downstream applications of hPSCs as they create variability and may introduce harmful pathogens. The search for improved and chemically defined conditions for hPSCs growth revealed their dependency on fibroblast growth factor (FGF) and Activin/Nodal signaling.24,25 Bone morphogenetic protein 4 (BMP4) is an inductive signal for differentiation of hPSCs,26 in contrast to its role in blocking differentiation of mouse PSCs.27 These and additional findings provided a basis for development of chemically defined media which generally use high concentrations of FGF2 or FGF2 in combination with transforming growth factor beta 1 (TGFβ1), Activin A, or a BMP antagonist.28–32 Chemically defined supportive substrates, such as laminin 521 and vitronectin, have also been successfully used to replace non-defined substrates such as Matrigel or MEFs.28,33 The development of chemically defined conditions will be tremendously important for efforts aimed at discerning the molecular cues that underpin stem cell fates. Nonetheless, the conventional two-dimensional cultures of adherent hPSCs may not be suitable for efficient production of large numbers of cells for drug discovery or clinical applications. Large-scale culture systems could be further improved by optimizing three-dimensional bioreactor-based propagation of hPSCs in suspension cultures.34–36
Characterization of hPSCs is based on a number of morphological and molecular features. Undifferentiated hPSCs display a high nucleus-to-cytoplasm ratio, and they express a repertoire of molecular markers. In addition to core pluripotency transcription factors POU5F1 (POU domain, class 5, transcription factor 1; also known as octamer-binding transcription factor 4 [OCT4]) and NANOG, hPSCs also express a series of cell-surface antigens, including globoseries glycolipid antigens, stage-specific embryonic antigens 3 and 4 (SSEA3 and SSEA4), and glycoprotein antigens TRA-1-60, TRA-1-81, Thy-1, and GCTM2.37,38 Although the use of cell-surface antigens provides the possibility of isolating desired populations using magnetic beads or fluorescence-activated single-cell sorting, none of the markers identified as yet are truly specific for hPSCs. Hence, multiple markers must be used in combinations to achieve purification of undifferentiated stem cells from their differentiated progeny.
Signaling and directed differentiation in hPSCs
Harnessing the immense developmental potential of hPSCs in applications such as drug discovery relies on the ability to direct differentiation of hPSCs to particular specialized cell types. The specification of hPSCs is controlled by the interaction and balance of signaling pathways that can be manipulated through the use of chemical inhibitors and/or recombinant proteins (Figure 2). The first stage in differentiating hPSCs toward a cell type of interest is the selection of one of the three primary germ layers (endoderm, ectoderm, and mesoderm). In the mouse, it is understood that cells destined for definitive endoderm or mesoderm transition through a stage known as mesendoderm within the primitive streak.39 These cells migrate throughout the embryo and depending upon their location will form definitive endoderm and mesoderm. Mesendoderm germ layer specification relies on the activation and interaction of the TGFβ, FGF, BMP, and Wnt signaling pathways.40 In hESCs, D’Amour et al showed that TGFβ activation through recombinant Activin A, in combination with low serum concentrations, was sufficient to induce up to 80% of cells expressing the endodermal marker SOX17.41 FGF signaling through mitogen-activated protein kinase (MAPK) was later found to further improve definitive endoderm specification.42 Furthermore, FGF2, through MEK/ERK, has been shown to switch BMP4-induced differentiation to the mesendoderm lineage through the maintenance of NANOG expression.43 BMP4 functions to aid the rapid downregulation of stem cell markers, in particular SOX2 (a neural marker), and an enrichment of definitive endoderm markers EOMES and SOX17. Mesoderm similarly relies on the same signaling pathways but has shown to be temporally dependent upon BMP4 exposure.44 Using Brachyury as a pan-mesoderm marker, Zhang et al demonstrated that a short exposure time of BMP4 to hESCs leads to maximal Brachyury expression.44 A seemingly fundamental difference between endoderm and mesoderm specification is the activation levels of TGFβ/Activin/Nodal signaling, whereby a low concentration of Activin A (10–50 ng/mL) effectively induces the mesodermal markers Goosecoid45 and MIXL146 in reporter cell lines. The generation of ectoderm requires the inhibition of the aforementioned signaling pathways. Initial studies in the frog demonstrated that inhibitors of the BMP pathway through Noggin were imperative for neural induction.47,48 This effect was later confirmed in mammalian cells, and it is now used in hPSC neural induction protocols.49 The generation of chemical inhibitors, including dorsomorphin, then provided a cost-effective way to potently inhibit the BMP pathway and further improve neural specification in hPSCs.50 However, the inhibition of BMP alone is not entirely sufficient. These protocols also require the inhibition of the TGFβ/Activin/Nodal pathway, using the chemical inhibitor SB431542.51 Using a combination of Noggin and SB431542, Chambers et al demonstrated much higher level of PAX6+ cell derivation than either inhibitor singly.52 Further differentiation protocols then allow the specification of pharmaceutically relevant cell types. For example, embryoid bodies have shown spontaneous hepatocyte specification through upregulation of markers such as albumin and AFP53 as well as cardiomyocyte specification54 using markers such as NKX2.5 and TNNI3, as reviewed by Matsa et al and Denning and Anderson.55,56
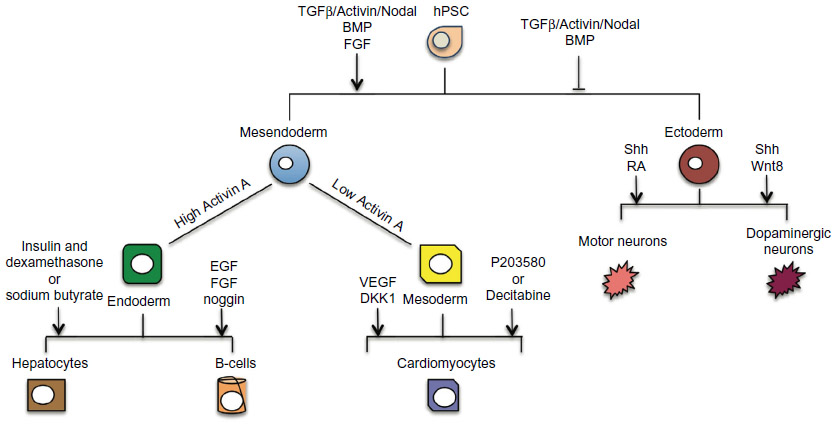 | Figure 2 hPSCs can differentiate to derivatives of all three embryonic germ layers (ectoderm, mesoderm, endoderm). |
Directed differentiation offers a much more powerful approach than spontaneous differentiation for deriving particular cell types due to reproducibility, homogeneity, and efficiency. Hepatocyte specification using insulin with dexamethasone57 and sodium butyrate58 has been reported. Similarly, cardiomyocytes have been specified through Activin A and bFGF treatment54 using a stepwise differentiation protocol using Activin A, BMP4, FGF, vascular endothelial growth factor, and DKK1,59 as well as the use of chemical inhibitors such as P203580 (P38 MAPK inhibitor)60 and 5-aza-2′-deoxy-cytidine to enhance specification.61 Insulin-producing cells have been derived through epidermal growth factor, FGF, and Noggin treatment.62,63 From early neural cells, dopaminergic neurones have been derived through the manipulation of sonic hedgehog (Shh) and Wnt864,65 and motor neurones through Shh and retinoic acid addition.66 Improved differentiation protocols are continually appearing allowing the potential use of many more cell types in drug discovery/toxicology, and this continued ability to derive new cell types will facilitate the identification and screening of new drugs.
hPSC-derived cells for disease modeling and drug discovery
Prior to 2006, disease modeling using hPSCs was based around either the genetic modification of hESC lines or the generation of a new line from embryos exhibiting monogenic diseases. Inevitably, few diseases have been investigated this way due to severe restrictions of these methods.67 Since the generation of hiPSCs, disease modeling has exploded and is regarded as one of the most exciting applications of hPSCs. hiPSCs from diseased patients can be used to follow disease progression as well as allow testing of compounds to alleviate or even cure specific diseases. The first reported instance of the use of hiPSCs in disease modeling was in the case of spinal muscular atrophy.68 Ebert et al were able to show that a mutation within the spinal muscular atrophy gene did not affect motor neuron specification; however, motor neuron production was hindered, and degeneration increased at later time points. Additionally, the compounds valproic acid and tobramycin were able to partially rescue the decrease in SMN protein production within diseased hiPSCs.68 The use of hiPSCs in amyotrophic lateral sclerosis (ALS) modeling yielded kenpaullone as a candidate compound to reduce the levels of mutant SOD1 protein within affected motor neurons.69 Kenpaullone, a GSK3β inhibitor, was the only one of many GSK3β inhibitors tested to have this protective effect; therefore, the mechanism of action presumably did not lie solely within GSK3β inhibition. The authors identified that kenpaullone also inhibits HPK1/GCK-like kinase (also known as MAP4K4), which prevents the activation of an apoptotic pathway involving phospho-c-Jun.69 To screen for new candidate drugs for treatment of ALS, Egawa et al produced spinal motor neurons from ALS-hiPSC lines derived from ALS patients with mutations in Tar DNA-binding protein-43. The ALS motor neurons were then tested against a panel of four compounds, and anacardic acid was identified as a drug that alleviated the disease phenotype of ALS motor neurons.70 Although performed on a limited set of compounds, the screen by Egawa et al demonstrated feasibility of using patient-specific hiPSC-derived cells in drug discovery for ALS therapies.
Another first was the use of hPSCs in modeling familial Alzheimer’s disease (FAD) that is caused by mutations primarily in the PS1 gene, although PS2 mutations are also documented.71,72 Pathologically, this leads to neural loss and accumulation of amyloid fibril plaques, mainly β-amyloid. hiPSCs were generated and differentiated to neurons to explore pathological events in FAD. Neuronal development was not hindered by PS1 or PS2 mutant hiPSC lines, but the cells did excrete higher levels of β-amyloid. Cell lines from both mutations were respondent to several chemicals which could reduce the levels of β-amyloid, and these lines could be particularly useful in drug screening for the treatment of FAD.73 Similarly, Lee et al derived hiPSC lines from patients suffering familial dysautonomia,74 caused by a point mutation in the IKBKAP gene and subsequent mis-splicing.75 They found particularly high levels of the mis-spliced version of IKBKAP in endodermal precursors, which they attribute to the debilitating gastrointestinal defects of affected individuals, as well as in neural crest, which also showed a significant decrease in genes for neurogenesis, neuronal differentiation, and migration.74 In a subsequent study, Lee et al have utilized hiPSC-derived neural crest precursors from familial dysautonomia patients in a primary screen of 6,912 small-molecular weight compounds and identified several hits that rescue IKBKAP expression and may represent candidates for developing therapies.76 A similar high-throughput screening effort of 3,131 compounds was performed on hiPSC-derived hepatic cells from a patient suffering from a liver disorder due to alpha-1 antitrypsin deficiency.77 The use of a high-throughput approach in these studies is an important step toward scaling up the hiPSC-based drug discovery to screening of more comprehensive chemical libraries.76,77
The understanding of disease mechanisms also allows for a much more informed prediction for drug treatment of specific diseases. For example, schizophrenia is a genetically and pathologically complicated disease with wide-ranging symptoms. One possible cause lies with the reduction of GAD6778,79 and GAT180 leading to a reduction of GABA synthesis and reuptake of GABA in approximately 25%–30% of GABA neurons. Drugs whose function causes the augmentation of the release of GABA, or enhances the response to GABA, such as the GABAA α2-selective benzodiazepine, may prove to be specific and effective in alleviating disease symptoms.81 hiPSC lines have been derived from schizophrenic patients82 and may therefore prove to be useful in validating and discovering such drugs.
Aside from neurological disorders, cardiac diseases have been modeled using hiPSCs. Cardiomyocytes and smooth muscles cells derived from hiPSCs of patients with cardiovascular diseases generally demonstrate the disease phenotype in vitro. For example, cardiomyocytes produced by differentiation of hiPSCs of LEOPARD syndrome patients exhibited increased cell size, indicative of hypertrophic cardiac state.83 Similarly, cardiomyocytes have been derived from hiPSCs of patients suffering from familial dilated cardiomyopathy caused by a mutation in the gene encoding cardiac troponin T. hiPSC-derived cardiomyocytes from patients exhibited a number of features characteristic of the disease phenotype, including altered regulation of calcium ion, decreased contractility, and changes in the distribution of sarcomeric proteins.84 Another notable example is long-QT syndrome, a channelopathy characterized by a delayed ventricular repolarization. Principally involving mutations in myocyte ion-channels, long-QT syndrome can lead to sudden arrhythmic death.85 hiPSC lines generated from affected patients displayed prolonged action-potential duration as well as early-after depolarizations, representative of human disease pathology.86 Upon the addition of nifedipine, action-potential duration and early-after depolarizations were reversed and abolished, respectively.86 Strikingly, however, the prolonged exposure of nifedipine leads to the cessation of beating within some embryoid bodies.86 Long-QT syndrome hiPSCs can thus also act as a platform for predictive cardiotoxicity. Therefore, disease modeling allows greater understanding of the pathogenesis of specific diseases, but also allows a detailed interrogation of the mechanisms behind disease phenotypes. This allows the identification of existing drug candidates in ameliorating disease symptoms, but will also allow the development of new, novel drugs targeted against specific proteins or signaling pathways. Nonetheless, an often overlooked caveat of the hiPSC approach for disease modeling is the use of appropriate controls. The controls for known, monogenic diseases can be created relatively easily by replacing the mutated gene with its wild-type form.87,88 However, the modeling of more complex diseases or diseases with an unknown etiology is complicated by the fact that even sibling controls may be inappropriate as the genetic background will be different and may influence the cell phenotype, thus warranting a careful consideration of experimental controls.89
The use of hPSCs as models for developmental toxicology
Some of the most devastating consequences of adverse drug effects include birth anomalies caused by drug exposure of a developing fetus. These effects are potentially preventable, but the issue of fetal exposure is compounded by the fact that pregnant women may be ingesting drugs before they realize they are pregnant, and drugs that manifest no adverse reactions in adults may still cause defects during prenatal development. On the other hand, avoidance of all drugs during pregnancy may be detrimental for women with preexisting medical conditions or medical problems. In order to assess the effects of a compound on the developing conceptus, the developmental safety of a compound is traditionally evaluated using in vivo studies in pregnant animals which generally include a rodent species (usually rat) and a non-rodent species (usually rabbit), usually with preterm evaluation of fetuses.90 Compound exposure is conducted during embryo organogenesis between the stage of implantation of the conceptus and the closure of the hard palate. Assessment at the end of gestation just prior to parturition is conducted for developmental external, visceral, and skeletal endpoints on fetuses. The traditional in vivo testing method based on extrapolating across dose and species to human has been regarded as effective91 ever since the thalidomide disaster in the 1960s. However, there is a widespread desire to develop alternate (nonmammalian) methods that can provide data more quickly, using less compound and reducing in vivo testing.
Ordinarily in the pharmaceutical industry, efficacy and general toxicity are assessed much earlier than developmental toxicity. In the last decade, efforts have been made to try to find alternative in vitro methods, which would allow screening of many compounds early in the pharmaceutical pipeline to evaluate developmental endpoints prior to the mammalian assays. Some of these in vitro methods have the potential to screen thousands of compounds for their effects on complex pathways relevant to developmental processes and toxicities.92 Many possible models have been explored, including hydra regeneration,93 chick embryo neural retina cells,91,94 embryonic palatal mesenchymal cells,95 mouse ovarian tumor cell attachment,96 chick embryos,97 whole rat, mouse or rabbit embryo culture in vitro,98–101 mouse palatal cultures,102 mouse limb bud reaggregates,103 in vivo larval zebrafish assays,104,105 and ESCs.106,107
In some laboratories, murine ESCs were the model of choice for developmental toxicology studies. A murine ESC platform based on adherent-cell differentiation culture108 monitored both cytotoxicity and myosin heavy-chain protein expression as a marker of cardiomyocyte differentiation that is dependent upon diverse cellular interactions across different primary germ layer lineages. Alternatively, in the “embryonic stem cell test” (EST), the use of murine ESCs (D3 cell line) cultured to form embryoid bodies focusing on the differentiation of beating cardiomyocytes as a visual endpoint marker, and additionally the relative effects on D3 and adult fibroblast cells (3T3) cytotoxicity, was shown to have the ability to predict the developmental toxicity of 78% of 20 compounds in a test panel.109,110 Both of these assays, the EST and murine ESC platforms,108 may provide important information about chemical effects on complex differentiation pathways in murine systems. This information should be, to a degree, translatable to a human, as seen with in vivo animal models, providing an insight into the potential developmental effects seen at a cellular level. However, the use of hESCs should alleviate species-specific differences between mouse and humans and therefore is expected to increase the predictive power of the developmental toxicity testing. Several proof-of-concept studies exposed hESCs to known developmental toxicants and ascertained the validity of this model.111,112
Profiling hESCs for their secreted metabolites has been proposed as an alternative testing platform for identifying compounds with developmental activity.113,114 Metabolomics detects dynamic variations in small molecule abundance, assessing functional changes in biochemical pathways and cellular metabolic response due to chemical exposure. Taking this into consideration, the profile of intermediary metabolites and small molecules released by hESCs to their environment (“secretome”) is therefore potentially a direct or indirect indicator of chemical disruptions that could lead to identification of the extent of adverse outcome pathways in the developing embryo. On the whole, the metabolome for stem cells is characterized by changes in metabolites involved in cellular respiration. The metabolome of hiPSCs has been shown to share a pluripotent metabolomic signature with hESCs that is distinct from their parental cells.115 However, some metabolites do differ between hiPSCs and hESCs, which reveal novel metabolic pathways that play a critical role in regulating somatic cell reprogramming.116 The identification of specific small molecule biomarkers of chemical exposure or effect could provide valuable mechanistic information and pinpoint sensitive pathways in early human embryogenesis.117,118 In addition, the application of metabolomics to developmental toxicity testing is also possible.113 The application of the hESC secretome to predictive developmental toxicity (devTOX platform – Stemina) has been described.114 The devTOX platform was shown to accurately predict 88% of compounds with known developmental toxicity out of a test set of eight teratogenic compounds.114
Approaches to high-throughput and high-content screening of hPSCs and their derivatives in drug discovery
Maintenance of genetically stable undifferentiated hPSCs in culture and their differentiation to relevant cell types is an important first step in addressing the lack of suitable in vitro models for drug discovery. However, challenges additional to the availability of appropriate cellular models need to be addressed in order for this resource to provide significant improvements in drug discovery campaigns. In particular, screening assays have to be sensitive and robust, but also suitable for high-throughput analyses. The screening assay has to be carefully selected from a range of methods, such as immunocytochemistry, reverse-transcriptase polymerase chain reaction, dot-blot analysis, and luminescence, depending on the nature of the signal that needs to be assessed. Plate reader-based assays, albeit rapid, have significant shortfalls for cellular phenotyping as they usually provide only a single readout from a population of cells.119 This type of a readout assumes a homogeneous population of cells and average population data. However, it is becoming increasingly apparent that even genetically identical populations of cells can have heterogeneous phenotypes, and that such heterogeneity has functional implications.120–122 Thus, drug screening assays have to be able to detect potentially differential response of genetically identical cells to treatments. This requires cell-by-cell analysis that can unmask the heterogeneity obscured by a single average population readout.120 Flow cytometry-based assays offer this possibility through single-cell analysis of cells labeled with a range of antibodies and stains, resulting in a multivariate profiling of each cell. Nonetheless, flow cytometry requires dissociation of adherent cells, potentially losing valuable information on cell morphology. As corollary of a limited readout and/or lack of morphological measurements and spatial signals, unanticipated effects of drugs’ effects on cells will go undetected until a later stage of drug discovery, possibly contributing to high rates of late-stage attrition. Extensive evaluation of new lead compounds early on in a drug development process would significantly reduce the overall cost and efforts, and would allow selection of candidates with the best prospect of success in the clinical stages of drug discovery.
High-content imaging assays (Figure 3) have emerged as a powerful tool for extensive assessment of cell phenotypes through simultaneous, quantitative measurements of a variety of cell parameters.123 In a typical high-content chemical screening assay, cells are plated into multi-well plates and treated with control compounds or drugs from a chosen drug library for a predetermined period of time. Cells are then fixed and stained with a number of antibodies and/or cellular dyes. Fluorescent imaging of labeled cells and the subsequent image analyses allow numerous readouts and hence multiparametric assessment of cells, including the presence of antigen(s) of interest and their subcellular localization, cell numbers, size, and shape. Indeed, the term “high content” was coined to reflect such a large amount of information obtained from imaging of individual cells. Unlike the target-based approach, high-content cell-based assays make no a priori predictions as to the targets involved. This type of a hypothesis-free screening allows an unbiased assessment of cell phenotype and detection of changes in features that may be missed in an assay with just a single readout. Importantly, due to the fact that a single high-content assay integrates various readouts, which would otherwise require testing in multiple individual assays, high-content analysis significantly improves the speed and reduces the cost of cellular phenotyping. Both the cost and speed of high-content assays are even further improved by automating majority of steps, from cell plating to image analysis. Automated image analysis also contributes to achieving robust and objective readouts. The sensitivity of high-content assays stems from the fact that the data are derived from single-cell measurements and can detect subtle changes within a cell population.124–126
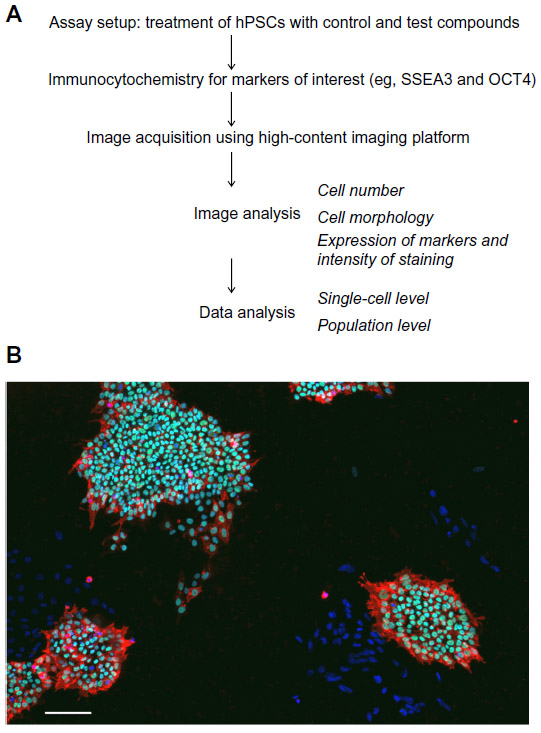 | Figure 3 High-content imaging of hPSCs allows detection of cell phenotypes based on cell numbers, morphology, and marker staining. |
Given the power of the high-content approach, it is not surprising that high-content assays have not only become an integral part of drug discovery screens but are also utilized by academic researchers investigating various aspects of cell biology. Indeed, high-content assays have been successfully applied toward gaining a better understanding of the signaling pathways involved in survival and differentiation of hPSCs. An illustrious example is a screen by Desbordes et al, which assessed effects of 2,880 small-molecular weight compounds on hPSCs. The screen utilized imaging of POU5F1-immunostained cells as a readout of the differentiation-inducing abilities of compounds, and identified four inducers and ten inhibitors of differentiation.127 Similarly, Andrews et al performed a high-content screen on hPSCs to identify compounds that enhance cell survival.128 Although high-content platforms for imaging of cells were used in these screens, the readouts were limited to the numbers of cells and/or cells positive for a marker of undifferentiated state, without fully exploiting the power of high-content screening. Barbaric et al devised a high-content assay on hPSCs that, in addition to the number of cells and cells positive for TRA-1-60 marker of undifferentiated state, assessed number of hPSC colonies, their area, and shape as well as the intensity of staining.129,130 This type of analysis enabled stratification of hits that induce the cell differentiation. For example, although all-trans-retinoic acid and steroid compounds induced a reduction in TRA-1-60 levels to a similar extent, a significant difference in the morphology of the hPSC colonies indicated that the differentiation phenotypes were likely to be different, and this was indeed confirmed by gene expression analyses.129 Ultimately, the high-content assays should be able to predict which molecular pathways are activated or disrupted upon chemical treatment of cells, based on the cell phenotype. To demonstrate the feasibility of this approach Perlman et al used 96 compounds with known targets, three compounds with unknown mechanism, and a compound with multiple known targets. High-content multivariate phenotyping of cells enabled clustering of molecules based on the phenotypic effects they induced in the cells.131 A pertinent question in high-content screening is how many and which cellular features should be measured from images? Given that high-content imaging offers the opportunity to extract hundreds of phenotypic measurements of each cell, the temptation may be to include as many features as possible; nonetheless, large datasets produced would not only be computationally challenging but may in fact reduce the sensitivity of the assay. Loo et al addressed this issue by reducing the set of approximately 300 features to approximately 20 readouts that could reliably discriminate between different sets of compounds on cancer cells.124 Similar analyses on hPSCs and their differentiated derivatives should be performed to define the minimal feature sets that would allow fast yet sensitive and reliable identification of compounds that perturb the cell phenotype.
Most high-content assays are performed as fixed-end point assays, and as such, they may not be suitable for detecting dynamic temporal changes in cellular behavior. The addition of time dimension offers the opportunity to acquire additional information on cell behavior (Figure 4). A case in point is cell motility, a feature that plays a significant role in the metastatic behavior of cancer cells and may be a particularly crucial readout in screens for anticancer drugs.132 Time lapse-based assays have been employed in screening of neural cells,133 but the widespread use of this method awaits improvement in automation of cell-tracking protocols, data storage, and analysis.134
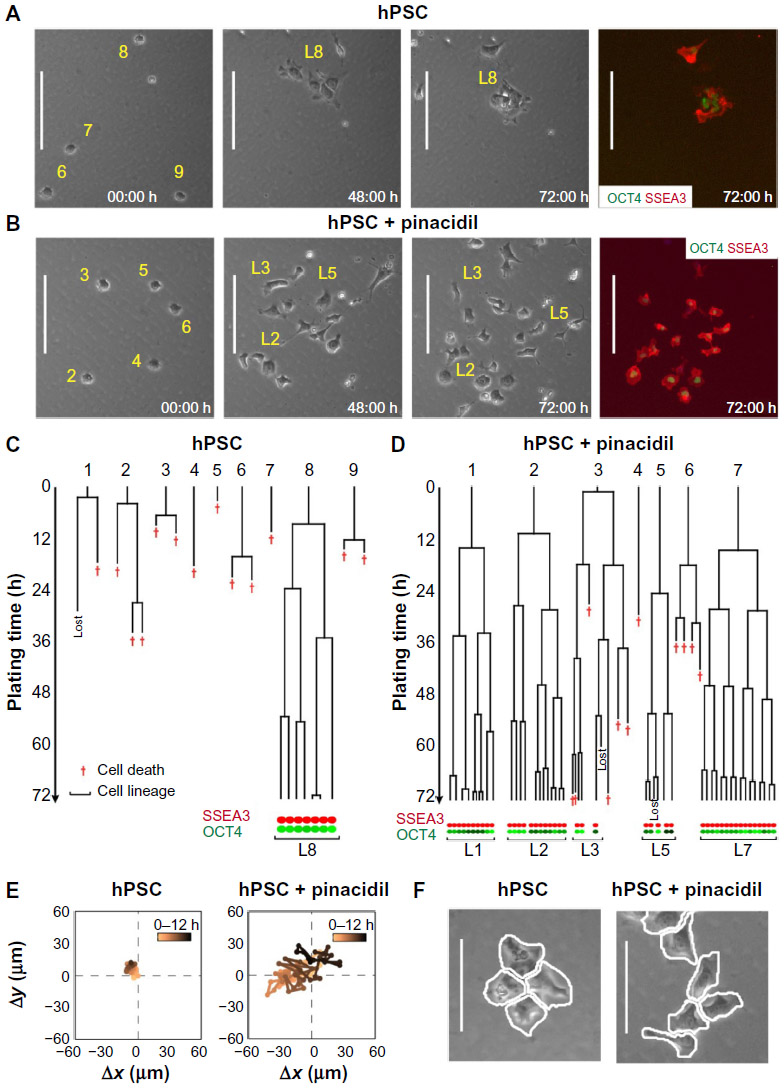 | Figure 4 Time-lapse imaging allows detailed analysis of hPSC survival, proliferation, motility, morphology, and spatial distribution of cells in colonies. |
Conclusion and future perspectives
Identification of suitable preclinical models has emerged as a critical element in improving drug discovery. The availability of hPSCs offers unprecedented opportunities for successful screening of new chemical entities. Perhaps, the most exciting prospect is the ability to obtain patient-specific cells for disease modeling and screening for drugs that correct the disease phenotype, thus paving the way to individualized treatments.135 Nonetheless, leveraging the enormous potential of hPSCs will require optimization of differentiation protocols and in-depth characterization of the resulting differentiated cells. One aspect that must be addressed is the immaturity of cell types derived from hPSCs, and whether these can appropriately read out toxicity that would manifest within an adult. Implementing methods to age cells in vitro may aid in resolving this issue and allow modeling of late-onset diseases.136 Moreover, further developments are warranted to ensure that culture systems accurately recapitulate the in vivo environment of cells. Indeed, traditional two-dimensional cell culture systems generally poorly represent mechanical, chemical, and cell–cell interaction cues that cells encounter in their native tissues.137 A number of approaches are being developed to address the drawbacks of two-dimensional cell culture, including the fabrication of scaffolds with either synthetic polymers or natural materials.138 Ultimately, hPSC-derived organs-on-chips will provide physiologically relevant models mimicking different organs amalgamated into a single system to mimic the tissue–tissue interaction, and drug absorption, metabolism, transport, and clearance.139 Final conclusions on the usefulness of such models in preclinical drug discovery await further validation studies. Nonetheless, examples of hPSC applications in disease modeling and drug testing reviewed in this article provide a tantalizing hint that we may be at the dawn of a new era in drug discovery.
Disclosure
PWA receives a share of royalties from the Wistar Institute from their licensing of the TRA series of antibodies. The other authors report no conflicts of interest in this work.
References
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32(1):40–51. | |
Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–715. | |
Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32(1):56–67. | |
Cook D, Brown D, Alexander R, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419–431. | |
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. | |
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. | |
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | |
Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. | |
Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–680. | |
Barbaric I, Biga V, Gokhale PJ, et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem cell Reports. 2014;3(1):142–155. | |
Ohgushi M, Matsumura M, Eiraku M, et al. Molecular pathway and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell. 2010;7(2):225–239. | |
Chen G, Hou Z, Gulbranson DR, Thomson JA. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7(2):240–248. | |
Dumitru R, Gama V, Fagan BM, et al. Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell. 2012;46(5):573–583. | |
Byrne AT, Southgate J, Brison DR, Leese HJ. Analysis of apoptosis in the preimplantation bovine embryo using TUNEL. J Reprod Fertil. 1999;117(1):97–105. | |
Desmarais JA, Hoffmann MJ, Bingham G, Gagou ME, Meuth M, Andrews PW. Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells. 2012;30(7):1385–1393. | |
International Stem Cell Initiative, Amps K, Andrews PW, et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29(12):1132–1144. | |
Baker DE, Harrison NJ, Maltby E, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25(2):207–215. | |
Draper JS, Smith K, Gokhale P, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22(1):53–54. | |
Enver T, Soneji S, Joshi C, et al. Cellular differentiation hierarchies in normal and culture-adapted human embryonic stem cells. Hum Mol Genet. 2005;14(21):3129–3140. | |
Avery S, Hirst AJ, Baker D, et al. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Reports. 2013;1(5):379–386. | |
Herszfeld D, Wolvetang E, Langton-Bunker E, et al. CD30 is a survival factor and a biomarker for transformed human pluripotent stem cells. Nat Biotechnol. 2006;24(3):351–357. | |
Fazeli A, Liew CG, Matin MM, et al. Altered patterns of differentiation in karyotypically abnormal human embryonic stem cells. Int J Dev Biol. 2011;55(2):175–180. | |
Werbowetski-Ogilvie TE, Bossé M, Stewart M, et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat Biotechnol. 2009;27(1):91–97. | |
Amit M, Carpenter MK, Inokuma MS, et al. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227(2):271–278. | |
Vallier L, Alexander M, Pedersen RA. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118(pt 19):4495–4509. | |
Xu RH, Chen X, Li DS, et al. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat Biotechnol. 2002;20(12):1261–1264. | |
Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115(3):281–292. | |
Chen G, Gulbranson DR, Hou Z, et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8(5):424–429. | |
Furue MK, Na J, Jackson JP, et al. Heparin promotes the growth of human embryonic stem cells in a defined serum-free medium. Proc Natl Acad Sci U S A. 2008;105(36):13409–13414. | |
Lu J, Hou R, Booth CJ, Yang SH, Snyder M. Defined culture conditions of human embryonic stem cells. Proc Natl Acad Sci U S A. 2006; 103(15):5688–5693. | |
Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA. Feeder-independent culture of human embryonic stem cells. Nat Methods. 2006;3(8):637–646. | |
Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods. 2005;2(3):185–190. | |
Rodin S, Antonsson L, Niaudet C, et al. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nat Commun. 2014;5:3195. | |
Amit M, Chebath J, Margulets V, et al. Suspension culture of undifferentiated human embryonic and induced pluripotent stem cells. Stem Cell Rev. 2010;6(2):248–259. | |
Olmer R, Haase A, Merkert S, et al. Long term expansion of undifferentiated human iPS and ES cells in suspension culture using a defined medium. Stem Cell Res. 2010;5(1):51–64. | |
Steiner D, Khaner H, Cohen M, et al. Derivation, propagation and controlled differentiation of human embryonic stem cells in suspension. Nat Biotechnol. 2010;28(4):361–364. | |
International Stem Cell Initiative, Adewumi O, Aflatoonian B, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25(7):803–816. | |
Draper JS, Pigott C, Thomson JA, Andrews PW. Surface antigens of human embryonic stem cells: changes upon differentiation in culture. J Anat. 2002;200(pt 3):249–258. | |
Tam PP, Behringer RR. Mouse gastrulation: the formation of a mammalian body plan. Mech Dev. 1997;68(1–2):3–25. | |
Kimelman D. Mesoderm induction: from caps to chips. Nat Rev Genet. 2006;7(5):360–372. | |
D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23(12):1534–1541. | |
Seguin CA, Draper JS, Nagy A, Rossant J. Establishment of endoderm progenitors by SOX transcription factor expression in human embryonic stem cells. Cell Stem Cell. 2008;3(2):182–195. | |
Yu P, Pan G, Yu J, Thomson JA. FGF2 sustains NANOG and switches the outcome of BMP4-induced human embryonic stem cell differentiation. Cell Stem Cell. 2011;8(3):326–334. | |
Zhang P, Li J, Tan Z, et al. Short-term BMP-4 treatment initiates mesoderm induction in human embryonic stem cells. Blood. 2008;111(4):1933–1941. | |
Tada S, Era T, Furusawa C, et al. Characterization of mesendoderm: a diverging point of the definitive endoderm and mesoderm in embryonic stem cell differentiation culture. Development. 2005;132(19):4363–4374. | |
Davis RP, Ng ES, Costa M, et al. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood. 2008;111(4):1876–1884. | |
Smith WC, Harland RM. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell. 1992;70(5):829–840. | |
Sasai Y, Lu B, Steinbeisser H, Geissert D, Gont LK, De Robertis EM. Xenopus chordin: a novel dorsalizing factor activated by organizer-specific homeobox genes. Cell. 1994;79(5):779–790. | |
Lee H, Shamy GA, Elkabetz Y, et al. Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells. 2007;25(8):1931–1939. | |
Morizane A, Doi D, Kikuchi T, Nishimura K, Takahashi J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J Neurosci Res. 2011;89(2):117–126. | |
Smith JR, Vallier L, Lupo G, Alexander M, Harris WA, Pedersen RA. Inhibition of Activin/Nodal signaling promotes specification of human embryonic stem cells into neuroectoderm. Dev Biol. 2008;313(1):107–117. | |
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. | |
Itskovitz-Eldor J, Schuldiner M, Karsenti D, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000;6(2):88–95. | |
Burridge PW, Anderson D, Priddle H, et al. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells. 2007;25(4):929–938. | |
Matsa E, Denning C. In vitro uses of human pluripotent stem cell-derived cardiomyocytes. J Cardiovasc Transl Res. 2012;5(5):581–592. | |
Denning C, Anderson D. Cardiomyocytes from human embryonic stem cells as predictors of cardiotoxicity. Drug Discov Today Ther Strateg. 2009;5:223–232. | |
Shirahashi H, Wu J, Yamamoto N, et al. Differentiation of human and mouse embryonic stem cells along a hepatocyte lineage. Cell Transplant. 2004;13(3):197–211. | |
Rambhatla L, Chiu CP, Kundu P, Peng Y, Carpenter MK. Generation of hepatocyte-like cells from human embryonic stem cells. Cell Transplant. 2003;12(1):1–11. | |
Yang L, Soonpaa MH, Adler ED, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453(7194):524–528. | |
Graichen R, Xu X, Braam SR, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008;76(4):357–370. | |
Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91(6):501–508. | |
D’Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–1401. | |
Jiang J, Au M, Lu K, et al. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25(8):1940–1953. | |
Fasano CA, Chambers SM, Lee G, Tomishima MJ, Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6(4):336–347. | |
Perrier AL, Tabar V, Barberi T, et al. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A. 2004;101(34):12543–12548. | |
Li XJ, Du ZW, Zarnowska ED, et al. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23(2):215–221. | |
Colman A, Dreesen O. Pluripotent stem cells and disease modeling. Cell Stem Cell. 2009;5(3):244–247. | |
Ebert AD, Yu J, Rose FF Jr, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457(7227):277–280. | |
Yang YM, Gupta SK, Kim KJ, et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12(6):713–726. | |
Egawa N, Kitaoka S, Tsukita K, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4(145):145ra104. | |
Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. | |
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | |
Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. | |
Lee G, Papapetrou EP, Kim H, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461(7262):402–406. | |
Slaugenhaupt SA, Blumenfeld A, Gill SP, et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68(3):598–605. | |
Lee G, Ramirez CN, Kim H, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 2012;30(12):1244–1248. | |
Choi SM, Kim Y, Shim JS, et al. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology. 2013;57(6):2458–2468. | |
Akbarian S, Kim JJ, Potkin SG, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52(4):258–266. | |
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57(3):237–245. | |
Volk D, Austin M, Pierri J, Sampson A, Lewis D. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry. 2001;158(2):256–265. | |
Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6(4):312–324. | |
Chiang CH, Su Y, Wen Z, et al. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry. 2011;16(4):358–360. | |
Carvajal-Vergara X, Sevilla A, D’Souza SL, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465(7299):808–812. | |
Sun N, Yazawa M, Liu J, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4(130):130ra147. | |
Goldenberg I, Zareba W, Moss AJ. Long QT syndrome. Curr Probl Cardiol. 2008;33(11):629–694. | |
Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–229. | |
Soldner F, Laganière J, Cheng AW, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–331. | |
Liu GH, Suzuki K, Qu J, et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. 2011;8(6):688–694. | |
Grieshammer U, Shepard KA. Proceedings: consideration of genetics in the design of induced pluripotent stem cell-based models of complex disease. Stem Cells Transl Med. 2014;3(11):1253–1258. | |
Wise LD. The ICH S5(R2) guideline for the testing of medicinal agents. Methods Mol Biol. 2013;947:1–11. | |
Daston GP. Genomics and developmental risk assessment. Birth Defects Res A Clin Mol Teratol. 2007;79(1):1–7. | |
Hartung T. Toxicology for the twenty-first century. Nature. 2009; 460(7252):208–212. | |
Johnson EM, Dansky LA. A hydra assay as a pre-screen for teratogenic potential. Food Chem Toxicol. 1987;25(8):637–638. | |
Moscona A. Rotation-mediated histogenetic aggregation of dissociated cells. A quantifiable approach to cell interactions in vitro. Exp Cell Res. 1961;22:455–475. | |
Pratt RM, Grove RI, Willis WD. Prescreening for environmental teratogens using cultured mesenchymal cells from the human embryonic palate. Teratog Carcinog Mutagen. 1982;2(3–4):313–318. | |
Braun AG, Emerson DJ, Nichinson BB. Teratogenic drugs inhibit tumour cell attachment to lectin-coated surfaces. Nature. 1979; 282(5738):507–509. | |
Tickle C. Positional signalling by retinoic acid in the developing chick wing. Prog Clin Biol Res. 1983;110(pt A):89–98. | |
Steele VE, Morrissey RE, Elmore EL, et al. Evaluation of two in vitro assays to screen for potential developmental toxicants. Fundam Appl Toxicol. 1988;11(4):673–684. | |
Sadler TW, Horton WE, Warner CW. Whole embryo culture: a screening technique for teratogens? Teratog Carcinog Mutagen. 1982; 2(3–4):243–253. | |
Van Maele-Fabry G, Delhaise F, Picard JJ. Morphogenesis and quantification of the development of post-implantation mouse embryos. Toxicol In Vitro. 1990;4(2):149–156. | |
Pitt JA, Carney EW. Development of a morphologically-based scoring system for postimplantation New Zealand White rabbit embryos. Teratology. 1999;59(2):88–101. | |
Abbott BD, Harris MW, Birnbaum LS. Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology. 1989;40(6):533–553. | |
Kistler A. Limb bud cell cultures for estimating the teratogenic potential of compounds. Validation of the test system with retinoids. Arch Toxicol. 1987;60(6):403–414. | |
Gustafson AL, Stedman DB, Ball J, et al. Inter-laboratory assessment of a harmonized zebrafish developmental toxicology assay – progress report on phase I. Reprod Toxicol. 2012;33(2):155–164. | |
Ball JS, Stedman DB, Hillegass JM, et al. Fishing for teratogens: a consortium effort for a harmonized zebrafish developmental toxicology assay. Toxicol Sci. 2014;139(1):210–219. | |
Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. | |
Seiler AE, Buesen R, Visan A, Spielmann H. Use of murine embryonic stem cells in embryotoxicity assays: the embryonic stem cell test. Methods Mol Biol. 2006;329:371–395. | |
Barrier M, Jeffay S, Nichols HP, et al. Mouse embryonic stem cell adherent cell differentiation and cytotoxicity (ACDC) assay. Reprod Toxicol. 2011;31(4):383–391. | |
Genschow E, Spielmann H, Scholz G, et al. The ECVAM international validation study on in vitro embryotoxicity tests: results of the definitive phase and evaluation of prediction models. European Centre for the Validation of Alternative Methods. Altern Lab Anim. 2002;30(2):151–176. | |
Genschow E, Spielmann H, Scholz G, et al. Validation of the embryonic stem cell test in the international ECVAM validation study on three in vitro embryotoxicity tests. Altern Lab Anim. 2004;32(3):209–244. | |
Adler S, Lindqvist J, Uddenberg K, Hyllner J, Strehl R. Testing potential developmental toxicants with a cytotoxicity assay based on human embryonic stem cells. Altern Lab Anim. 2008;36(2):129–140. | |
Krug AK, Kolde R, Gaspar JA, et al. Human embryonic stem cell-derived test systems for developmental neurotoxicity: a transcriptomics approach. Arch Toxicol. 2013;87(1):123–143. | |
Cezar GG. Can human embryonic stem cells contribute to the discovery of safer and more effective drugs? Curr Opin Chem Biol. 2007;11(4):405–409. | |
West PR, Weir AM, Smith AM, Donley EL, Cezar GG. Predicting human developmental toxicity of pharmaceuticals using human embryonic stem cells and metabolomics. Toxicol Appl Pharmacol. 2010;247(1):18–27. | |
Munoz J, Low TY, Kok YJ, et al. The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol Syst Biol. 2011;7:550. | |
Panopoulos AD, Yanes O, Ruiz S, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22(1):168–177. | |
Sabatine MS, Liu E, Morrow DA, et al. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation. 2005; 112(25):3868–3875. | |
Tan DX, Manchester LC, Reiter RJ, et al. A novel melatonin metabolite, cyclic 3-hydroxymelatonin: a biomarker of in vivo hydroxyl radical generation. Biochem Biophys Res Commun. 1998;253(3):614–620. | |
Sundberg SA. High-throughput and ultra-high-throughput screening: solution – and cell-based approaches. Curr Opin Biotechnol. 2000; 11(1):47–53. | |
Altschuler SJ, Wu LF. Cellular heterogeneity: do differences make a difference? Cell. 2010;141(4):559–563. | |
Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. 2009;4(5):387–397. | |
Niepel M, Spencer SL, Sorger PK. Non-genetic cell-to-cell variability and the consequences for pharmacology. Curr Opin Chem Biol. 2009;13(5–6):556–561. | |
Carpenter AE. Image-based chemical screening. Nat Chem Biol. 2007; 3(8):461–465. | |
Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nat Methods. 2007;4(5):445–453. | |
Giuliano KA, Haskins JR, Taylor DL. Advances in high content screening for drug discovery. Assay Drug Dev Technol. 2003;1(4):565–577. | |
Taylor DL, Giuliano KA. Multiplexed high content screening assays create a systems cell biology approach to drug discovery. Drug Discov Today. 2005;Suppl:13–18. | |
Desbordes SC, Placantonakis DG, Ciro A, et al. High-throughput screening assay for the identification of compounds regulating self-renewal and differentiation in human embryonic stem cells. Cell Stem Cell. 2008;2(6):602–612. | |
Andrews PD, Becroft M, Aspegren A, et al. High-content screening of feeder-free human embryonic stem cells to identify pro-survival small molecules. Biochem J. 2010;432(1):21–33. | |
Barbaric I, Gokhale PJ, Jones M, Glen A, Baker D, Andrews PW. Novel regulators of stem cell fates identified by a multivariate phenotype screen of small compounds on human embryonic stem cell colonies. Stem Cell Res. 2010;5(2):104–119. | |
Barbaric I, Gokhale PJ, Andrews PW. High-content screening of small compounds on human embryonic stem cells. Biochem Soc Trans. 2010;38(4):1046–1050. | |
Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306(5699):1194–1198. | |
Carragher NO. Profiling distinct mechanisms of tumour invasion for drug discovery: imaging adhesion, signalling and matrix turnover. Clin Exp Metastasis. 2009;26(4):381–397. | |
Danovi D, Falk A, Humphreys P, et al. Imaging-based chemical screens using normal and glioma-derived neural stem cells. Biochem Soc Trans. 2010;38(4):1067–1071. | |
Eliceiri KW, Berthold MR, Goldberg IG, et al. Biological imaging software tools. Nat Methods. 2012;9(7):697–710. | |
Inoue H, Yamanaka S. The use of induced pluripotent stem cells in drug development. Clin Pharmacol Ther. 2011;89(5):655–661. | |
Miller JD, Ganat YM, Kishinevsky S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13(6):691–705. | |
Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci. 2012;125(pt 13):3015–3024. | |
Nam KH, Smith AS, Lone S, Kwon S, Kim DH. Biomimetic 3D tissue models for advanced high-throughput drug screening. J Lab Autom. Epub 2014. | |
Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011;21(12):745–754. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.