Back to Journals » International Journal of Nanomedicine » Volume 12
Human mesenchymal stem cell-derived iron oxide exosomes allow targeted ablation of tumor cells via magnetic hyperthermia
Authors Altanerova U, Babincova M, Babinec P, Benejova K, Jakubechova J, Altanerova V, Zduriencikova M, Repiska V, Altaner C ![]()
Received 28 June 2017
Accepted for publication 9 September 2017
Published 27 October 2017 Volume 2017:12 Pages 7923—7936
DOI https://doi.org/10.2147/IJN.S145096
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Altanerova et al
Views: 1162
U Altanerova,1 M Babincova,2 P Babinec,2 K Benejova,1 J Jakubechova,1 V Altanerova,1 M Zduriencikova,3 V Repiska,4 C Altaner1,3
1Stem Cell Preparation Department, St Elisabeth Cancer Institute, Bratislava, Slovakia; 2Department of Nuclear Physics and Biophysics, Comenius University, Bratislava, Slovakia; 3Cancer Research Institute, Biomedical Center, Slovak Academy of Sciences, Bratislava, Slovakia; 4Institute of Medical Biology, Genetics and Clinical Genetics, Faculty of Medicine, Comenius University in Bratislava, Slovakia
Abstract: Magnetic hyperthermia, or the heating of tissues using magnetic materials, is a promising approach for treating cancer. We found that human mesenchymal stem cells (MSCs) isolated from various tissues and MSCs expressing the yeast cytosine deaminase::uracil phosphoribosyl transferase suicide fusion gene (yCD::UPRT) can be labeled with Venofer, an iron oxide carbohydrate nanoparticle. Venofer labeling did not affect cell proliferation or the ability to home to tumors. All Venofer-labeled MSCs released exosomes that contained iron oxide. Furthermore, these exosomes were efficiently endocytosed by tumor cells. Exosomes from Venofer-labeled MSCs expressing the yCD::UPRT gene in the presence of the prodrug 5-fluorocytosine inhibited tumor growth in a dose-dependent fashion. The treated tumor cells were also effectively ablated following induction of hyperthermia using an external alternating magnetic field. Cumulatively, we found that magnetic nanoparticles packaged into MSC exosomes are efficiently endocytosed by tumor cells, facilitating targeted tumor cell ablation via magnetically induced hyperthermia.
Keywords: mesenchymal stem cells, iron oxide labeling, Venofer, yCD::UPRT-exosomes, yCD::UPRT-MSCs/Fe exosomes, magnetic hyperthermia
Introduction
Hyperthermia mediated by magnetic materials is a promising minimally invasive strategy for eliminating tumors that are at present difficult to treat.1–3 This approach involves targeting magnetic nanoparticles to tumors and then inducing cytotoxic hyperthermia by applying an alternating magnetic field (AMF). Iron oxide nanoparticles are optimal for inducing hyperthermia, and their effectiveness as a cancer therapeutic should be evaluated according to the following criteria: 1) they should selectively target tumor cells, 2) they should easily penetrate tumor cells, 3) they should be internalized by tumor cells, 4) they should be biocompatible and nontoxic across a wide range of concentrations, 5) they should be easy to administer in the clinic, 6) they should be likely to be approved for clinical use, and 7) they should be targetable to specific tumor types or to organs with metastases. Currently, superparamagnetic iron oxide nanoparticles (SPIONs) have been widely investigated in the clinic as magnetic hyperthermia reagents because SPIONs form a stable suspension and their highly active surface is available for a wide range of biofunctionalization. Depending on the size of the SPIONs and the frequency of the AMF, heat is generated through either Néel or Brownian relaxation.4
Human mesenchymal stem cells (MSCs) have the ability to home to tumors. We, as well as others, have shown that systemically administered MSCs migrate to and incorporate into tumors.5,6 The tumor-specific tropism of MSCs inspired us to develop a targeted prodrug gene therapy mediated by MSCs engineered to express suicide genes.7,8 Specifically, we engineered MSCs to express the yeast cytosine deaminase::uracil phosphoribosyl transferase fusion gene (yCD::UPRT), which allows cells to convert nontoxic 5-fluorocytosine (5-FC) into cytotoxic 5-fluorouracil (5-FU). We found that genetically modified MSCs maintained their ability to migrate to tumors and inhibit their growth in the presence of the prodrug 5-FC. This prodrug gene therapy mediated by MSCs has effectively inhibited the growth of human colon carcinoma,7 melanoma,9 and prostate carcinoma10,11 in nude mice. MSCs from the adipose tissue (AT-MSCs) engineered to express yCD::UPRT gene were shown to induce curative therapeutic effect in a substantial number of rats with intracranial glioblastoma in a preclinical model.12,13 All these results were supported by the evidence of migration of these cell to tumors and the inhibition of tumor growth as a bystander effect of 5-FU formation at the tumor site. Later on, we found release of exosomes possessing the mRNA of suicide gene in their cargo, thus expanding the interpretation to combined action of bystander effect and internalized exosomes.14,15
We and others have shown that MSCs labeled with SPIONs display no differences in cell proliferation or survival, compared to control MSCs. Iron oxide-labeled cells migrate to and integrate into tumors.3,15 Recently, we reported a simple procedure to label MSCs of the human dental pulp (DP-MSCs) and DP-MSCs expressing the yCD::UPRT fusion gene with iron oxide (Venofer). We found that both Venofer-labeled and Venofer-unlabeled DP-MSCs and yCD::UPRT-DP-MSCs released exosomes into the conditioned medium (CM). Furthermore, the CM from unlabeled and labeled yCD::UPRT-DP-MSCs in the presence of the prodrug 5-FC triggered tumor cell death in a dose-dependent manner.14
In this study, we extended these Venofer labeling experiments to other types of MSCs and found that Venofer particles were successfully incorporated as the cargo of exosomes released from labeled MSCs. The iron oxide-rich materials exocytosed from Venofer-labeled MSCs were found to be natural nanoparticles that could be internalized by the tumor cells. This subsequently facilitated the ablation of these tumor cells via AMF-induced hyperthermia.
Materials and methods
MSC isolation, culture, and maintenance
All experimental protocols involving cells of human subjects were approved by the ethical committee of the St Elisabeth Cancer Institute. Four types of human MSCs were used. All donors of the adipose tissue, bone marrow, dental pulp, and umbilical cords used for isolation and propagation of MSCs were informed about the nature of the study, and each provided written informed consent. The same was true for patients with primary brain tumors who supplied tissue samples. All MSC isolation procedures have been previously described. Briefly, human AT-MSCs were isolated from lipo-aspirates using a collagenase type VII digestion and plastic adherence technique, as described previously.7 This tissue was obtained from healthy individuals undergoing elective liposuction. Bone marrow MSCs (BM-MSCs) were isolated by density gradient centrifugation (Percoll separating solution, density 1.077 g/mL).15 DP-MSCs were isolated from dental pulp tissue fragments that adhered to plastic tissue culture dishes.16,17 The same technique was used to obtain umbilical cord MSCs (UBC-MSCs). All experiments were carried out in accordance with relevant guidelines and regulations.
To expand each type of tissue-specific MSC, cells were seeded into plastic dishes (Corning Life Sciences) at 4,000 cells/cm2 and grown with medium exchanges every 2–3 days. Adherent cells were split after they reached confluence with 0.05% trypsin/EDTA (Thermo Fisher Scientific). All MSCs used for experiments were passaged five times or less. MSCs were cultured in DMEM with low glucose (1 g/L) supplemented with 4% human platelet extract (PE) and a solution containing an antibiotic and antimycotic (Thermo Fisher Scientific) at 37°C in a humidified incubator under 5% CO2. Cells were then frozen in aliquots of 1 mL in liquid nitrogen resuspended to a concentration of 10×106 cells/mL in a medium containing 10% dimethyl sulfoxide and 10% human serum albumin.
Each type of tissue-specific MSC was engineered to express the yCD::UPRT fusion gene, as described previously.7 These transfected cell lines were designated as therapeutic stem cells (yCD::UPRT-AT-MSCs, yCD::UPRT-BM-MSCs, yCD::UPRT-DP-MSCs, and yCD::UPRT-UBC-MSCs). Briefly, MSCs were infected with a retrovirus prepared in helper cells. Transfected cells were selected based upon resistance to G-418 (0.4 mg/mL). All therapeutic stem cells were expanded in low-glucose (1 g/L) DMEM supplemented with 4% PE.
Labeling of MSCs with Venofer
Labeling with Venofer (Vifor, France SA, Paris) was performed, as described previously.16 Briefly, heparin and protamine sulfate were mixed together to form a stable complex. Venofer was added at indicated concentrations to this stable complex in PE-free culture medium at room temperature. The labeling complex was added to the MSCs plated in dishes 24 hours before the experiments began, and they were incubated for 4 hours at 37°C under 5% CO2. The culture medium was supplemented with 4% PE, and the cells were incubated overnight at 37°C under 5% CO2. After 24 hours, MSCs were washed with DMEM only, followed by DMEM containing heparin (10 U/mL), and then passaged. Medium conditioned for 24 hours by labeled cells was prepared in PE-free tissue culture fluid and harvested on day 3 after cell labeling. Cell debris was removed by sequential centrifugation and passed through a 0.45 μM syringe filter. Aliquots of the samples were stored at −80°C.
Prussian blue staining of Venofer-labeled MSCs
Prussian blue staining was performed using the Accustain iron staining kit (Sigma-Aldrich, St Louis, MO, USA), according to the manufacturer’s protocol. Venofer-labeled MSCs were washed with PBS to remove any free Venofer nanoparticles and fixed in 10% formaldehyde for 1 hour. Fixed samples were then washed three times in deionized water and incubated with Pearls’ reagent (4% potassium ferrocyanide/12% HCl, 1:1 v/v) for 10–20 minutes at room temperature. Samples were observed on an inverted optical microscope. The labeling efficiency of MSCs for each concentration of Venofer was assessed visually.
Nanoparticle tracking analysis
The concentration and size distribution of the nanoparticles in the CM of Venofer-labeled MSCs were measured using a NanoSight NS500 (Malvern Instruments Ltd., Malvern, UK) equipped with an sCMOS Trigger camera and a 405 nm laser. Data were analyzed using NTA 2.3 analytical software. Nanoparticle tracking analysis was based on capturing the Brownian motion and light scattering to obtain particle size distributions and concentrations. Each sample was diluted in PBS prior to the measurements to optimize the number of particles in the field of view to a concentration of 108–109 particles per milliliter. Samples were measured in triplicate in 60-second videos with manual shutter and gain adjustments. All measurements were performed at room temperature.
Magnetic hyperthermia
An AMF with a frequency of 3.5 MHz and an induction of 1.2 kA/m (1.5 mT) was generated using an experimental setup with water-cooled three-turn coil, as described previously.18,19 To measure the temperature changes over time during exposure to the AMF, we used an optic fiber thermometer FOB101 with automatic registration (Omega Engineering, Norwalk, CT, USA). Briefly, the tumor cells (5×105) were plated 1 day before AMF exposure in 35 mm Petri dishes and maintained in the growth medium after the addition of 400 μL of CM from MSCs/Fe or yCD::UPRT-MSCs/Fe. The total volume of the culture in each dish was 2 mL. Dishes were incubated at 37°C for 18 hours and then exposed to the AMF for 20 minutes. Cell viability was measured using the CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT), according to the manufacturer’s instructions. Data represent the average of at least three independent experiments.
Separation of the exosomes from secretome by size-exclusion chromatography
The CM from yCD::UPRT-DP-MSCs/Fe was separated on a Sephacryl 500 HR column (55×15 mm) pre-equilibrated with PBS. Each fraction (0.5 mL) was sequentially collected using the peristaltic pump P-1 (Pharmacia Lkb, Uppsala, Sweden). Fractions were tested for proliferation (based on the absence of 5-FC) and growth inhibition (based on the presence of 5-FC) in PC3 and HeLa cells. The concentration of iron in all fractions was estimated using the Colorimetric Iron Assay Kit (Sigma-Aldrich), according to the manufacturer’s instructions. The number of exosomes in each fraction was quantified using a Nanosight instrument.
Statistical analysis
Unless noted otherwise, all experiments were repeated at least two times to enable statistical analysis and all results were similar between replicates. All figures indicate statistical significance of P<0.05 as *, P<0.01 as **, and P<0.001 as ***. Data for bar graphs were calculated using an unpaired Student’s t-test. Error bars represent the standard error of the mean from independent samples within each experiment. All statistical analyses were performed using GraphPad Prism 4 software.
Results
Schematic overview of all steps performed in this study is presented in Figure 1. MSCs prepared from various human tissues were engineered to express suicide yCD::UPRT gene. CM from yCD::UPRT gene-transduced cells contain exosomes carrying in their cargo mRNA of the suicide gene. The exosomes were easily internalized by the tumor cells and in the presence of 5-FC, they caused their death in a dose-dependent manner. When the yCD:UPRT-MSCs were labeled with Venofer, we found that the Venofer nanoparticles were included in the exosomes released into the CM. These exosomes allow targeted ablation of tumor cells by three distinct strategies. Magnetic hyperthermia, addition of 5-FC, and the combination of both approaches caused tumor cell death.
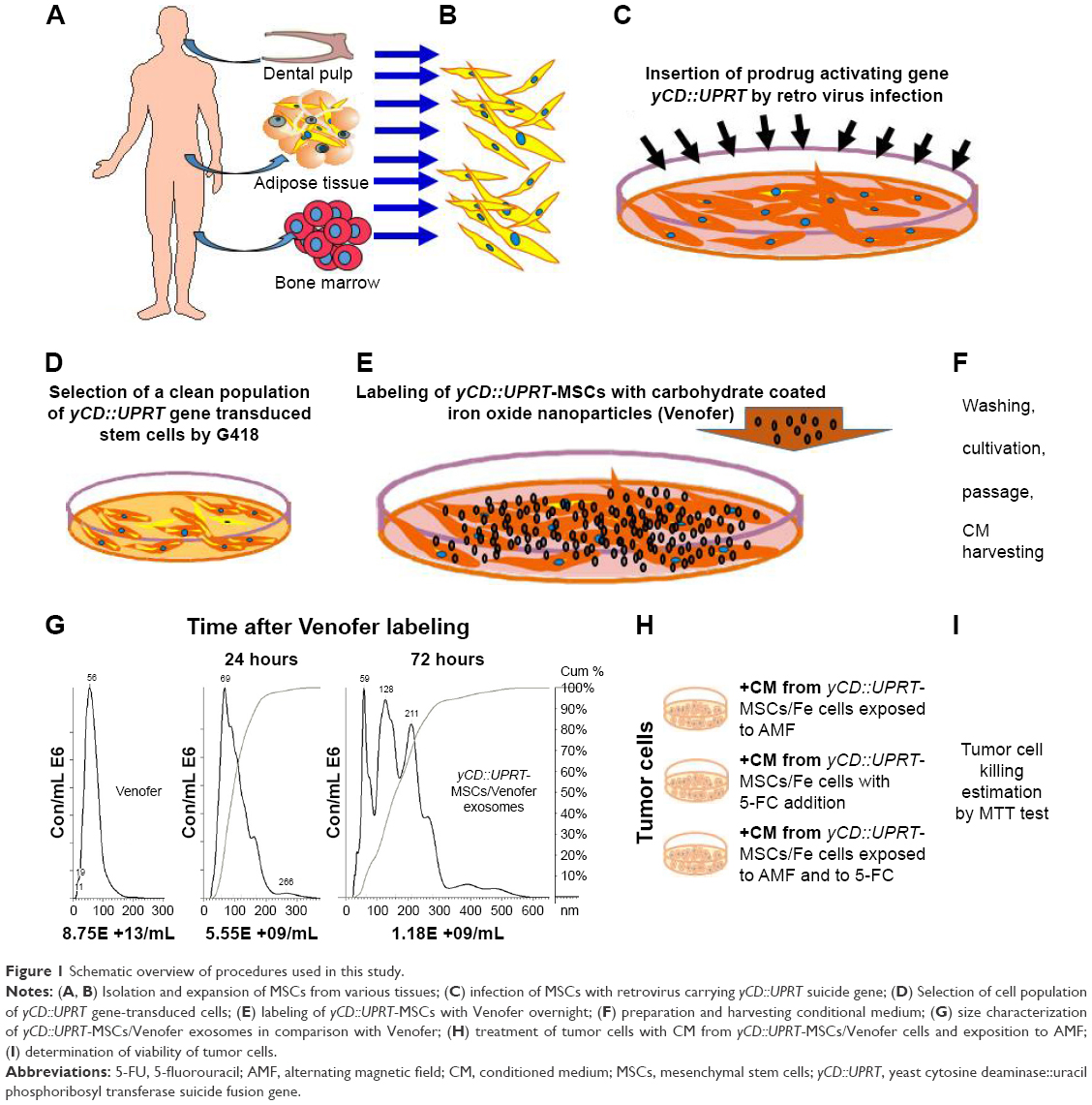 | Figure 1 Schematic overview of procedures used in this study. |
Labeling the MSCs and yCD::UPRT-MSCs with carbohydrate-coated ultrasmall SPIONs (Venofer) did not influence cell physiology
Recently, we described an optimized Venofer labeling procedure in DP-MSCs.16 Using the same technique, we extended our studies to AT-MSCs, BM-MSCs, and UBC-MSCs. We labeled both wild-type and yCD::UPRT gene-transduced cells with Venofer, which we then designated as MSCs/Fe and yCD::UPRT-MSCs/Fe, respectively. The labeled MSCs retained the ability to differentiate into adipocytes, osteoblasts, and chondrocytes. These Venofer-labeled MSCs were still positive for CD44, CD73, CD90, and CD105 and negative for CD45 and CD34, as we recently reported.16 The proliferation of labeled wild-type and yCD::UPRT-expressing DP-MSCs was not affected by a broad concentration range of Venofer. In addition, cell death was induced in yCD::UPRT-DP-MSCs following treatment with the prodrug 5-FC. This indicated that Venofer did not inhibit the expression of the yCD::UPRT fusion enzyme, which converts 5-FC into cytotoxic 5-FU (Figure 2A–C).
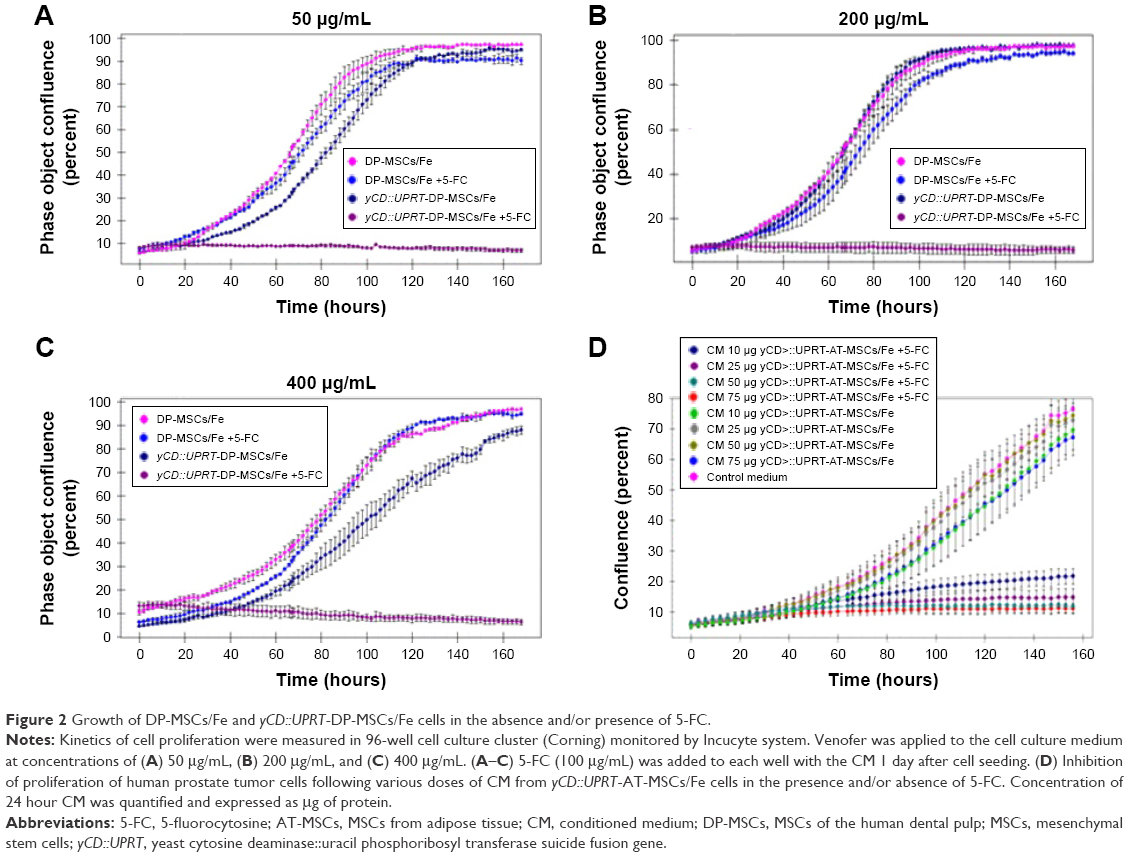 | Figure 2 Growth of DP-MSCs/Fe and yCD::UPRT-DP-MSCs/Fe cells in the absence and/or presence of 5-FC. |
MSCs/Fe and yCD::UPRT-MSCs/Fe release exosomes
We previously found that all MSCs and yCD::UPRT-MSCs secreted exosomes into the CM, regardless of whether they were labeled with Venofer (MSCs/Fe and yCD::UPRT-MSCs/Fe). These exosomes are hereafter referred to as yCD::UPRT-MSCs/Fe-Exos. We have previously shown that exosomes from yCD::UPRT-MSCs also contained mRNA of the suicide fusion gene as cargo. These exosomes were easily internalized by the tumor cells and the mRNA was translated to produce a fused yCD::UPRT enzyme that converts 5-FC into cytotoxic 5-FU.14 Medium conditioned for 24 hours by the presence of yCD::UPRT-AT-MSCs/Fe inhibited the proliferation of PC3 tumor cells in the presence of 5-FC (Figure 2D). The Venofer labeling of yCD::UPRT-DP-MSCs cells did not influence the expression of suicide gene. As shown in Video S1, the presence of 5-FC in the tissue culture fluid induced cell death. The cytotoxic effects of the CM containing yCD::UPRT-MSCs/Fe-Exos in the presence of 5-FC were found to be equivalent among the three human tumor cell lines tested, which included uterine cervical carcinoma HeLa cells, the prostate cancer cell line PC3, and the human brain glioma cell line U-118.
Characterization of nanoparticles released from DP-MSCs/Fe and yCD::UPRT-DP-MSCs/Fe
Exosomes frequently contain compounds foreign to cells. We determined whether iron oxide was accumulated in MSCs/Fe-Exos and yCD::UPRT-MSCs/Fe-Exos. Our data demonstrated that exosomes containing Venofer nanoparticles were released from the labeled DP-MSCs and yCD::UPRT-DP-MSCs. The process of formation of DP-MSC-Venofer nanoparticles took 3 days of cell cultivation. Nanosight analysis of nanoparticles released from the labeled cells showed gradually increasing size of the particles with time, reaching a peak 3 days after cell labeling. The size of the released nanoparticles was found to be a heterogeneous population in the range of 120–210 nM in diameter. Previously, we determined the size of Venofer as a homogenous population of 65 nM diameter particles.16 The number and size of the Venofer nanoparticles subsequently diminished as the labeled cells divided (Figure 3).
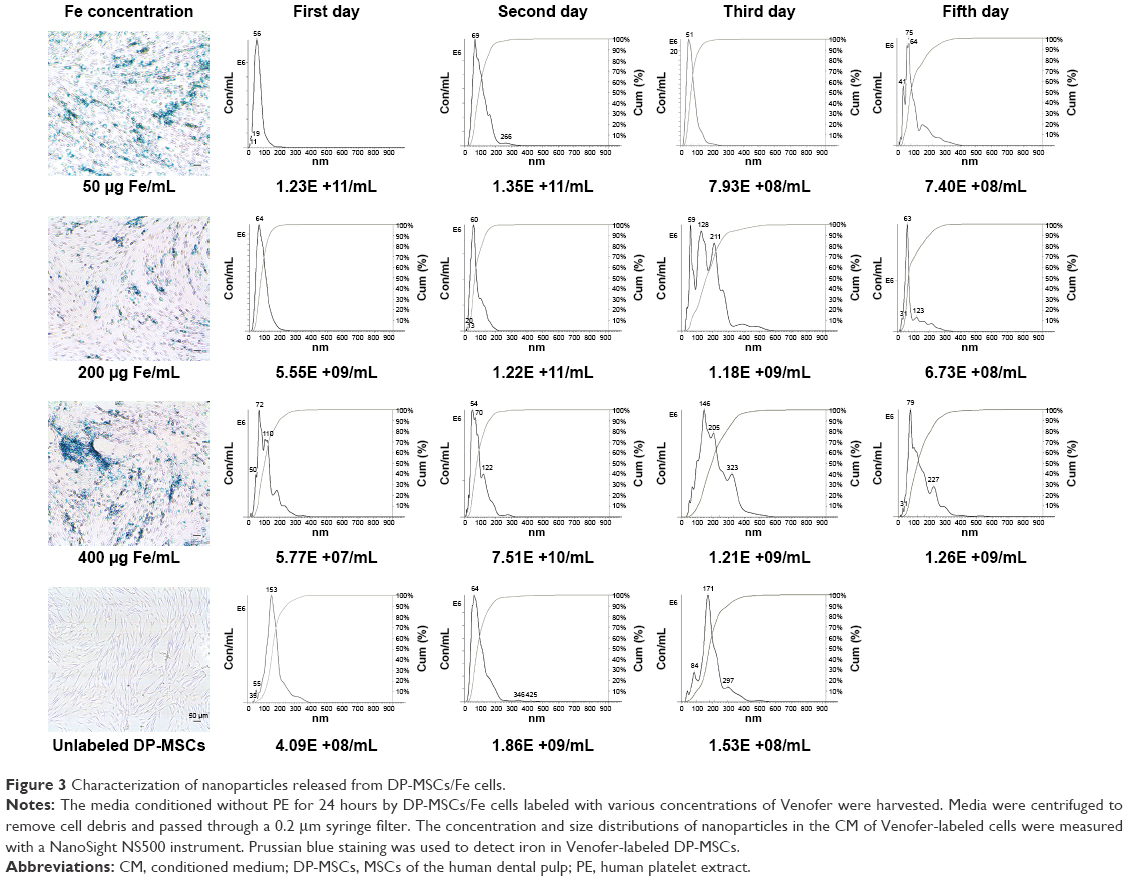 | Figure 3 Characterization of nanoparticles released from DP-MSCs/Fe cells. |
Tumor cell inhibition correlated with the presence of iron oxide-containing MSC exosomes
The CM of MSCs contains a rich secretome of soluble factors and exosomes. To discriminate between the biologic activities of the secretome versus the exosomes, we fractionated the CM harvested from cultured yCD::UPRT-DP-MSCs/Fe using size-exclusion chromatography on a Sephacryl 500 HR column. All fractions obtained were tested for their effects on proliferation of PC3 and HeLa cells in the presence or absence of 5-FC. The concentration of exosomes was also determined. Tumor cell proliferation was inhibited most effectively by treatment with fractions containing nanoparticles, for which Fe was quantified (Figure 4A).
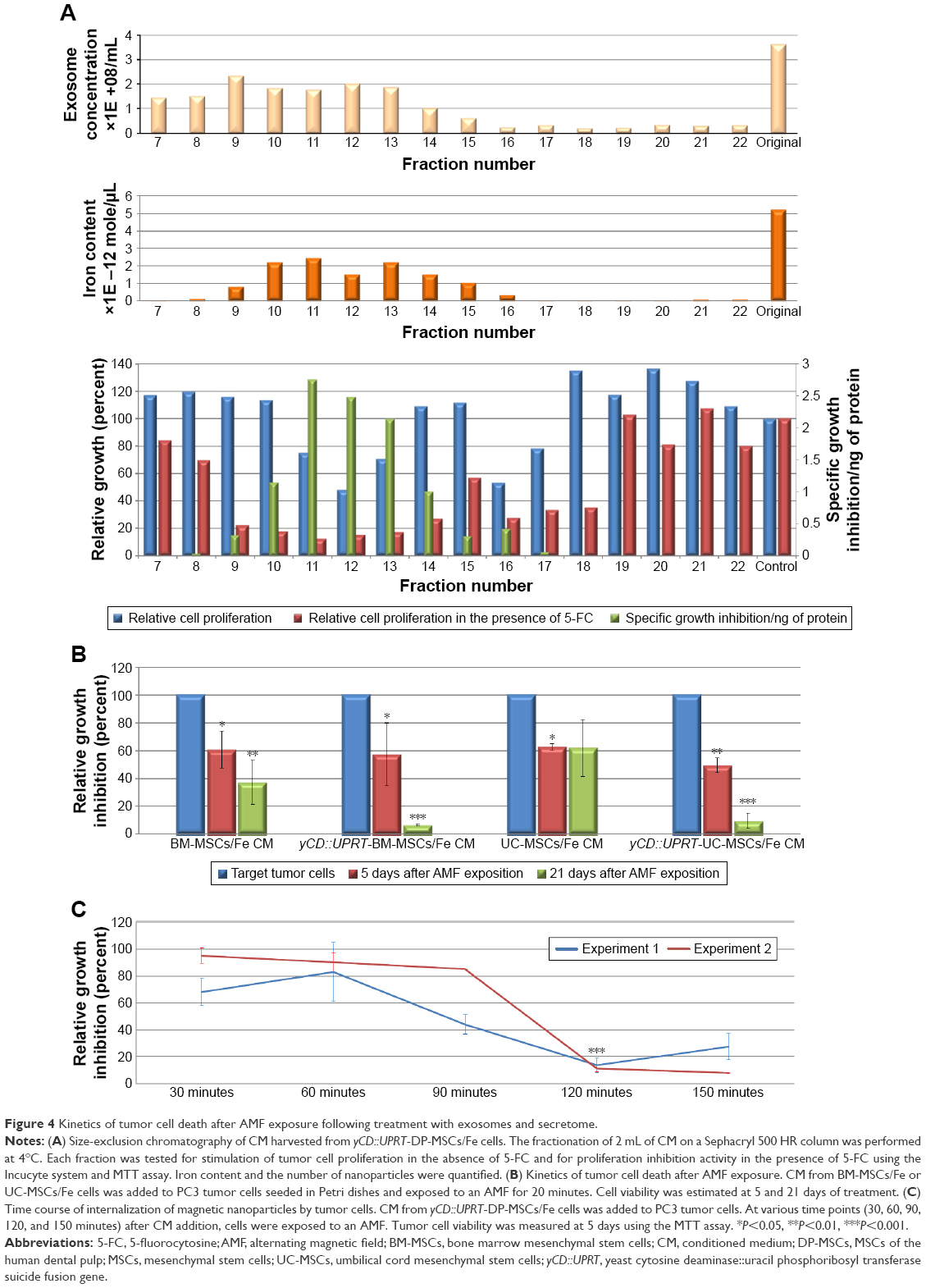 | Figure 4 Kinetics of tumor cell death after AMF exposure following treatment with exosomes and secretome. |
Kinetics of tumor cell death following exposure to an AMF
Hyperthermia-induced tumor cell death mediated by internalized yCD::UPRT-MSCs/Fe exosomes was dose dependent and variable, especially when the CM from different tissue-specific MSCs was used. This was determined by measuring cell death kinetics. MSCs/Fe exosomes were less effective at inducing cell death than yCD::UPRT-MSCs/Fe exosomes. Most of the tumor cells were dead by 5 days after AMF exposure, and all cells were dead by 21 days after AMF exposure. The tissue of origin of the MSCs/Fe exosomes also influenced these kinetics. For example, CM with exosomes of umbilical cord MSCs/Fe was less effective at inducing tumor cell death than CM with exosomes from bone marrow MSCs/Fe (Figure 4B).
Nanoparticles released from Venofer-labeled MSCs are taken up by tumor cells
In order to study the uptake kinetics of yCD::UPRT-AT-MSC/Fe exosomes by the tumor cells, we treated PC3 cells with the same dosage of yCD::UPRT-AT-MSC/Fe CM as previously described and exposed the cultures to an AMF. We found that 120 minutes was sufficient for the PC3 cells to take up the exosomes and undergo cell death when excess yCD::UPRT-AT-MSC/Fe CM was administered (Figure 4C).
Exosomes released from yCD::UPRT-MSCs/Fe kill tumor cells via AMF-induced intracellular hyperthermia
An example of the heating curve of Venofer suspension at a concentration of 200 μg/mL is shown in Figure 5. In the control experiments, heating of the sample without Venofer was found to be negligible. From measurements of time-dependent temperature curves, the temperature-to-time variation for the start of heating (ΔT/Δt) was calculated from the regression line. For thermally equilibrated samples with steady temperatures before exposure to an AMF-specific absorbed rate (SAR), an important parameter characterizing the heating capabilities of magnetic nanoparticles was calculated by the following formula:
SAR = (cms ΔT/Δt)/mv |
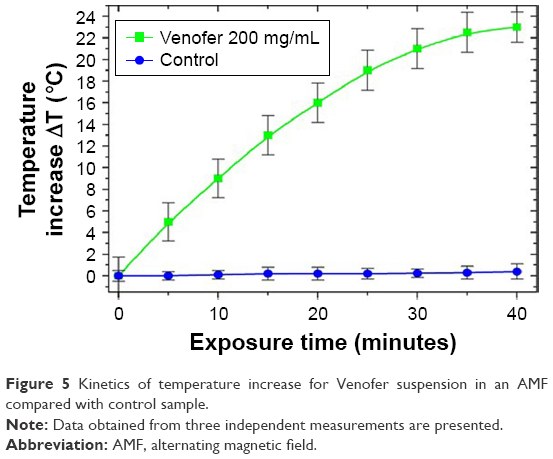 | Figure 5 Kinetics of temperature increase for Venofer suspension in an AMF compared with control sample. |
where c is the specific heat capacity of the sample, mv is the total Venofer mass in the suspension, ms is the mass of the sample mixture, T is the temperature, and t is time. From the measured values, we obtained for Venofer, SAR =297 W/g at a frequency 3.5 MHz and magnetic field 1.2 kA/m. For the hyperthermia experiments, we selected exosomes incubated with different amounts of Venofer. Heating curves of these samples are shown in Figure 6. For all three concentrations of Venofer, hyperthermic temperature of 42°C–45°C is attained in an interval of 20 minutes; therefore, this exposure time was used in all experiments.
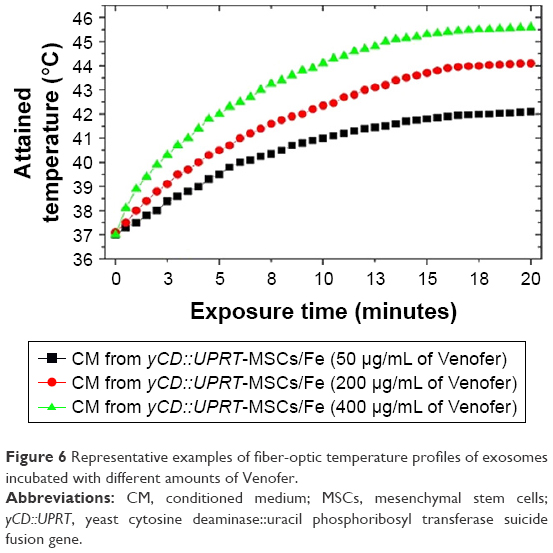 | Figure 6 Representative examples of fiber-optic temperature profiles of exosomes incubated with different amounts of Venofer. |
To determine whether MSCs/Fe or yCD::UPRT-MSCs/Fe are cytotoxic, we exposed a mixture of tumor cells and Venofer-labeled MSCs to an AMF. We found that tumor cells in the vicinity of Venofer-labeled MSCs preferentially underwent cell death (data not shown). In contrast, when the CM from MSCs/Fe or yCD::UPRT-MSCs/Fe was added to the culture containing the tumor cells and an AMF was applied for 20 minutes, tumor cell death was observed in a dose-dependent manner. Therefore, the exosomes behaved as magnetic nanoparticles. The nanoparticles present in the CM were internalized by the tumor cells, which resulted in cell death via intracellular hyperthermia. The nanoparticles in CM from Venofer-labeled yCD::UPRT-MSCs contain mRNA of the suicide gene in their cargo.16 When CM was added to tumor cells in the presence of a prodrug, the tumor cells died due to the intracellular process of mRNA translation to an enzyme converting 5-FC to 5-FU. The intracellular internalization of yCD::UPRT-MSCs/Fe exosomes is a condition sine qua non for the tumor cell killing effect. Besides other components, the cargo of yCD::UPRT-MSCs/Fe exosomes contains Venofer nanoparticles and suicide gene mRNA. This circumstantial evidence supports the conclusion that intracellular hyperthermia was the mechanism of tumor cell killing. As expected, the efficacy of tumor cell ablation was dependent on the concentration of Venofer used for cell labeling. The highest concentration tolerated by the cells was 400 μg/mL of Venofer, which yielded a higher tumor cell ablation efficiency compared to 200 or 50 μg/mL concentration. PC3 and HeLa cell lines treated with nanoparticles from yCD::UPRT-DP-MSCs/Fe and exposed to an AMF underwent hyperthermia-induced cell death in a dose-dependent manner very effectively (Figure 7A). When 5-FC was added immediately following AMF treatment, an increase in cytotoxicity was observed. When 5-FC was added 3 days later to the tumor cells that survived AMF treatment, no additional cell death was observed. Furthermore, an increase in cytotoxicity occurred when the tumor cells surviving AMF exposure were subsequently treated with the CM from yCD::UPRT-MSCs in the presence of the prodrug 5-FC (Figure 7A). On the other hand, a partial killing effect of AMF exposition under the same conditions was observed with the human glioma cell line U118 (data not shown). Human primary glioblastoma and recurrent astrocytoma cells treated with nanoparticles from yCD::UPRT-DP-MSCs/Fe and exposed to an AMF differed in the sensitivity to hyperthermia-induced cell death (Figure 7B). While most glioblastoma cells died within 5 days after AMF, the remainder died during subsequent days. The response of the recurrent astrocytoma cells was particularly noteworthy, as only a proportion of the cells (approximately 50%) underwent hyperthermia-induced cell death at later time points following AMF exposure (Figure 7B).
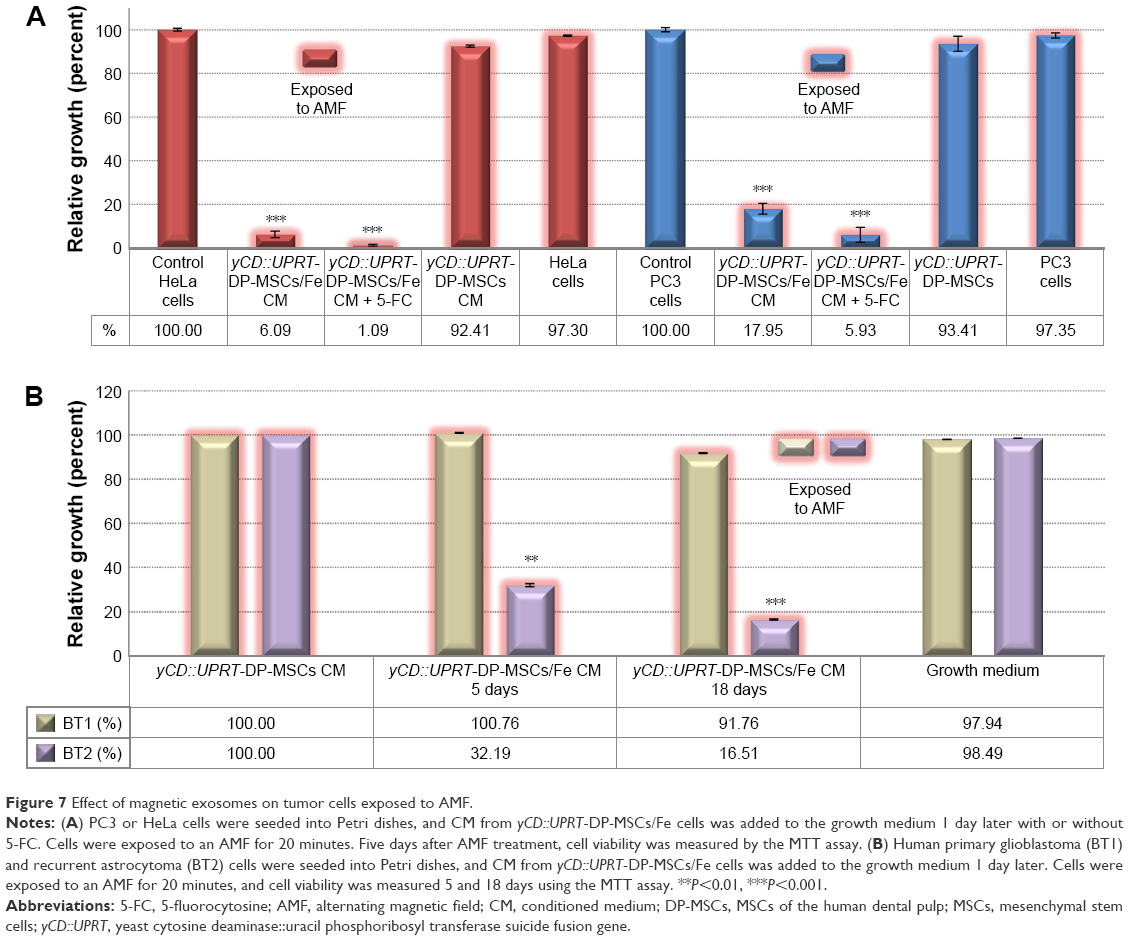 | Figure 7 Effect of magnetic exosomes on tumor cells exposed to AMF. |
Discussion
Tumor targeting using an external AMF has been previously proposed. We and others have shown that iron-labeled magnetoliposomes can be targeted to specific sites in tissues.18,20 Since then, significant progress has been achieved with respect to using nanoparticles in experimental cancer therapies. The discovery of adult stem cells responsible for the homeostasis and repair of aged and damaged tissues in the body has been of great medical interest.21 The discovery of MSC-derived exosomes that act in a paracrine/endocrine fashion is an important step toward cell-free MSC therapy.22 MSCs and their exosomes are known to recognize tumors and metastases as wounds, migrate to them and integrate into the tumor stroma. Stoff-Khalili et al have shown that intravenous injection of human MSCs loaded with conditionally replicating adenoviruses targeted MDA-MB-231 mouse model of metastatic breast cancer in vivo. The treatment led to extended mouse survival compared to mice treated with conditionally replicating adenoviruses alone.23 MSCs expressing and secreting tumor necrosis factor-related apoptosis-inducing ligand using systemic MSC-mediated delivery were shown to suppress metastatic growth of pancreatic carcinoma.24 Furthermore, Toro et al have shown an efficient treatment of the metastatic dissemination of the ovarian tumor by yCD-UPRT gene-modified AT-MSCs in nude mouse model.25 The authors did not consider existence of MSC-exosomes with suicide gene mRNA; therefore, the explanation was linked to the bystander effect. Combined action of bystander effect and exosome internalization of metastases was likely involved. Kalber et al reported migration and integration of systemically administered SPION-labeled MSCs in ovarian tumor nude mouse model.3 They found that even though the nanocarrier was able to accumulate into tumors, the amount of magnetic nanoparticles was not necessarily enough to promote hyperthermia in order to treat cancer exclusively by heat delivery 14 days after the administration of labeled MSCs. Studies of biodistribution, migration, and homing of systemically applied MSCs revealed that 80% of cells are immediately entrapped in the lung tissue and then cleared to the liver within 1 day.26 Our data suggest the importance of intracellular localization of iron oxide nanoparticles for efficient magnetic hyperthermia.
A significant number of clinical trials using MSCs for the treatment of several diseases such as bone defects, myocardial infarction, and autoimmune diseases such as, for instance, Crohn’s disease, and diabetes have shown beneficial therapeutic effect. Homing of cells to the tissue injury sites and the capability to secrete a large amount of trophic factors were attributed to the therapeutic effect. Difficulty to find cells shortly after even local administration to the site of the injury and appearance of therapeutic effect several weeks later were clarified by the discovery of nanoparticles responsible for the therapeutic effect. MSCs act through paracrine/endocrine mechanisms to trigger these regenerative processes. The most important components within the secretome are the exosomes, which transfer bioactive molecules (such as mRNAs and microRNAs) between cells.27,28 The exchange of genetic information together with microRNAs leads to reprogramming of recipient cells, allowing them to regenerate damaged tissue.29,30 In our preclinical studies where human melanoma cells,9 prostate cancer cells10 were implanted subcutaneously to immunocompromised nude mice, a significant tumor growth inhibition was observed when suicide gene-engineered MSCs were intravenously injected. These data were not compatible with the known biodistribution of intravenously administered cells. This discrepancy was explained when we found that suicide gene-transduced MSCs release exosomes with mRNA of the suicide gene in their cargo.14,16 We and others found that MSCs or yCD::UPRT-MSCs can be labeled with iron oxide nanoparticles without affecting cell proliferation or survival.16,31 Riegler et al previously showed that MSCs labeled with SPIONs migrate to and integrate into vascular injuries in vivo within 1 hour. They also demonstrated that the size and chemical coating of the SPIONs influence the uptake of MSCs in humans.32 Our in vitro data support this observation. Since exosomes are released from yCD::UPRT-transduced cells, this allows the testing of their biologic activity using the prodrug 5-FC to assay dose-dependent tumor cytotoxicity. Our results indicate that the accumulation of Fe-containing nanoparticles in MSCs required a minimum of 3 days following initial treatment to achieve optimal labeling. Using size-exclusion gel chromatography, we found that tumor cell death in the presence of the prodrug correlated with the presence of nanoparticles and Fe in MSC exosomes. The size of most yCD::UPRT-MSCs/Fe nanoparticles suggests that Venofer nanoparticles were taken up as cargo in MSC exosomes. However, the heterogeneity of MSCs/Fe nanoparticles with regard to cytotoxicity following AMF treatment does not exclude the possibility that some Venofer nanoparticles were de-coated. Nanoparticles released from MSCs labeled with Venofer currently represent clinically acceptable tools for cancer therapy using magnetic-mediated hyperthermia. Venofer, a drug approved for treating anemia, is Food and Drug Administration approved, in addition to all other reagents needed for MSC labeling (protamine sulfate and heparin). This is advantageous for advancing AMF treatments into formal clinical testing. MSCs/Fe nanoparticles possess several properties that make them preferable to artificially prepared nanoparticles for treating cancer with hyperthermia. MSCs/Fe nanoparticles preferentially accumulate in tumors. They are efficiently taken up by the tumor cells, and they are nontoxic and nonimmunogenic. They can be applied intravenously, and further chemical modifications are possible. Despite obtaining promising results for tumor cell killing by yCD::UPRT-AT-MSCs/Venofer exosomes via AMF in vitro, only future experiments with tumor-bearing animals exposed to AMF could reveal the utility of this therapeutic approach. The human tumor cells PC3 and HeLa used in our AMF experiments were found to be highly sensitive. On the other hand, with the glioma cell line U118, only a partial killing effect was observed (data not shown). The results presented with two different primary brain tumors revealed difference in the efficiency of the CD::UPRT-AT-MSCs/Venofer exosome/AMF system on comparing human glioblastoma versus astrocytoma. These results suggest existence of some sort of selected uptake of yCD::UPRT-AT-MSCs/Venofer exosomes by the tumor cells probably on a receptor/ligand level. More studies addressing this issue are needed that could discover the tumor-specific targeting of these exosomes. Quick biodistribution of exosomes applied systemically and their elimination in 1–6 hours after administration33–35 suggests that AMF exposition should be applied shortly after the magnetoexosome application.
The therapeutic potential of human MSCs for treating glioma, either as cellular vehicles for prodrug gene therapy or as producers of microparticles, is of great interest.36–39 We have previously shown the curative effects of the yCD::UPRT-AT-MSCs/5-FC gene therapy system mediated by MSCs in an animal model of glioblastoma.12,13,40 Magnetic nanoparticles delivered by MSCs could be an excellent tool for glioma ablation via AMF-induced hyperthermia. Using tissue-specific MSCs, neural stem cells, or antibody-modified MSCs, highly specific tumor-targeted magnetic nanoparticles might be established.41 The efficacy of magnetic nanoparticles in cancer therapy might be further enhanced by using the prodrug suicide gene-transduced MSCs for magnetic nanoparticle production. We have recently shown that DP-MSCs can migrate to intracerebral glioblastomas after intranasal application.16 Magnetic nanoparticles from yCD::UPRT-DP-MSCs/Fe could potentially be used to treat pontine brainstem glioma, the most malignant brain tumor in children. Genetically modified Human -AT-MSCs that express rabbit carboxylesterase enzyme, which can efficiently convert the prodrug CPT-11 (irinotecan-7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin) into the active drug SN-38 (7-ethyl-10-hydroxycamptothecin), were found to be effective in brainstem glioma-bearing rats.42 Furthermore, MSCs/Fe cells and/or MSCs/Fe exosomes can be regarded as nanotheranostics.43,44 They can help visualize tumor localization by magnetic resonance imaging and eradicate tumors using hyperthermia, and the modified yCD::UPRT-MSCs/Fe cells might inhibit tumor growth via the expression of the prodrug suicide gene. All of these procedures mentioned are minimally invasive and do not produce systemic toxicity.
MSCs are known to take up various types of chemicals that could facilitate targeted drug delivery. MSCs were previously suggested to be efficient mass producer of exosomes for drug delivery.45 Pascucci et al have shown that MSCs treated with Paclitaxel incorporate this drug into exosomes.46 Similarly, MSCs primed with Paclitaxel kill leukemia cells, inhibit angiogenesis, and improve the survival of leukemia-bearing mice.47 Wilhelm et al reviewed the nanoparticle delivery literature from the past decade and estimated that the median delivery efficiency is low. Only 0.7% of an injected dose of nanoparticles ends up in a tumor.48 Since MSCs have a remarkable tendency to home to tumors, their exosomes loaded with chemotherapeutic drugs may retain the tumor-homing properties. Whether the delivery of Paclitaxel through MSC exosomes will produce better therapeutic effect in comparison with Paclitaxel administered via nanoparticles remains to be determined. MSC exosomes can be easily stored in comparison to cells that are used for prodrug gene therapy for cancer. We found that MSC exosomes can be preserved by lyophilization without losing bioactivity. A previous clinical study using magnetic iron oxide nanoparticles injected into tumors combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme improved overall survival compared to conventional therapies.38 Thus, treating tumors with MSC-derived magnetic nanoparticles to cause AMF-induced hyperthermia may be more effective than simply injecting iron oxide nanoparticles alone into tumors. Recently, an AMF applicator for humans, the Nanoactivator® with magnetic nanoparticles NanoTherm® (MagForce AG, Berlin, Germany), has received approval from the German Federal Institute for Drug and Medical Devices to start a clinical study in glioblastoma patients.
For clinical applications, it is important to realize that in the human body water is a conductor, and hence, eddy currents can be induced causing a damaging effect by AMF. Therefore, there must be an upper limit for the allowed magnetic field and frequency that can be safely applied to living organisms.49 In 1984, Atkinson et al50 performed some clinical tolerance tests on healthy volunteers. They conducted the test using a single-turn induction coil, which was placed around the thorax of the volunteer. Atkinson et al found that field intensities up to 35.8 A·turns/m at a frequency of 13.56 MHz could be thermally tolerated for extended periods of time. This clinical tolerance is not known to be repeated, and the results reported by Atkinson et al became accepted as “Atkinson–Brezovich criterion”, where the product of magnetic field intensity and frequency H × f should not exceed 4.85×109 A/m/s. This criterion is considered at best an upper limit when applying a uniform field over an entire thorax of an adult. In practice, smaller coils are used with inhomogeneous fields and off-axis field directions, which are significantly different conditions than those used by Atkinson et al.50 These factors are expected to reduce eddy current heating. Hence, this criterion should not be considered as the only one. In a more recent study,52 the authors suggested that the upper limit for H × f should be 5×109 A/m/s. In our study, we have used H =1.2×103 A/m and f =3.5×106 s−1, therefore our product was H × f =4.2×109 A/m/s. Nevertheless, the proposed method is not limited to a particular value of either magnetic field intensity or the frequency, and the usage of higher frequencies in our experiments was motivated by the physical effect.53 At lower frequencies, the heating is mainly caused by viscous losses and at higher frequencies, magnetic nanoparticles are thermally driven to magnetodynamic region, leading to enhanced SAR. Magnetic hyperthermia was introduced and tested by Gilchrist et al as a means to heat lymph nodes 60 years ago.54 Since then, hundreds of studies applied this approach for the treatment of cancer. Most of the studies in vivo are using direct injection of magnetic nanoparticles into the center of tumor.55 More elegant approach is to use high-gradient external magnets for the targeting of magnetic nanoparticles to the tumors.56 Unfortunately, this is possible only for treatment of superficial tumors, for example, melanoma.57 In 1993, we proposed58 that AMF can not only be used for hyperthermia, but the generated heat can also be exploited for controlled release of drugs encapsulated in magnetic carriers. This approach is also widely used for combined cancer hyperthermia and chemotherapy.59
Conclusion
MSCs and yCD::UPRT-MSCs labeled with carbohydrate-coated iron oxide nanoparticles incorporate these into exosomes. We discovered the release of exosomes possessing magnetic nanoparticles and mRNA of suicide gene in their cargo. Magnetic nanoparticles packaged either into MSC exosomes or yCD::UPRT-MSC exosomes were efficiently endocytosed by tumor cells. Tumor cells treated with the MSCs/Fe or yCD::UPRT-MSCs/Fe exosomes internalized them and are triggered to apoptosis by AMF-induced intracellular hyperthermia or by exposure to the prodrug 5-FC. These exosomes possess dual tumor cell killing activity in a dose-dependent manner. MSCs/Fe nanoparticles are nontoxic and nonimmunogenic. They can be administered intravenously, and further chemical modifications are possible. These results have major implications for developing MSC-based therapies for cancer patients possessing tumors that are difficult to treat.
Acknowledgment
This study was supported by a grant awarded to CA by the Slovak League against Cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Sawdon A, Weydemeyer E, Peng CA. Antitumor therapy using nanomaterial-mediated thermolysis. J Biomed Nanotechnol. 2014;10(9):1894–1917. | ||
Maier-Hauff K, Ulrich F, Nestler D, et al. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol. 2011;103(2):317–324. | ||
Kalber TL, Ordidge KL, Southern P, et al. Hyperthermia treatment of tumors by mesenchymal stem cell-delivered superparamagnetic iron oxide nanoparticles. Int J Nanomedicine. 2016;11:1973–1983. | ||
Suriyanto, Ng EY, Kumar SD. Physical mechanism and modeling of heat generation and transfer in magnetic fluid hyperthermia through Néelian and Brownian relaxation: a review. Biomed Eng Online. 2017;16(1):36. | ||
Altaner C. Stem Cell-Mediated Prodrug Gene Therapy of High-Grade Brain Tumors, In: K. Shah, editor. Stem Cell Therapeutics for Cancer. New Jersey, USA: Wiley-Blackwell; 2013:57–72. | ||
Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623. | ||
Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67(13):6304–6313. | ||
Cihova M, Altanerova V, Altaner C. Stem cell based cancer gene therapy. Mol Pharm. 2011;8(5):1480–1487. | ||
Kucerova L, Matuskova M, Hlubinova K, Altanerova V, Altaner C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. 2010;9:129. | ||
Cavarretta IT, Altanerova V, Matuskova M, Kucerova L, Culig Z, Altaner C. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol Ther. 2010;18(1):223–231. | ||
Abrate A, Buono R, Canu T, et al. Mesenchymal stem cells expressing therapeutic genes induce autochthonous prostate tumor regression. Eur J Cancer. 2014;50(14):2478–2488. | ||
Altanerova V, Cihova M, Babic M, et al. Human adipose tissue-derived mesenchymal stem cells expressing yeast cytosinedeaminase::uracil phosphoribosyltransferase inhibit intracerebral rat glioblastoma. Int J Cancer. 2012;130(10):2455–2463. | ||
Altaner C, Altanerova V, Cihova M, et al. Complete regression of glioblastoma by mesenchymal stem cells mediated prodrug gene therapy simulating clinical therapeutic scenario. Int J Cancer. 2014;134(6):1458–1465. | ||
Altaner C. Prodrug gene therapy for cancer mediated by mesenchymal stem/stromal cells engineered to express yeast cytosinedeaminase::uracil phosphoribosyltransferase. J Stem Cell Res Ther. 2015;5:2157–2633. | ||
Altaner C, Altanerova V, Cihova M, et al. Characterization of mesenchymal stem cells of “no-options” patients with critical limb ischemia treated by autologous bone marrow mononuclear cells. PLoS One. 2013;8(9):e73722. | ||
Altanerova U, Benejova K, Altanerova V, et al. Dental pulp mesenchymal stem/stromal cells labeled with iron sucrose release exosomes and cells applied intra-nasally migrate to intracerebral glioblastoma. Neoplasma. 2016;63(6):925–933. | ||
Stanko P, Kaiserova K, Altanerova V, Altaner C. Comparison of human mesenchymal stem cells derived from dental pulp, bone marrow, adipose tissue, and umbilical cord tissue by gene expression. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158(3):373–377. | ||
Babincova M, Cicmanec P, Babinec P, et al. Magnetoliposome mediated local electromagnetic tumor hypothermia. Radioengineering. 2000;9(2):12–13. | ||
Babincova M, Altanerova V, Altaner C, Bergemann C, Babinec P. In vitro analysis of cisplatin functionalized magnetic nanoparticles in combined cancer chemotherapy and electromagnetic hyperthermia. IEEE Trans Nanobioscience. 2008;7(1):15–19. | ||
Ito A, Hibino E, Shimizu K, et al. Magnetic force-based mesenchymal stem cell expansion using antibody-conjugated magnetoliposomes. J Biomed Mater Res B Appl Biomater. 2005;75(2):320–327. | ||
Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16(3):381–390. | ||
Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells. 2017;35(4):851–858. | ||
Stoff-Khalili MA, Rivera AA, Mathis JM, et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat. 2007;105(2):157–167. | ||
Mohr A, Albarenque SM, Deedigan L, et al. Targeting of XIAP combined with systemic mesenchymal stem cell-mediated delivery of sTRAIL ligand inhibits metastatic growth of pancreatic carcinoma cells. Stem Cells. 2010;28(11):2109–2120. | ||
Toro L, Bohovic R, Matuskova M, Smolkova B, Kucerova L. Metastatic ovarian cancer can be efficiently treated by genetically modified mesenchymal stromal cells. Stem Cells Dev. Epub 2016 Aug 16. | ||
Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res Ther. 2016;7:7. | ||
Timmers L, Lim SK, Arslan F, et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2007;1(2):129–137. | ||
Rani S, Ryan AE, Griffin MD, Ritter T. Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther. 2015;23(5):812–823. | ||
Chen TS, Lai RC, Lee MM, Choo AB, Lee CN, Lim SK. Mesenchymal stem cell secretes microparticles enriched in pre-microRNAs. Nucleic Acids Res. 2009;38(1):215–224. | ||
Baglio SR, Rooijers K, Koppers-Lalic D, et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res Ther. 2015;1(6):127. | ||
Thu MS, Bryant LH, Coppola T, et al. Self-assembling nanocomplexes by combining ferumoxytol, heparin and protamine for cell tracking by magnetic resonance imaging. Nat Med. 2012;18(3):463–467. | ||
Riegler J, Liew A, Hynes SO, et al. Superparamagnetic iron oxide nanoparticle targeting of MSCs in vascular injury. Biomaterials. 2013;34(8):1987–1994. | ||
Lai CP, Mardini O, Ericsson M, et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporte. ACS Nano. 2014;8(1):483–494. | ||
Lai CP, Kim EY, Badr CE, et al. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun. 2015;6:7029. | ||
Grange C, Tapparo M, Bruno S, et al. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int J Mol Med. 2014;33(5):1055–1063. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Delcroix GJ, Jacquart M, Lemaire L, et al. Mesenchymal and neural stem cells labeled with HEDP-coated SPIO nanoparticles: in vitro characterization and migration potential in rat brain. Brain Res. 2009;1255:18–31. | ||
Roger M, Clavreul A, Venier-Julienne MC, et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials. 2010;31(32):8393–8401. | ||
Silva AC, Oliveira TR, Mamani JB, et al. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int J Nanomedicine. 2011;6:591–603. | ||
Matuskova M, Hlubinova K, Pastorakova A, et al. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010;290(1):58–67. | ||
Balyasnikova IV, Ferguson SD, Sengupta S, Han Y, Lesniak MS. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. PLoS One. 2010;5(3):e9750. | ||
Choi SA, Lee JY, Wang KC, et al. Human adipose tissue-derived mesenchymal stem cells: characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur J Cancer. 2012;48(1):129–137. | ||
Luo S, Yang X, Sh C. Newly emerging theranostic agents for simultaneous cancer targeted imaging and therapy. Curr Med Chem. 2016;23(5):483–497. | ||
Ansari C, Tikhomirov GA, Hong SH, et al. Development of novel tumor-targeted theranostic nanoparticles activated by membrane-type matrix metalloproteinases for combined cancer magnetic resonance imaging and therapy. Small. 2014;10(3):566–575. | ||
Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341. | ||
Pascucci L, Coccè V, Bonomi A, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262–270. | ||
Pessina A, Coccè V, Pascucci L, et al. Mesenchymal stromal cells primed with Paclitaxel attract and kill leukaemia cells, inhibit angiogenesis and improve survival of leukaemia-bearing mice. Br J Hematol. 2013;160(6):766–778. | ||
Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(16014):1–12. | ||
Ortega D, Pankhurst QA. Magnetic hyperthermia. In: O’Brien P, editor. Nanoscience. Volume 1: Nanostructures Through Chemistry. Cambridge, UK: Royal Society of Chemistry; 2013:60–88. | ||
Atkinson WJ, Brezovich IA, Chakraborty DP. Usable frequencies in hyperthermia with thermal seeds. IEEE Trans Biomed Eng. 1984;31(1):70–75. | ||
Obaidat IM, Issa B, Haik Y. Magnetic properties of magnetic nanoparticles for efficient hyperthermia. Nanomaterials (Basel). 2015;5(1):63–89. | ||
Hergt R, Dutz S. Magnetic particle hyperthermia-biophysical limitations of a visionary tumour therapy. J Magn Magn Mater. 2007;311(1):187–192. | ||
Usov NA, Liubimov BY. Dynamics of magnetic nanoparticle in a viscous liquid: application to magnetic nanoparticle hyperthermia. J Appl Phys. 2012;112(2):023901. | ||
Gilchrist RK, Medal R, Shorey WD, Hanselman RC, Parrott JC, Taylor CB. Selective inductive heating of lymph nodes. Ann Surg. 1957;146(4):596–606. | ||
Babincová M, Altanerová V, Altaner C, Cicmanec P, Babinec P. In vivo heating of magnetic nanoparticles in alternating magnetic field. Med Phys. 2004;31(8):2219–2221. | ||
Babincová M, Altanerová V, Lampert M, et al. Site-specific in vivo targeting of magnetoliposomes using externally applied magnetic field. Z Naturforsch C. 2000;55(3–4):278–281. | ||
Babinec P, Babincová M, Sourivong P, Leszczynska D. Efficient treatment of pigmented B16 melanoma using photosensitized long-circulating magnetofullerenosomes. J Magn Magn Mater. 2005;293(1):394–397. | ||
Babincová M. Microwave induced leakage of magnetoliposomes. Possible clinical implications. Bioelectrochem Bioenerg. 1993;32(2):187–189. | ||
Babincová M, Čičmanec P, Altanerová V, Altaner C, Babinec P. AC-magnetic field controlled drug release from magnetoliposomes: design of a method for site-specific chemotherapy. Bioelectrochemistry. 2002;55(1–2):17–19. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.