")
Back to Journals » International Journal of Nanomedicine » Volume 12
Human cytotoxic T-lymphocyte membrane-camouflaged nanoparticles combined with low-dose irradiation: a new approach to enhance drug targeting in gastric cancer
Authors Zhang L, Li R, Chen H, Wei J, Qian H , Su S, Shao J, Wang L, Qian X , Liu B
Received 28 October 2016
Accepted for publication 11 February 2017
Published 17 March 2017 Volume 2017:12 Pages 2129—2142
DOI https://doi.org/10.2147/IJN.S126016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lianru Zhang, Rutian Li, Hong Chen, Jia Wei, Hanqing Qian, Shu Su, Jie Shao, Lifeng Wang, Xiaoping Qian, Baorui Liu
The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University and Clinical Cancer Institute of Nanjing University, Nanjing, People’s Republic of China
Abstract: Cell membrane-derived nanoparticles are becoming more attractive because of their ability to mimic many features of their source cells. This study reports on a biomimetic delivery platform based on human cytotoxic T-lymphocyte membranes. In this system, the surface of poly-lactic-co-glycolic acid nanoparticles was camouflaged using T-lymphocyte membranes, and local low-dose irradiation (LDI) was used as a chemoattractant for nanoparticle targeting. The T-lymphocyte membrane coating was verified using dynamic light scattering, transmission electron microscopy, and confocal laser scanning microscopy. This new platform reduced nanoparticle phagocytosis by macrophages to 23.99% (P=0.002). Systemic administration of paclitaxel-loaded T-lymphocyte membrane-coated nanoparticles inhibited the growth of human gastric cancer by 56.68% in Balb/c nude mice. Application of LDI at the tumor site significantly increased the tumor growth inhibition rate to 88.50%, and two mice achieved complete remission. Furthermore, LDI could upregulate the expression of adhesion molecules in tumor vessels, which is important in the process of leukocyte adhesion and might contribute to the localization of T-lymphocyte membrane-encapsulated nanoparticles in tumors. Therefore, this new drug-delivery platform retained both the long circulation time and tumor site accumulation ability of human cytotoxic T lymphocytes, while local LDI could significantly enhance tumor localization.
Keywords: cell membrane, drug delivery system, gastric cancer, low-dose irradiation, nanoparticles
Introduction
Gastric cancer is ranked as the fourth most common cancer and the second most common cause of cancer-related deaths worldwide,1 and is most prevalent in Eastern Asia.2 Since most patients with gastric cancer are asymptomatic at the early stages, the disease is often at an advanced stage when identified.3 Previous studies4,5 have demonstrated that systemic chemotherapy for palliation is advisable for this cohort to improve survival. Paclitaxel (PTX) is the most common hydrophobic, chemotherapeutic drug used in gastric cancer treatment,6 but it also exhibits significant adverse effects because of the drug itself and the co-administered solvent.7
Nanoparticles have been shown to effectively increase chemotherapeutic drug concentration at tumor sites and to decrease systemic exposure.8 The passive accumulation ability of nanoparticles can be attributed to the well-known enhanced permeability and retention effect.9 The nanoparticles can also be engineered to actively accumulate at tumor sites by incorporating targeting ligands on the nanoparticle surface.10 However, the clinical utility of synthetic nanoparticles is significantly limited by their relatively short circulation time11 and by the complexity of producing such actively targeted carriers.12 In addition, passive targeting must overcome the difficulty of heterogeneous and varied tumor vascularization and tumor permeability with different tumor types and stages.13 Furthermore, most synthetic targeting materials are not applicable in the clinic because of their unknown toxicity. Therefore, biomimetic materials are gaining popularity instead.14 Combining synthetic nanoparticles with natural biomaterials to create biomimetic delivery systems is gaining attention as a new engineering strategy.15–17 The concept of using cells as drug delivery vehicles is rapidly developing.18–20 The use of natural cellular membranes to camouflage synthetic polymeric nanoparticles has demonstrated significant potential.21–25 In this study, the membranes of human cytotoxic T lymphocytes (hCTLs) were chosen to produce cancer-targeting nanoparticles, because of the long blood circulation time and the ability to recruit and localize at tumor sites, of this cell type.22 Leukocytes have a variety of proteins on their membranes to detect inflammation and diseased tissues.26,27 hCTLs express higher levels of adhesion molecules than their naive counterparts28 and are more effective in targeting tumor sites.
Radiotherapy has a crucial role in the treatment of locally advanced gastric cancer in a preoperative, postoperative, or palliative setting.29 However, radiotherapy often leads to catastrophic side effects including liver injury, nausea, and diarrhea. Patient intolerance arises since the radiation dose to normal organs is high.30,31 Recently, low-dose radiotherapy has been reported to safely control gastric cancer-related symptoms, demonstrating significant clinical utility.29 Previous studies have demonstrated that local low-dose irradiation (LDI) alone resulted in an increase in intratumoral CD8+ T cells,32 which was shown to be correlated with the upregulated expression of adhesion molecules on tumor vasculature and the increased expression of chemoattractants.33 Furthermore, local LDI could promote the normalization of aberrant vasculature, which facilitated the entry of T lymphocytes into tumors.34
Therefore, in this study, radiotherapy was used as a tumor localization stimulus for PTX-loaded poly-lactic-co-glycolic acid (PLGA) nanoparticles, which were coated with cellular membranes isolated from hCTLs. More hCTL membrane-coated PLGA nanoparticles (TPNPs) are expected to localize in irradiated tumor sites upon exposure to local LDI, which induces the expression of adhesion molecules and chemoattractants.
Materials and methods
Cell lines and mice
The MKN-45 human gastric cancer cell line was obtained from the Shanghai Institute of Cell Biology. MKN-45 cells were cultured in Roswell Park Memorial Institute 1640 medium (Wisent, Nanjing, People’s Republic of China) with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA). Cultures were maintained at 37°C with 5% CO2 in a humidified incubator. Male Balb/c nude athymic mice at 4–6 weeks old were supplied by the Department of Experimental Animals, Yangzhou University. All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals approved by Nanjing Drum Tower Hospital Laboratory Animals Welfare & Ethical Committee, People’s Republic of China.
hCTL membrane isolation
The hCTLs were obtained as previously described.35 All donors signed an informed consent for scientific research statement. To harvest the membranes, source cells (109 cells) were washed using phosphate-buffered saline (PBS) twice and lysed in 10 mL of hypotonic lysis buffer containing 10 mM Tris–HCl (pH 7.5), 1 mM KCl, 1 mM MgCl2, and one phosphatase inhibitor cocktail tablet (Roche, Mannheim, Germany) per 10 mL of solution. The cells were disrupted using a hand-held Dounce homogenizer (20 passes while on ice). The homogenized cells were centrifuged at 500× g for 10 min at 4°C. The supernatant was removed and saved, while the pellet was resuspended in hypotonic lysis buffer and subjected to another 20 passes and centrifuged again. The process was repeated until there were no intact cells in the pellet, as confirmed using a Nikon Eclipse TI-SR microscope (Tokyo, Japan). The supernatants were subjected to discontinuous sucrose density gradient ultracentrifugation (30%, 40%, and 55%) in an Eppendorf 5404 centrifuge (Shanghai, People’s Republic of China) at 4°C.22 The isolated membranes were collected, lyophilized overnight, weighed, resuspended in a 0.9% normal saline solution, and stored at 4°C.
Dot-blot analysis
The distribution of proteins along the discontinuous sucrose density gradient was analyzed using a dot-blot procedure. Briefly, equal amounts of each fraction were spotted on a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). The membrane was blocked using 5% bovine serum albumin in TBS-T, followed by sequential incubation with primary antibodies (1:1,000 dilution) against: CD3z (Bioworld Technology, St Louis Park, MN, USA) and lymphocyte function-associated antigen-1 (LFA-1 or CD11a; Bioworld Technology), and horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin (Ig) G antibody (1:10,000 dilution; Sangon Biotech, Shanghai, People’s Republic of China). The blots were developed using the SuperSignal West Dura chemiluminescent substrate (Pierce, Rockford, IL, USA).
Preparation of PLGA nanoparticles
Briefly, 500 μL of dichloromethane containing PLGA (5 mg) and PTX (0.5 mg) was added dropwise to 4 mL of aqueous phase containing 1% poly(vinyl alcohol) (Aladdin, Shanghai, People’s Republic of China) and emulsified using probe sonication (VCX 130; Sonics & Materials, Inc., Newtown, CT, USA) for 10 min at an amplitude of 30%. The mixture was subsequently stirred in the open air at room temperature for 4 h. The nanoparticles were washed using ultra-pure water three times and filtered. For confocal microscopy imaging and in vivo particle tracking purposes, 10 μg of green dye (DiO; Beyotime, Shanghai, People’s Republic of China) was added to the PLGA dichloromethane solution prior to PLGA nanoparticle synthesis. To determine the encapsulation efficiency, PTX encapsulated in the PLGA was measured using HPLC (Agilent 1200 Series; Agilent, Santa Clara, CA, USA).
Fusion of hCTL membrane-derived vesicles with PLGA nanoparticles and nanoparticle characterization
hCTL membrane-derived vesicles were prepared as previously reported.21 Briefly, the collected hCTL membranes were sonicated for 5 min using a S300H bath sonicator (Elma, Munich, Germany) at a frequency of 37 kHz and a power of 100 W. The resulting vesicles were subsequently coated onto PLGA nanoparticles through co-extrusion using an Avanti mini extruder (Avanti Polar Lipids, Alabaster, AL, USA), as previously reported.21 The size distribution and polydispersity were determined using dynamic light scattering using a Mastersizer 2000 laser particle size analyzer (Malvern Instruments, Malvern, UK). All measurements were performed in triplicate. The surface morphology of the samples was observed using transmission electron microscopy (TEM, JEM-1011; JEOL, Tokyo, Japan). The in vitro release profile of PTX from the TPNPs was assessed using HPLC.
In vitro cellular uptake studies
The human THP-1 cell line was purchased from American Type Culture Collection and is a well-characterized phagocytic cell line to study particle phagocytosis. THP-1 cells were differentiated using phorbol 12-myristate 13-acetate (PMA) at 100 ng/mL for 48 h. The cells were cultured in 24-well plates and were incubated with nanoparticles (loaded with DiO) at 37°C for 30 min. Subsequently, the cells were washed using PBS, and the level of nanoparticle uptake was analyzed using a flow cytometer (BD, San Jose, CA, USA), with data obtained from 20,000 cells per sample.
In the confocal laser scanning microscopy (CLSM) imaging studies, MKN-45 cells were seeded on a four-chamber glass bottomed dish (Invitrosci, Mountain View, CA, USA). DiO-labeled PLGA nanoparticles were encapsulated in T-cell membranes and incubated with MKN-45 cells for 1 h at 37°C. The cells were then washed using PBS and were fixed using 10% formaldehyde for 10 min. The cell membranes and nuclei were stained using DiI (Beyotime) and DAPI (Beyotime), respectively. The corresponding fluorescence images were taken using a Zeiss LSM710 confocal microscope (Carl Zeiss, Oberkochen, Germany).
In vitro cytotoxicity assay
Three groups of MKN-45 cells were seeded (8×103 cells/well) in 96-well plates (Corning, Corning, NY, USA) and incubated overnight. Free PTX, PTX-encapsulated PLGA nanoparticles, and PTX-containing TPNPs were added to the first group and the third group, while the second group received only empty TPNPs. The third group was partially irradiated using 2 Gy irradiation using an electron generator (12MeV; Elekta, Stockholm, Sweden). After incubation for 24 and 48 h, 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide solution was added and incubated for 4 h. The absorbance at 490 nm was measured using a Biotek ELx800 plate reader (BioTek Instruments, Inc., Winooski, VT, USA). The relative cell viabilities were then normalized to the control, set as 100%.
In vivo near-infrared fluorescence imaging and in vivo immunofluorescence
DiR (Bridgen, Beijing, People’s Republic of China) was incubated with hCTL membrane-derived vesicles for 30 min to monitor the biodistribution of the nanoparticles in vivo. A subcutaneous xenograft model of gastric cancer was established by injecting 106 MKN-45 cells in 100 μL of PBS subcutaneously in male Balb/c nude mice. Treatment started when the tumor volume reached ~300 mm3. Three tumor-bearing Balb/c nude mice received LDI (2 Gy), and three did not. They were all injected intravenously with TPNPs the day after LDI. Then, the mice were anesthetized and scanned at different time intervals using a Maestro™ Automated In Vivo Imaging System. For resected organ imaging, the animals were euthanized, and the tumors and organs were excised and imaged.
DiO-loaded TPNPs were injected into the tail vein of tumor-bearing mice. After 24 h, the mice were sacrificed under deep anesthesia. Tumors were harvested and processed for immunostaining. Frozen sections were stained using rabbit anti-human CD31 (Abcam, Cambridge, UK) followed by secondary antibodies with Cy3-conjugated Goat Anti-rabbit IgG (H+L; Abcam). Tumor cells were stained using DAPI. Images were taken using a confocal microscope.
In vivo antitumor efficacy
PTX-loaded TPNPs were prepared. Treatment started when the tumor volume of the Balb/c nude mice reached 150 mm3 at day 0. The mice were randomized and divided into five groups (n=5 for each group). The five groups were treated using the following formulations through intravenous administration on days 0 and 7: irradiation (2 Gy) alone 1 day before administration; free PTX at 10 mg PTX/kg; PTX-loaded TPNPs (same quantities as above); PTX-loaded TPNPs (same quantities as above) plus irradiation (2 Gy); and PBS. Tumor volumes were measured every 2 days for 15 days and compared. The tumor volume (TV) was calculated according to the formula: TV = L × W2/2, where L and W are the major and minor dimensions, respectively. The mice were anesthetized and irradiated using an electron generator. Irradiation was directed at the tumor region by shielding other areas using a 10-mm lead apron.
Interaction between LDI and TPNPs
The tumor growth inhibition rate (IR)36 was calculated as IR (%) = (1− T/C) × 100, where T and C are the tumor volume of the treated and control groups, respectively. In addition, the following formula37 was adopted to analyze the interaction between LDI and PTX-loaded TPNPs: Q = Ea+b/(Ea + Eb − Ea × Eb), where Ea+b, Ea, and Eb are the average effects of combination treatment, drug A only, and drug B only, respectively. The interaction between the two drugs could be classified as an antagonistic effect (Q ≤0.85), an additive effect (0.85≤ Q <1.15), and a synergistic effect (Q ≥1.15).
Histology analysis
On day 16 after injection, one mouse from each group was randomly selected and sacrificed for histological analysis. Organs including the heart, liver, spleen, lung, and kidney were collected; fixed in 10% neutral-buffered formalin, processed in paraffin; sliced into 10-μm-thick sections; stained using hematoxylin–eosin (H&E); and examined using a microscope.
Immunohistochemistry study
Tumor samples were fixed in 10% neutral-buffered formalin and embedded in paraffin. After deparaffinization, rehydration, antigen retrieval, endogenous peroxidase blockade, and non-specific binding site blockade, immunohistochemical staining was performed. Briefly, the anti-intercellular adhesion molecule-1 (ICAM-1) antibody (Proteintech, Wuhan, People’s Republic of China) was diluted 1:200, applied to the samples, and incubated at 4°C overnight. The HRP-conjugated goat anti-rabbit IgG antibody (1:200 dilution; Abcam) was applied and incubated at room temperature for 50 min. Coloration was performed by incubating the slides with 3,3′-diaminobenzidine (Dako, Santa Clara, CA, USA) at room temperature without light. Hematoxylin (Sigma, St Louis, MO, USA) was used to counterstain the slides for 3 min.
Statistical analysis
Data were analyzed using the SPSS software 17.0 (SPSS, Inc.). Quantitative variables are presented as mean and standard deviation (SD) unless otherwise noted. A comparative analysis was performed using Student’s t-test. Statistical significance was set as follows: *P<0.05, **P<0.01, ***P<0.005, and ****P<0.001.
Results
Physical characterization of hCTL membrane-coated PLGA nanoparticles
The preparation process of the TPNPs involved two major steps: the extraction of hCTL membrane-derived vesicles from hCTLs and the vesicle-particle fusion (Figure 1). First, the membranes were collected using a combination of hypotonic lysing (Figure 2A and B) and mechanical membrane disruption, and were further purified through a discontinuous density sucrose gradient.22 Three lipid layers were localized at the interfaces of the different sucrose layers after ultracentrifugation. Two leukocyte plasmacellular membrane marker proteins (LFA-1 and CD3z) were used to confirm the existence of plasmacellular membranes through immunoblotting and flow cytometry (Figure 2C and D). As a result, the membranes were collected from the interface between 30% and 40%. Second, the isolated membranes were washed and extruded through a 200-nm porous polycarbonate membrane to form hCTL membrane-derived vesicles. To synthesize the TPNPs, PLGA nanoparticles were prepared using a modified nanoprecipitation method. Then, the two components were coextruded through an apparatus with 200-nm pores. The encapsulation efficiency and the drug-loading efficiency of PTX-loaded PLGA were 51.20% and 5.07%, respectively. The average diameter of the final PTX-loaded TPNPs was 165.9±1.0 nm (PDI =0.232±0.008; Figure 3A). Furthermore, TEM imaging revealed a membrane coating around the polymeric core (Figure 3B). The in vitro release behavior of PTX from the TPNPs is shown in Figure 3C. The release exhibited a biphasic pattern characterized by a fast-initial release during the first 48 h, and then a slow and continuous release subsequently.
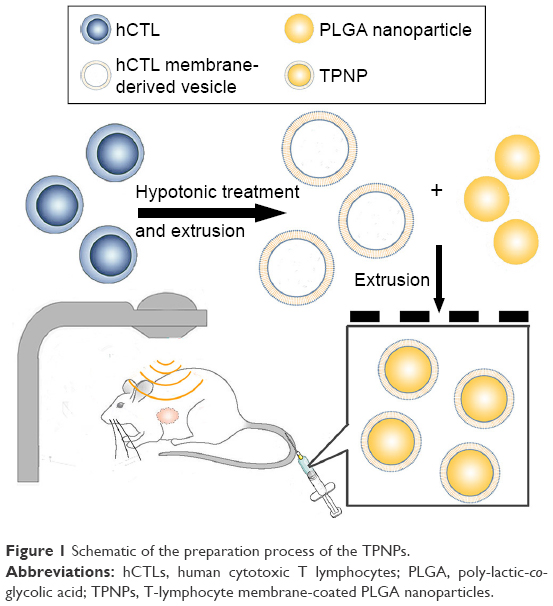 | Figure 1 Schematic of the preparation process of the TPNPs. |
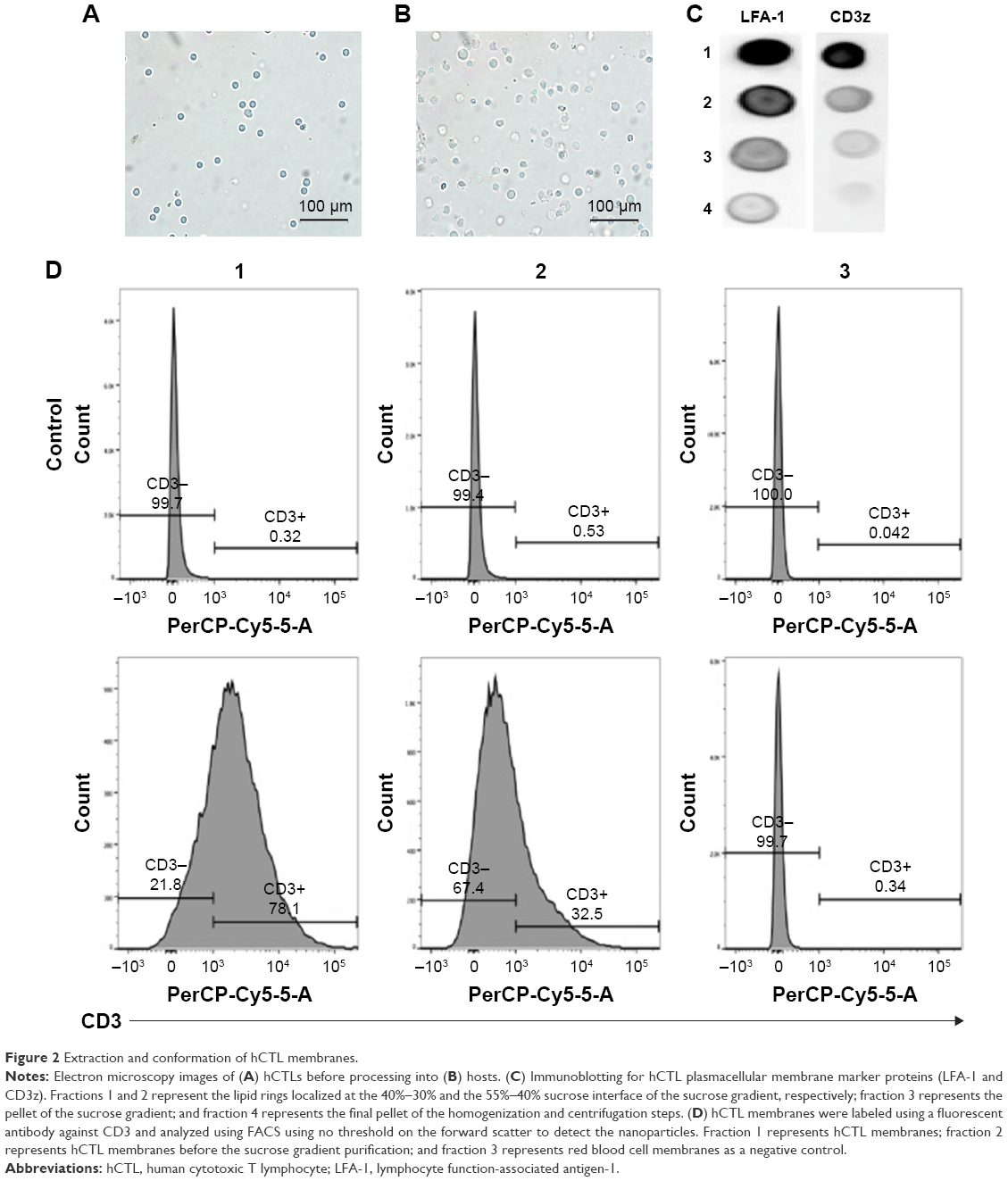 | Figure 2 Extraction and conformation of hCTL membranes. |
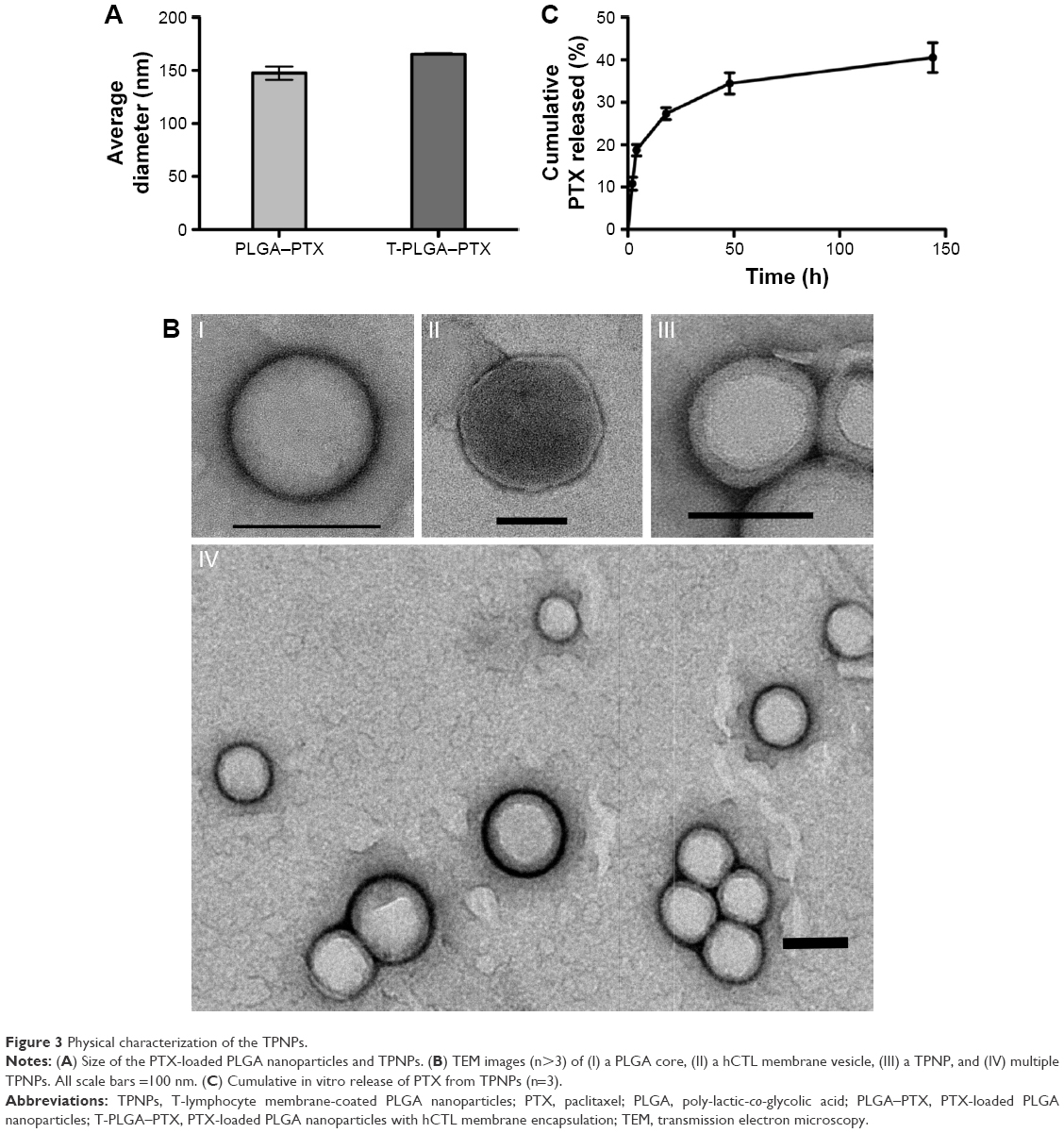 | Figure 3 Physical characterization of the TPNPs. |
Reduced particle phagocytosis
The stealth performance of the hCTL membrane-cloaked PLGA nanoparticles was compared with the uncloaked nanoparticles. As shown in Figure 4A and B, a decreased cellular uptake rate of 23.99% was observed in the presence of hCTL membranes, confirming the satisfactory stealth performance of the hCTL membranes (P=0.002). This suggested that camouflaging with hCTL membranes would prolong the circulation time of the TPNPs.
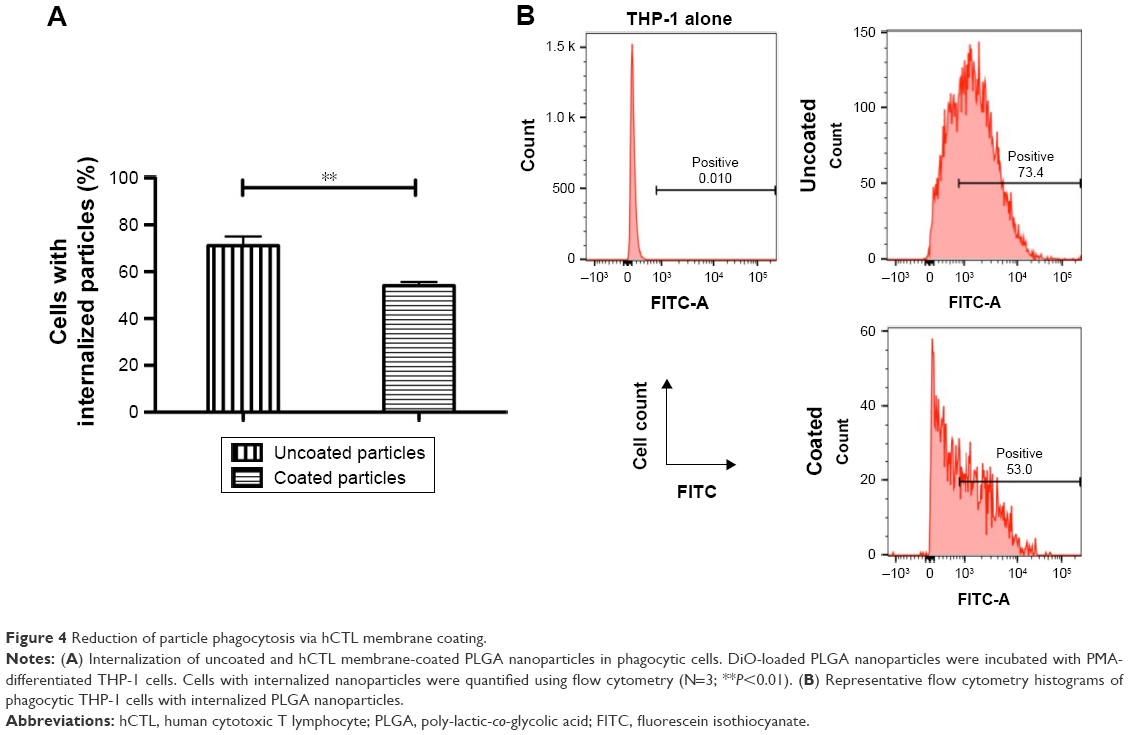 | Figure 4 Reduction of particle phagocytosis via hCTL membrane coating. |
In vitro targeting and cytotoxicity of PTX-loaded TPNPs
PLGA cores were loaded with DiO dye, while the membranes were tagged with DiI dye. The CLSM images demonstrated that the membranes were coated around the polymeric cores, and the TPNPs underwent uptake by the MKN-45 cancer cells within 1 h, localizing around the nuclei in the cytoplasm (Figure 5A).
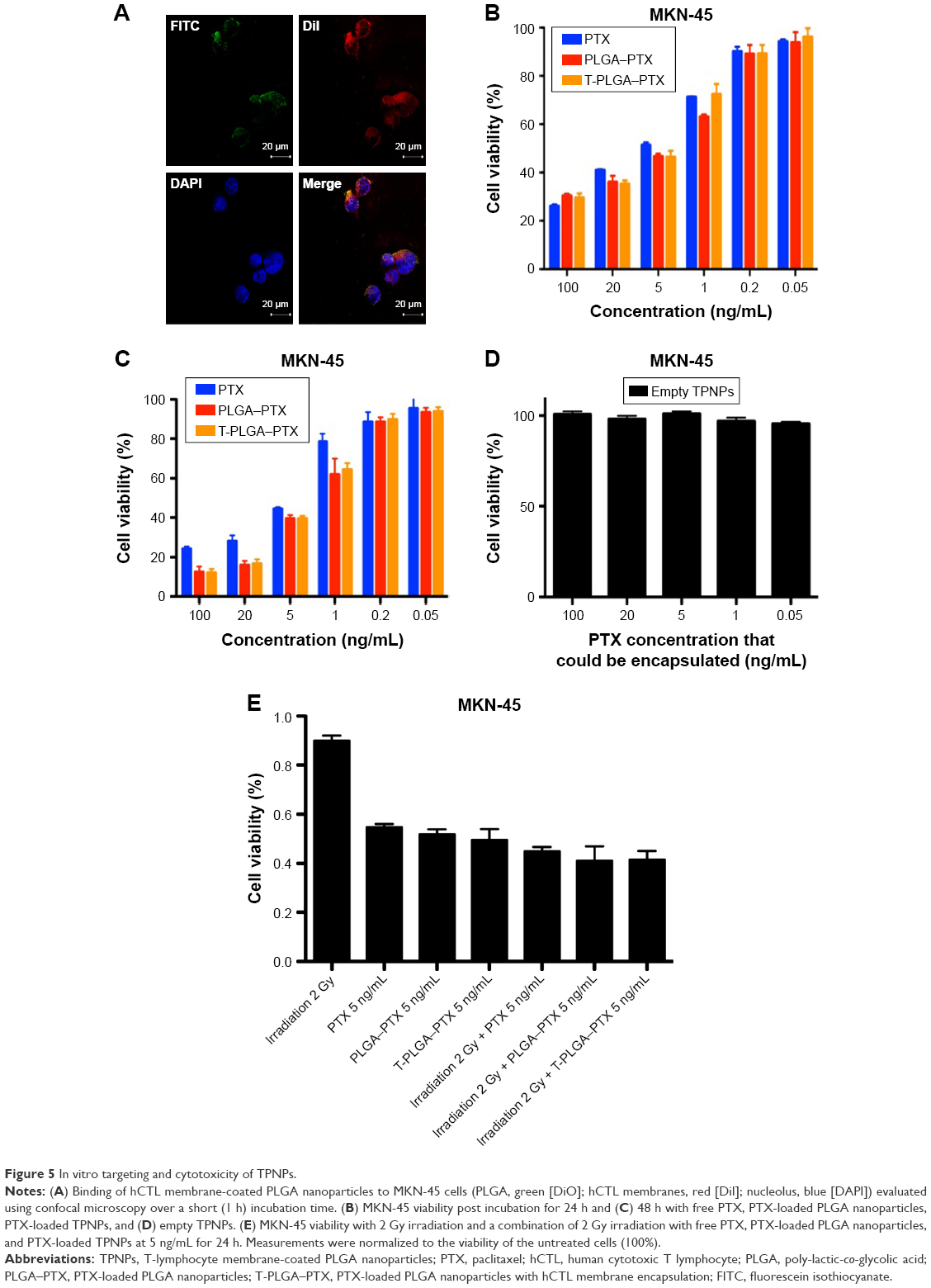 | Figure 5 In vitro targeting and cytotoxicity of TPNPs. |
The IC50 (half maximal inhibitory concentration) was 8.663 μg/mL (free PTX, 24 h), 6.787 μg/mL (PTX-loaded PLGA, 24 h), 7.717 μg/mL (PTX-loaded TPNPs, 24 h), 6.130 μg/mL (free PTX, 48 h), 2.620 μg/mL (PTX-loaded PLGA, 48 h), and 2.822 μg/mL (PTX-loaded TPNPs, 48 h). Free PTX and PTX-loaded TPNPs exhibited a dose-dependent cytotoxicity in MKN-45 cells and were not significantly different (Figure 5B and C). Empty TPNPs displayed no cytotoxicity, even at a high concentration that could encapsulate 100 ng/mL PTX (Figure 5D), indicating their good biocompatibility. Combining 2 Gy irradiation with TPNPs could raise apoptosis in the MKN-45 cell line from 50.4% (related to using 2 Gy irradiation alone) to 58.5% (Figure 5E). There was no significant difference in cytotoxicity between PTX, PTX-encapsulated PLGA nanoparticles, and PTX-loaded TPNPs, when combined with LDI.
In vivo tumor targeting, antitumor efficacy, and safety of PTX-loaded TPNPs
PLGA cores were loaded with DiO dye and then were camouflaged with hCTL membranes. It was examined whether intravenously injected TPNPs targeted subcutaneous tumors after 24 h. Immunofluorescence demonstrated that the TPNPs had better tumor specificity than the PLGA nanoparticles, and LDI significantly enhanced tumor localization (Figure 6A).
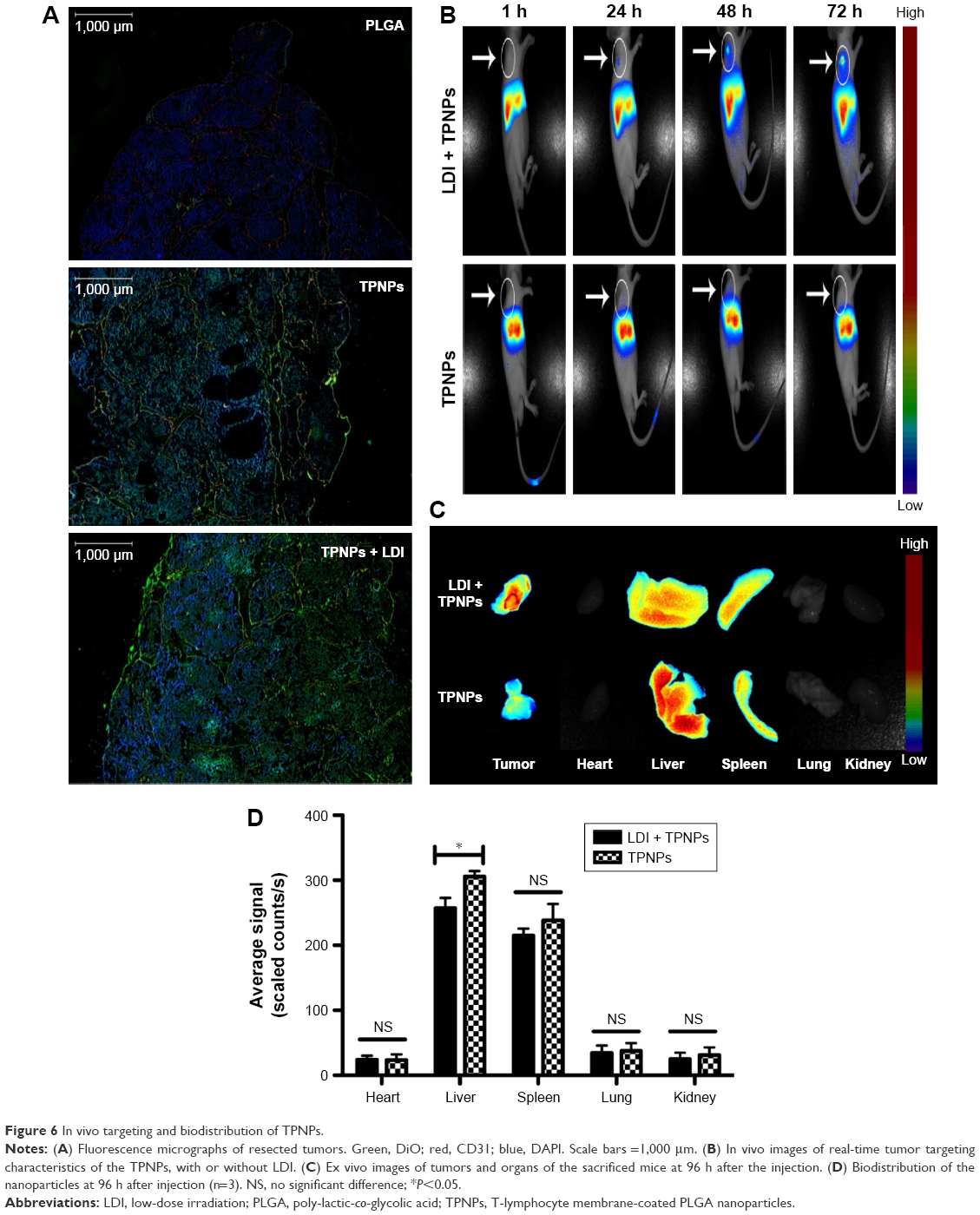 | Figure 6 In vivo targeting and biodistribution of TPNPs. |
To ascertain whether the TPNPs could efficiently accumulate at tumor sites and whether LDI could enhance their efficiency, their route in a xenograft mouse model was assessed (Figure 6B–D). The TPNPs mainly accumulated in the liver within 96 h. After the induction of LDI, the fluorescence signal was clearly visualized and gradually increased at the tumor sites.
PTX-loaded TPNPs were injected into tumor-bearing nude mice (10 mg PTX/kg) on days 0 and 7 (Figure 7A). Significant inhibition of tumor growth was observed in mice treated with PTX-loaded TPNPs when compared with the untreated mice (P<0.001), and the mice treated with free PTX (P<0.001, Figure 7B and C). The tumor growth inhibition rate of PTX-loaded TPNPs was 56.68%. This antitumor efficacy was improved by the addition of irradiation. A combined treatment of tumor LDI and a subsequent TPNP injection resulted in more efficient inhibition of tumor outgrowth than a TPNP injection alone (P=0.002), while irradiation alone had only a minor effect on tumor progression in this experiment (P=0.032). The tumor growth inhibition rate of the combined treatment was ~88.50%. Two mice achieved complete remission (CR) in the TPNP plus LDI group. LDI was, therefore, required for the accumulation of the tumor-targeting TPNPs.
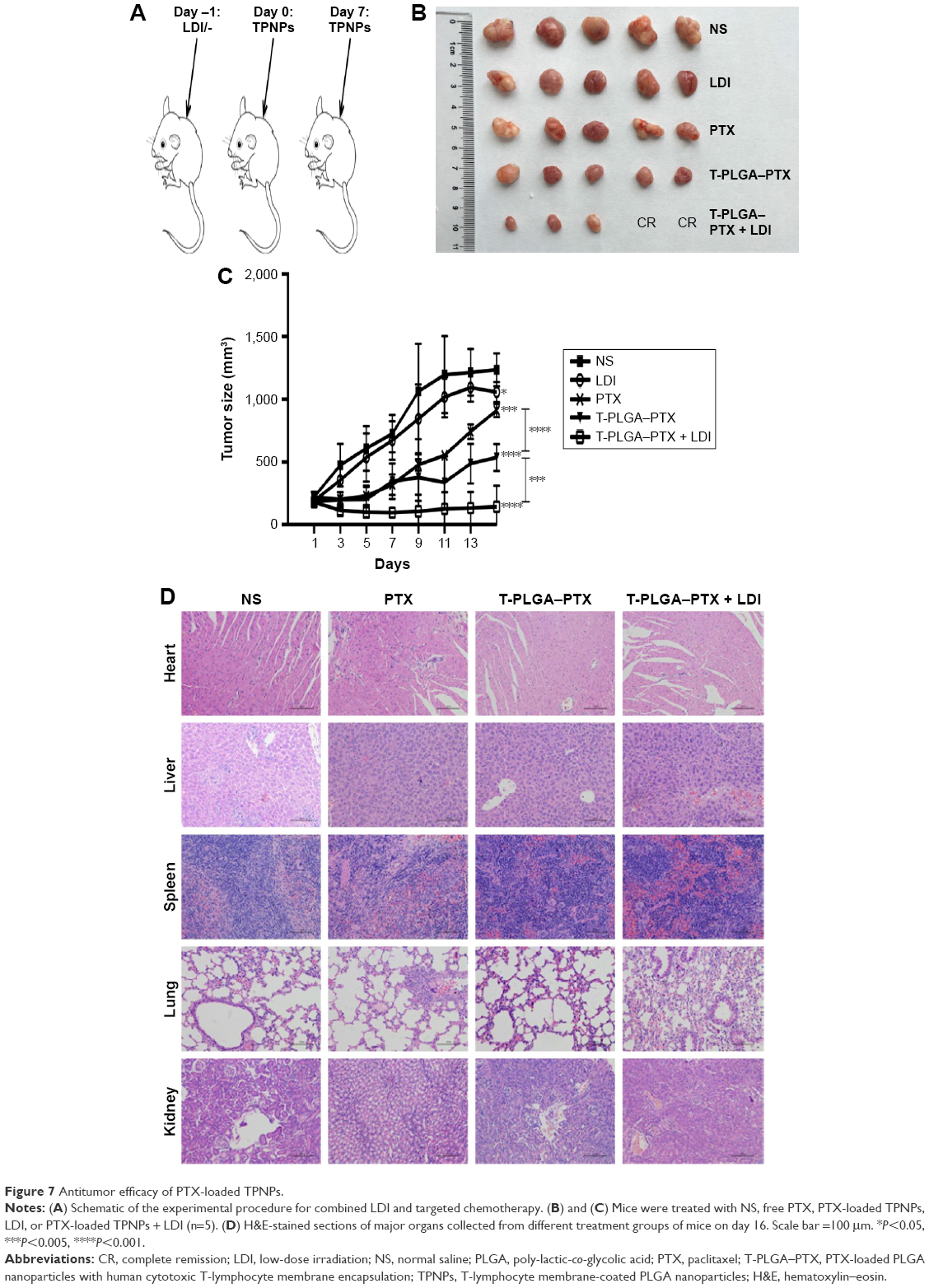 | Figure 7 Antitumor efficacy of PTX-loaded TPNPs. |
In the interaction analysis of antitumor efficacy, the average Q value was 1.4, demonstrating a synergistic effect between LDI and PTX-loaded TPNPs.
Furthermore, no noticeable sign of organ damage was observed in the H&E-stained slices (Figure 7D), which suggests a good in vivo biocompatibility of the hCTL membrane-coated nanoparticles.
Immunohistochemical analysis of mouse tumors after LDI
The expression level of ICAM-1 was relatively low without LDI and increased significantly at the tumor vessel sites following LDI.
Discussion
Leukocytes and tumor cells share similarities including their transport within blood, adhesion to vessel walls, and migration into inflammatory sites.38 In recent years, it has been shown that leukocytes are effective in carrying drugs and nanoparticles for cancer therapy,39,40 while leukocyte cell membrane-camouflaged nanoparticles preserve the surface properties of parent leukocytes and provide a platform for biomimetic delivery.22,41–43 ICAM-1 is a ligand for LFA-1, which is found on vascular endothelium, squamous cell carcinoma, fibroblasts, and in other cells.44 The interaction between LFA-1 and ICAM-1 is important in the process of leukocyte adhesion and may contribute to the localization of leukocytes in tumors. T-lymphocytes, serving as a kind of leukocyte, express the LFA-1-binding site for the ICAM-1 ligand.45 Notably, the expression level of the LFA-1 adhesion molecule is two- to fourfold higher on hCTLs than on naive T lymphocytes, which enables hCTLs to bind more effectively to target cells.28 Therefore, this study explored the role of hCTL membranes as a drug delivery vehicle. Numerous studies have also identified that local LDI induces recruitment of tumor-specific T lymphocytes to the tumor site, but focused only on the cell behavior.32,34,46 Therefore, this is the first study to use local irradiation, which is one of the most commonly used treatment modalities for gastric cancer, as a chemoattractant for a hCTL membrane-based drug delivery system.
The in vivo near-infrared (NIR) fluorescence imaging studies of DiR-labeled nanoparticles in mice revealed a gradual increase in tumor accumulation following local LDI (Figure 6B–D). After 1 h, the signal was focused at the liver sites. The signal at the tumor sites was visualized with the guidance of LDI after 24 h and then enhanced with time. However, the signal from the TPNPs accumulated mostly in the liver without LDI. In the in vitro cellular uptake study, PLGA nanoparticles were taken up by the THP-1 human phagocytic cell at a decreased level when they were camouflaged with hCTL membranes (Figure 4A). Therefore, in the mouse model, TPNPs derived from humans were expected to be phagocytosed by mouse macrophages. The DiR dye was incubated with hCTL membrane-derived vesicles, which included some other membrane fragments. From the in vivo images, it was assumed that the membrane fragments might also undergo uptake by the liver. The immunofluorescence results showed that the TPNPs accumulated more specifically than the PLGA nanoparticles at the tumor sites (Figure 6A). Therefore, although the liver signal of the TPNPs in the in vivo NIR fluorescence imaging was too strong for the tumor to be visualized, the targeting ability of TPNPs was still confirmed.
Compared with the saline-treated group, PTX alone produced only a slight antitumor effect at a dosage of 10 mg/kg because of a low concentration at the tumor site. This efficacy was improved significantly by encapsulation with hCTL membranes. When LDI was added, the tumor growth was further suppressed, and two mice in the TPNP plus LDI group achieved complete remission (Figure 7B). This improved effectiveness might enable both effective treatment for advanced gastric cancer patients and lower side effects because of a decreased dose of PTX. The efficacy of the current system exceeded that of previously reported PLGA nanoparticles containing PTX tested on mammary adenocarcinoma and melanoma.47,48 The clinical advantage of the current delivery system is even more definitive when compared with previously reported cell membrane-based nanoparticles or nanocapsules.24,43,49 The use of local LDI as a new chemoattractant and sensitizer for drug delivery is expected to be of significant utility for the clinical treatment of gastric cancer.
ICAM-1 has been reported as routinely expressed at a very low level; however, it is upregulated substantially by cytokine stimulation, enhancing the adhesion of leukocytes to endothelial cells at sites of infection or injury.50 According to the current immunohistochemistry study, LDI could upregulate ICAM-1 expression on tumor vessels (Figure 8), demonstrating that LDI could enhance the tumor targeting ability of TPNPs. The interaction analysis proved the existence of a synergistic effect between LDI and PTX-loaded TPNPs. Further studies focusing on the mechanism of LDI enhancement of the migration of TPNPs to the tumor are needed.
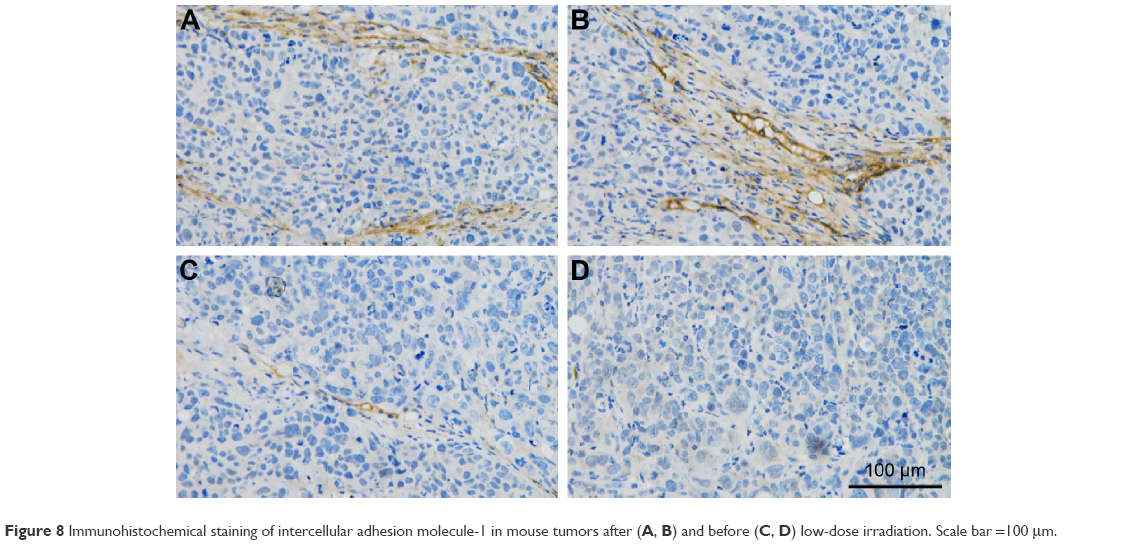 | Figure 8 Immunohistochemical staining of intercellular adhesion molecule-1 in mouse tumors after (A, B) and before (C, D) low-dose irradiation. Scale bar =100 μm. |
Stimuli-responsive drug delivery systems have attracted considerable attention during recent decades.51 External stimuli, including variations in temperature, magnetic fields,52,53 and NIR light,54,55 have been well employed in drug delivery systems in previous studies. However, compared with the LDI in this study, externally placed magnets have no antitumor efficacy themselves, while NIR light or thermotherapy are not widely applied. In addition, most NIR-resonant nanomaterials, paramagnetic materials, and thermosensitive nanomaterials are difficult to use in clinical practice. The in vitro cytotoxicity experiments and H&E-stained slices of the major organs together confirmed the safe nature of the hCTL membrane-based drug delivery vehicle. Therefore, the LDI-guided drug delivery system demonstrates significant advantages as a drug delivery system.
This study demonstrated the significant tumor targeting ability of a LDI-guided hCTL membrane-based drug delivery system. Challenges and opportunities still lie ahead in the translation of these hCTL membrane-coated nanoparticles into a viable clinical drug delivery vehicle. Whether TPNPs will work well in humans remains unclear. A determination of the short- and long-term toxicity of these nanoparticles needs more systematic work. The LDI was used only to enhance the migration of TPNPs to the tumor site in the in vivo study and, therefore, a group combining both free PTX and exposure to LDI was not investigated. The encapsulation and drug-loading efficiencies of PTX-loaded PLGA were only 51.20% and 5.07% in this study, respectively. Although PLGA is a successfully developed biodegradable polymer and the in vivo biocompatibility of the hCTL membrane-coated nanoparticles was promising, the relatively low encapsulation and drug-loading efficiencies must be improved in further studies. Nevertheless, this biomimetic drug delivery platform has significant potential for personalized medicine whereby the drug delivery nanocarrier is made from the hCTLs of individual patients with little risk of immunogenicity.
In conclusion, a new LDI-guided hCTL membrane-based drug delivery system was engineered. In this platform, hCTL membranes isolated from hCTLs were reconstructed into vesicles and then fused onto PLGA nanoparticles. Combining ex vivo experiments with in vivo experiments, it was found that TPNPs can avoid opsonization and localize at the tumor site with the helpful enhancement in targeting ability produced by LDI. This LDI-guided biomimetic drug delivery platform is expected to be further expanded to cancer immunotherapy, photothermal therapy, and diagnosis in the near future.
Acknowledgment
This work was funded by grants from the National Natural Science Foundation of China (Grant Nos 81472216, 81672398, and 81302053).
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2015;51(9):1201–1202. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Van CE, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–2664. | ||
Wagner AD, Grothe W, Behl S, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2005;(2):CD004064. | ||
Murad AM, Santiago FF, Petroianu A, Rocha PR, Rodrigues MA, Rausch M. Modified therapy with 5-fluorouracil, doxorubicin, and methotrexate in advanced gastric cancer. Cancer. 1993;72(1):37–41. | ||
Sakamoto J, Matsui T, Kodera Y. Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancer. 2009;12(2):69–78. | ||
Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst. 1990;82(15):1247–1259. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Farokhzad OC. Using ligands to target cancer cells. Clin Adv Hematol Oncol. 2012;10(10):543–544. | ||
Srivastava DK, Mallik S. Controlled release liposomes and methods of use. United States patent US20160000724; 2006 Jan. | ||
Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10(9):3223–3230. | ||
Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. | ||
Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10(7):521–535. | ||
Langer R, Tirrell DA. Designing materials for biology and medicine. Nat. 2004;428(6982):487–492. | ||
Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12(11):978–990. | ||
Wegst UG, Bai H, Saiz E, Tomsia AP, Ritchie RO. Bioinspired structural materials. Nat Mater. 2015;14(1):23–36. | ||
Trinidad AJ, Hong SJ, Peng Q, Madsen SJ, Hirschberg H. Combined concurrent photodynamic and gold nanoshell loaded macrophage-mediated photothermal therapies: an in vitro study on squamous cell head and neck carcinoma. Lasers Surg Med. 2014;46(4):310–318. | ||
Bahmani B, Bacon D, Anvari B. Erythrocyte-derived photo-theranostic agents: hybrid nano-vesicles containing indocyanine green for near infrared imaging and therapeutic applications. Sci Rep. 2013;3(6142):144–150. | ||
Baek SK, Makkouk AR, Krasieva T, Sun CH, Madsen SJ, Hirschberg H. Photothermal treatment of glioma; an in vitro study of macrophage-mediated delivery of gold nanoshells. J Neurooncol. 2011;104(2):439–448. | ||
Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proce Natl Acad Sci U S A. 2011;108(27):10980–10985. | ||
Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. | ||
Toledano Furman NE, Lupu-Haber Y, Bronshtein T, et al. Reconstructed stem cell nanoghosts: a natural tumor targeting platform. Nano Lett. 2013;13(7):3248–3255. | ||
Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. | ||
Piao JG, Wang L, Gao F, You YZ, Xiong Y, Yang L. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS Nano. 2014;8(10):10414–10425. | ||
Springer TA. Traffic signal for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–314. | ||
Baggiolini M. Chemokines and leukocyte traffic . Nature. 1998;392(6676):565–568. | ||
Punt J. Cancer immunotherapy (Sencond Edition). Anticancer Research. 2013:41–53. | ||
Coccolini F, Montori G, Ceresoli M, et al. Advanced gastric cancer: what we know and what we still have to learn. World J Gastroenterol. 2016;22(3):1139–1159. | ||
Li G, Wang J, Hu W, Zhang Z. Radiation-induced liver injury in 3-dimensional radiation therapy (3D-RT) for postoperative or locoregional recurrent gastric cancer: the risk factors and dose limitation. PLoS One. 2015;10(2):e0136288. | ||
Zhu WG, Xua DF, Pu J, et al. A randomized, controlled, multicenter study comparing intensity-modulated radiotherapy plus concurrent chemotherapy with chemotherapy alone in gastric cancer patients with D2 resection. Radiother Oncol. 2012;104(3):361–366. | ||
Draghiciu O, Walczak M, Hoogeboom BN, et al. Therapeutic immunization and local low-dose tumor irradiation, a reinforcing combination. Int J Cancer. 2014;134(4):859–872. | ||
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180(5):3132–3139. | ||
Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. | ||
Su S, Hu B, Shao J, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6:20070. | ||
Okuno S, Harada M, Yano T, et al. Complete regression of xenografted human carcinomas by camptothecin analogue-carboxymethyl dextran conjugate (T-0128). Cancer Res. 2000;60(11):2988–2995. | ||
Yang Y, Lv QJ, Du QY, Yang BH, Lin RX, Wang SQ. Combined effects of Cantide and chemotherapeutic drugs on inhibition of tumor cells’ growth in vitro and in vivo. World J Gastroenterol. 2005;11(16):2491–2496. | ||
Mitchell MJ, King MR. Leukocytes as carriers for targeted cancer drug delivery. Expert Opin Drug Deliv. 2015;12(3):375–392. | ||
Choi MR, Bardhan R, Stanton-Maxey KJ, et al. Delivery of nanoparticles to brain metastases of breast cancer using a cellular Trojan horse. Cancer Nanotechnol. 2012;3(1–6):47–54. | ||
Choi M-R, Stanton-Maxey KJ, Stanley JK, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–3765. | ||
Gao C, Wu Z, Lin Z, Lin X, He Q. Polymeric capsule-cushioned leukocyte cell membrane vesicles as a biomimetic delivery platform. Nanoscale. 2016;8(6):3548–3554. | ||
Xuan M, Shao J, Dai L, Li J, He Q. Macrophage cell membrane camouflaged au nanoshells for in vivo prolonged circulation life and enhanced cancer photothermal therapy. ACS Appl Mater Interfaces. 2016;8(15):9610–9618. | ||
Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthc Mater. 2015;4(11):1645–1652. | ||
Yamanaka R, Tanaka R, Yoshida S. Effects of irradiation on the expression of the adhesion molecules (NCAM, ICAM-1) by glioma cell lines. Neurol Med Chir (Tokyo). 1993;33(11):749–752. | ||
Dominguez GA, Anderson NR, Hammer DA. The direction of migration of T-lymphocytes under flow depends upon which adhesion receptors are engaged. Integr Biol (Camb). 2015;7(3):345–355. | ||
Lim JY, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8 + T cells. Cancer Immunol Immunother. 2014;63(3):259–271. | ||
Roy A, Singh MS, Upadhyay P, Bhaskar S. Nanoparticle mediated co-delivery of paclitaxel and a TLR-4 agonist results in tumor regression and enhanced immune response in the tumor microenvironment of a mouse model. Int J Pharm. 2013;445(1–2):171–180. | ||
Patil Y, Sadhukha T, Ma L, Panyam J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J Control Release. 2009;136(1):21–29. | ||
Fu Q, Lv P, Chen Z, et al. Programmed co-delivery of paclitaxel and doxorubicin boosted by camouflaging with erythrocyte membrane. Nanoscale. 2015;7(9):4020–4030. | ||
Bella J, Kolatkar PR, Marlor CW, Greve JM, Rossmann MG. The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc Natl Acad Sci U S A. 1998;95(8):4140–4145. | ||
Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. | ||
Shevtsov MA, Multhoff G. Recent developments of magnetic nanoparticles for theranostics of brain tumor. Curr Drug Metab. 2016;17(8):737–744. | ||
Mao H, Qian W, Wang L, et al. Functionalized milk-protein-coated magnetic nanoparticles for MRI-monitored targeted therapy of pancreatic cancer. Int J Nanomedicine. 2016;11:3087–3099. | ||
Lei Q, Qiu WX, Hu JJ, et al. Multifunctional mesoporous silica nanoparticles with thermal – responsive gatekeeper for NIR light – triggered chemo/photothermal – therapy. Small. 2016;12(31):4286–4298. | ||
Gao H, Bi Y, Chen J, et al. Near-infrared light-triggered switchable nanoparticles for targeted chemo/photothermal cancer therapy. ACS Appl Mater Interfaces. 2016;8(24):15103–15112. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.