Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 5
Hsp90 as a therapeutic target in endocrinology: current evidence
Authors Ratajczak T, Ward B, Walsh J, Cluning C
Received 6 July 2015
Accepted for publication 23 August 2015
Published 23 October 2015 Volume 2015:5 Pages 141—155
DOI https://doi.org/10.2147/RRED.S68546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Mingzhao Xing
Thomas Ratajczak,1,2 Bryan Kenneth Ward,1,2 John Peter Walsh,2,3 Carmel Cluning1,2
1Laboratory for Molecular Endocrinology, Harry Perkins Institute of Medical Research and The UWA Centre for Medical Research, The University of Western Australia, 2Department of Endocrinology and Diabetes, Sir Charles Gairdner Hospital, Hospital Avenue, 3School of Medicine and Pharmacology, The University of Western Australia, Nedlands, WA, Australia
Abstract: The ability of heat shock protein 90 (Hsp90) to modulate many growth and signaling pathways simultaneously makes it an attractive target in the field of cancer therapeutics and provided the initial impetus for significant efforts over the past decade to identify Hsp90 inhibitors, several of which are now showing promise in the clinic for cancer treatment. The four known human Hsp90 members are compartmentalized: Hsp90α and ß in the cytoplasm, GRP94 in the endoplasmic reticulum, and TRAP1 in the mitochondrial matrix. While these isoforms share a similar N-terminal domain adenosine triphosphate-binding pocket, structural variations allow unique interactions for inhibitors targeting this binding site, providing an avenue for the development of paralog-selective drugs with different biological effects applicable therapeutically to a wide range of diseases. At the same time, the conformational flexibility of the Hsp90 molecular chaperone has unveiled multiple small-molecule target sites within all subdomains of the protein, greatly expanding opportunities for viable drug development. This review summarizes the function, expression, and clinical significance of the Hsp90 isoforms and elaborates on the inhibitors and modulators that impact Hsp90 chaperone activity. Finally, the review focuses on the therapeutic utility of a range of Hsp90-modulating agents in the treatment of specific diseases associated with the endocrine system.
Keywords: heat shock protein 90, GRP94, TRAP1, Hsp90 inhibitors, endocrine disorders
Introduction
Protein folding defects associated with misfolding and aggregation can interfere with critical cellular functions of specific proteins, giving rise to numerous human diseases ranging from cystic fibrosis to cancer and neurodegeneration.1 Heat shock protein 90 (Hsp90) is a highly conserved, abundant molecular chaperone that is essential for the proper folding, turnover, and trafficking of a variety of client proteins.2 The wide array of Hsp90 substrates so far identified (http://www.picard.ch/downloads/Hsp90interactors.pdf) encompasses proteins involved in diverse signaling cascades including kinases, hormone-activated transcription factors such as steroid receptors, as well as other transcription factors and E3 ligases. Many of these Hsp90 substrates contain exposed hydrophobic surfaces that contribute to their inherent instability and require Hsp90 chaperoning to attain active conformations.3–5
Cytosolic Hsp90 functions as an adenosine triphosphatase (ATPase) and exists as a constitutive dimer, with the dimerization interface being located within the C-terminal domain, separated from a nucleotide-binding N-terminal region by a middle domain that accommodates client proteins.2 The N-terminal adenosine triphosphate (ATP)-binding site maintains an open dimerized conformation, but upon ATP binding, Hsp90 generates a closed conformation involving transient N-terminal domain dimerization favorable for ATP hydrolysis (Figure 1).2 Our current mechanistic understanding of Hsp90 chaperoning function is best derived from in vitro studies of steroid receptor complexes in which the receptor undergoes ordered, dynamic assembly with several chaperones during the reaction cycle to furnish a functionally mature receptor with a ligand-binding domain (LBD) ready for hormonal activation.6–9 Specific cochaperone components of the Hsp90 chaperone machine partner Hsp90 in this process (Figure 1). In a general model, the ATP-driven ordered assembly of steroid receptor complexes is initiated by heat shock protein 70 (Hsp70), followed by the loading of receptor from Hsp70 to Hsp90 via an intermediate complex involving Hsp70–Hsp90 organizing protein (Hop). Hop, together with additional cochaperones – FK506-binding protein 51 (FKBP51), FKBP52, protein phosphatase 5, and cyclophilin 40 – all interact through common tetratricopeptide repeat (TPR) domains with MEEVD structural motifs located at the C-terminus of the Hsp90 dimer.10,11 In the Hsp90 chaperone cycle, Hop facilitates client loading by binding to the open conformation to inhibit the ATPase. Entry of one of the TPR cochaperones into the Hsp90–client complex allows Hsp90 to convert to the closed ATP-bound conformation, resulting in the recruitment of the cochaperone, p23 to the N-terminal domain and stabilization of the mature receptor complex.7,12 Following hormone binding and ATP hydrolysis, p23, TPR cochaperones, and the activated receptor are released from Hsp90, allowing the receptor to fulfill its biological role as a ligand-activated transcription factor.
 | Figure 1 Regulation of Hsp90 conformation by nucleotide binding, its association with cochaperones and the competing modulating influences of inhibitory ligands. |
Recent in vitro studies with the glucocorticoid receptor (GR) have revealed that the functional dependence of the GR LBD on Hsp90 requires a tight coordination of chaperones within the assembled Hsp90:Hsp70:Hop:GR complex, with Hsp70 initially unfolding/inactivating GR and Hsp90 involved in GR refolding and enhancing its ligand binding affinity.13 Hop promotes this critical collaboration by delivering the GR LBD within the Hsp70:GR complex proximal to the Hsp90 client binding domain. In a similar fashion, the cell division cycle 37 (Cdc37) adaptor protein arrests the Hsp90 ATPase cycle to recruit protein kinases to the Hsp90 machinery.12,14 On the other hand, the stress-regulated cochaperone, Activator of Hsp90 ATPase 1 (Aha1), binds to the middle domain of Hsp90 and substantially increases the low ATPase rate of Hsp90.15,16
Often observed to be overexpressed in cancer cells, cytosolic Hsp90 was seen as a crucial facilitator of cancer cell growth and survival,17 acting by protecting key oncoprotein clients such as kinases and transcription factors from degradation.18,19 Its ability to modulate many growth and signaling pathways simultaneously has made Hsp90 an attractive target in cancer therapeutics. In this regard, derivatives of geldanamycin, a natural product Hsp90 inhibitor, have served as pathfinder molecules in animal models and early clinical trials by binding to the ATP-binding pocket to block the ATPase-coupled chaperone cycle resulting in the depletion of oncoproteins and subsequent cell cycle arrest and apoptosis.20,21 In addition to cytosolic Hsp90α and β isoforms, mammalian cells contain the mitochondrial and endoplasmic reticulum (ER) Hsp90 paralogs, TRAP1 (tumor necrosis factor receptor-associated protein 1), and GRP94 (glucose-regulated protein 94), respectively.22 Together, these Hsp90 homologs act on a range of client proteins and have differential control over many cellular processes including growth, cell cycling, apoptosis, cancer, stress response, endocrine function, immunity, and development. Structural differences between the Hsp90 isoforms, within their nucleotide-binding pockets and subpockets, have led to the development of paralog-selective Hsp90 inhibitors suitable for pharmacological intervention not only in diseases such as cancer and neurodegeneration, but also in endocrine disorders. In this review, we provide an overview of the functional and clinical significance of the Hsp90 paralogs and the mechanisms associated with their inhibition and focus on the application of Hsp90 inhibitors as a therapeutic toolset for the treatment of specific diseases linked to the endocrine system.
Function, expression, and clinical significance of Hsp90 isoforms
Hsp90α and Hsp90β
Hsp90α and Hsp90β, the major cytoplasmic isoforms of Hsp90, display ~86% amino acid sequence identity, with the α-form being more readily dimerized than its β counterpart. Although both are ubiquitously expressed, Hsp90α is heat-inducible in specific tissues, while Hsp90β typically has a more constitutive pattern of expression.22 These regulatory differences translate into varied responses to extracellular signals and stress events, with Hsp90α adopting a cytoprotective role by mediating a rapid response, whereas Hsp90β is associated with long-term cellular adaptation. Owing to its higher levels of expression, Hsp90β is the predominant Hsp90 isoform involved in normal cellular function, including the maintenance of cytoskeletal architecture, cellular transformation, and signal transduction. Multiple differences are apparent between the Hsp90α and β isoforms in cell differentiation and embryonic development.22 Additional evidence supports the isoform-specific participation of Hsp90α in antigen processing23 and in the negative regulation of caspase-2 activity.24 Regarding Hsp90 involvement in antigen processing and specifically in the MHC 1 (major histocompatibility complex 1) endogenous processing pathway, evidence suggests that newly synthesized, misfolded proteins targeted for degradation via the ubiquitin-dependent proteasome system are initially associated with Hsp90α (but not Hsp90β), then transferred to the E3 ligase CHIP (carboxyl terminus of Hsc70 interacting protein) for polyubiquitination.23 On the other hand, both Hsp90α and β were confirmed to play physiologically important roles in antigen cross-presentation, most likely through a mechanism of translocation of the internalized extracellular antigen within the endosome/phagosome to the cytosol for proteasomal degradation.25,26
The secretion of Hsp90α allows the protein to act as an extracellular molecular chaperone, where it promotes the maturation of MMP2 (matrix metalloproteinase 2) and tumor cell migration and invasiveness.27 Hsp90α is secreted as a C-terminal truncated form, and this secretion is tightly regulated by TPR protein interaction with the EEVD motif located at the Hsp90 extreme C-terminus and is dependent on Hsp90α phosphorylation status, which may determine a critical conformation.28 It is of interest that in rat models, both high glucose levels and diabetes mellitus have also been shown to induce Hsp90α translocation to the outside of aortic endothelial cells.29 This led to an impairment of endothelial nitric oxide synthase (eNOS) activity and reduced the bioavailability of nitric oxide, which is critical for the regulation of cardiovascular homeostasis. Since Hsp90α enhances eNOS activation and trafficking, the result implicates the extracellular secretion of Hsp90α in the pathological process.30 Within the cell, Hsp90α and Hsp90β have opposite effects on eNOS activation by differentially modulating eNOS phosphorylation by the serine/threonine kinase, Akt. Thus, although both isoforms were found to associate with the eNOS client protein, Hsp90α promoted a significant increase in eNOS-activating phosphorylation (at residue Ser1177), whereas Hsp90β produced an increase in eNOS-inactivating phosphorylation (at residue Thr495).31
Mouse oocytes almost exclusively express the Hsp90α isoform, and recent data suggest that Hsp90α activity is required for mouse oocyte meiosis mediated through a possible role in the G2/M transition.32 Hsp90α-deficient male mice, whose Hsp90β levels remain unaltered, have been shown to be sterile because of an inability to produce sperm.33 The Hsp90α isoform has been further linked to male fertility through evidence of its specific chaperoning of the androgen receptor (AR) of spermatogonia, a key factor for the initiation and maintenance of spermatogenesis and for the survival of spermatocytes in adult testis.34
Since the Hsp90β null mutation in mouse was early embryonic lethal, this isoform has specific developmental functions that cannot be fully supported by Hsp90α.35 Thus, while a redundancy may exist between the two Hsp90s in cell function involving protein folding and chaperoning, Hsp90β appears to have a specific responsibility for inducing labyrinthine trophoblast differentiation. Some of the biological differences between the Hsp90α and β isoforms are likely to be determined by client proteins and by cochaperones associated with Hsp90 chaperone machinery.12,22,36 It is intriguing therefore that the cochaperone, GCUNC45 (general cell UNC45), previously implicated in the Hsp90 chaperoning of the progesterone receptor,37 was found to bind preferentially to Hsp90β over Hsp90α, resulting in an efficient blockade of progesterone receptor chaperoning in vitro.38
The recent surprising observation that Hsp90β interacts within the extracellular medium with MMP3 (matrix metalloproteinase 3), an important regulator of branching morphogenesis in the mammary gland, adds a further dimension to the functions of this Hsp90 isoform.39 Expression of constitutively active MMP3 in mouse mammary epithelia eventually triggers tumor development.40 The extracellular presence of Hsp90β was shown to be selective and strongly correlated with the modulation of MMP3 functions essential for mammary epithelial invasion and morphogenesis. These findings identify Hsp90β as a crucial player in signaling pathways associated with MMP3-mediated invasion and branching.
Initial evidence suggested the involvement of Hsp90β in human ovarian autoimmunity, identifying the isoform as a significant causative factor in early ovarian failure.41 This was supported by results from a study of a female mouse model in which fertilization and embryo development were highly disrupted in animals immunized with in vitro-generated Hsp90 antibodies.42
TRAP1
Originally cloned from two independent yeast two-hybrid screens, the 704 amino acid human TRAP1 sequence bears an overall 50% similarity to Hsp90β and includes an N-terminal mitochondrion-targeting sequence, as well as a more highly conserved N-terminal ATP-binding domain that is ~60% similar to Hsp90. Unlike Hsp90, TRAP1 does not possess the charged linker region connecting the N-terminal and middle domains. This results in a more poorly conserved C-terminal region (~35% similar to Hsp90) that also lacks the MEEVD sequence found at the C-terminus of Hsp90.43 TRAP1 functions as a tight homodimer, binds ATP, and undergoes a two-step ATP hydrolysis cycle in which its ATPase activity was shown to be sensitive to the Hsp90 inhibitor, geldanamycin.43–45
Neither p23 nor Hop, cochaperones which are critical for the chaperoning functions of cytosolic Hsp90, form stable complexes with TRAP1 and TRAP1-associated cochaperones, as well as its client proteins, still remain to be identified.43,46 Within mitochondria, TRAP1 resides in the matrix and specifically in the inner membrane.47 Accumulating evidence supports a role for the mitochondrion-located Hsp90 analog in critical pathways that determine mitochondrial integrity, protect against reactive oxygen species-mediated apoptosis arising from oxidative stress, and safe-guard the mitochondrial protein-folding environment.46,48,49 TRAP1-mediated cytoprotective effects may require phosphorylation of TRAP1 by the mitochondrial-localized kinase, PTEN-induced putative kinase (PINK1),50 in addition to the stabilizing influences of Sorcin, a Ca2+-dependent regulator that binds TRAP1 and prevents its proteolytic degradation in the mitochondrial organelle.51
Deregulation of TRAP1 function has been noted in neurodegenerative diseases, in particular Parkinson’s disease, a disorder in which mitochondrial dysfunction, oxidative stress, and impaired clearance of misfolded proteins and damaged organelles play key roles.52,53 The PINK1 gene has been linked to early onset Parkinson’s disease, with most pathogenic PINK1 mutations impairing kinase activity.53 Together with the E3 ubiquitin ligase, Parkin, PINK1 acts as part of a quality control system to preserve mitochondrial integrity through the removal of damaged mitochondria.53 Further insight into the pathophysiology of TRAP1 stems from the recent identification of TRAP1 recessive mutations in congenital anomalies of the kidney and urinary tract.54 One such mutation (R469H), within the TRAP1 middle domain, might disrupt TRAP1 mitochondrial function, resulting in perturbation of normal kidney development.
In nonneoplastic settings, TRAP1 transcripts were shown to be expressed at different levels in a range of tissues,46 although a detailed analysis of TRAP1 tissue expression patterns has yet to be undertaken. The functions of TRAP1 have also been broadly linked to the development of cancer.46,48,49 In certain neoplasms such as prostate cancer, TRAP1 was differentially expressed, being elevated in the mitochondria of tumor cells and having very low expression levels in mitochondria of cells from matched normal tissue.49 These observations suggested that increased tumor expression of TRAP1 formed part of an adaptive response that protected cancer cells, favoring tumor progression and led to the implication of TRAP1 as an important cancer target.49
GRP94
GRP94 is the counterpart of cytoplasmic Hsp90 in the ER, the primary organelle within the cell for the synthesis of secreted and transmembrane proteins and for their folding to tertiary structures critical for their function (reviewed by Eletto et al,55 Marzec et al,56 and Lee57). GRP94 shares ~50% amino acid homology with Hsp90 and has a similar organizational structure that comprises an N-terminal domain containing a homologous nucleotide-binding pocket, a charged linker and a middle domain that cooperatively mediate conformational changes critical for GRP94 ATPase function, and a C-terminal domain important for constitutive dimerization. Notable differences from Hsp90 include the N-terminal domain peptide-binding site with a role in GRP94 immunological activity and the presence of Ca2+-binding sites within the charged linker that impacts GRP94 N-terminal domain function and substitution of the MEEVD sequence at the C-terminus by the KDEL ER retention signal.55,56 As for Hsp90, ATP hydrolysis is essential for the cellular activity of GRP94 which cycles through a series of nucleotide-driven conformations from the unliganded to the ATP-bound state.58 Evidence that ATP-bound GRP94 enters a lower energy ground state prior to transient dimerization of the N-terminal domain suggests mechanistic differences for ATPase activity between the GRP94 and Hsp90 chaperones.59
Physiological ER stress caused by environmental stressors such as glucose deprivation, disruption of intracellular Ca2+ stores, and inhibition of protein glycosylation leads to unfolded protein accumulation in the ER lumen.60 This initiates the unfolded protein response or UPR, which reduces the burden of newly synthesized protein to be folded by the ER, induces the transcription of ER-resident molecular chaperones such as GRP94, and removes misfolded proteins in the ER through proteasomal degradation.61 The most important biological activity of GRP94 is its role as a master molecular chaperone, directing the folding and/or assembly of secreted or membrane proteins. This essential role is dependent on GRP94 ATPase activity in vivo62 and results in the folding of a select group of client proteins – the insulin-like growth factors (IGFs) I–III, immunoglobulins, some integrins, and TLRs55 and lipoprotein receptor-related protein (LRP) 6.63 GRP94 protects cells against ER stress (which is induced in response to growth factor withdrawal on serum deprivation) through its chaperoning of clients such as the IGFs, the maturation, secretion, and activation of which is strongly linked to GRP94 antiapoptotic activity.64 Although GRP94 has no known cochaperones, its association with osteosarcoma amplified 9 (OS-9), a component of ER-associated degradation, provides an intriguing link between the protein-folding molecular chaperone role of GRP94 and the proteasomal targeting of misfolded proteins.65–67
GRP94 is an abundant ER protein whose expression is increased as a result of transcriptional induction related to ER stress and the UPR,61 with especially high levels evident in secretory tissues.55 GRP94 differs from the Hsp90α and β isoforms in that its expression is not induced by the heat shock response, but rather by signal transduction pathways activated by alterations in the physiological environment within the ER lumen.56
As an important component of the ER chaperone system, GRP94 has an essential antiapoptotic role that confers survival advantage to cells subjected to ER and chemotoxic stress. These prosurvival properties implicate the function of this molecular chaperone in cancer progression, partly owing to GRP94-controlled secretion of IGFs, mitogenic factors that bind to the IGF1 receptor leading to Akt activation.57 Homozygous knockout of this major ER chaperone results in embryonic lethality.68 The pathogenesis of human multiple myeloma, a plasma cell neoplasm characterized by the proliferation of malignant plasma cells within the bone marrow, is closely linked to a dysregulated UPR. The development and progression of myeloma was shown to be critically dependent on GRP94, which was recently implicated in controlling LRP6 cell surface expression.63 LRP6, in turn, is required for canonical Wnt signaling and functions as a coreceptor for the cell surface Wnt receptor, Frizzled.69 Genetic deletion of GRP94 attenuates multiple myeloma in a mouse model of the disease.70 Additionally, silencing GRP94 compromised human multiple myeloma cell growth.70 In the absence of GRP94, LRP6 is prevented from reaching the cell surface, thus inhibiting the proproliferative and prosurvival Wnt–β-catenin signaling pathway.57,70 A similar mechanism has been proposed for the attenuation of inflammatory colorectal cancer. High levels of GRP94 are linked to the control of macrophage-specific Toll-like receptors (TLRs), leading to the production of proinflammatory cytokines and promotion of inflammatory-associated cancer in the colon.57,71 The mechanism that drives this oncogenic program involves upregulation of the Wnt pathway. Taken together, these findings suggest that Wnt is regulated by UPR and identify GRP94 as a novel therapeutic target for both multiple myeloma and inflammatory colorectal cancer.
Inhibitors, modulators, and enhancers of Hsp90 ATPase activity
Functional analysis has revealed a dynamic interrelationship between the N-terminal, middle, and C-terminal domains of Hsp90 that is coordinated by nucleotide, cochaperone, and specific client protein interactions, which together determine Hsp90 conformation and therefore the activation state of the chaperone. Initial evidence suggested that, in tumor cells, Hsp90 exists primarily in a high affinity binding state for ATP/adenosine diphosphate (ADP) or nucleotide-blocking ligands in equilibrium with a low affinity conformation predominating in normal tissue.72,73 Chronic stress imposed on the cell in response to an altered environment, cancer, or disease may increase association between the Hsp90 chaperone and specifically altered client proteins, resulting in complexes prone to selective targeting by small molecules.74 Posttranslational modification of Hsp90 as well as its cochaperones (eg, through acetylation, phosphorylation) may give rise to distinct conformations that influence the chaperoning of protein substrates and determine outcomes for client proteins, including folding, activation, or degradation, and govern Hsp90 interaction with specific inhibitors.74 Interference with Hsp90 conformational changes during the dynamic ATPase cycle may inhibit Hsp90 function. There is now an emerging understanding that local conformational preferences for individual Hsp90 inhibitory ligands may convert to preferences at the wider tertiary level (eg, either for the open Hsp90 conformation or the client-bound Hsp90 closed conformation). Additionally, the effects of posttranslational modifications and varying cochaperones and adaptors may present a range of target complexes.74 Thus, although certain Hsp90 inhibitory small molecules have access to the same binding site, differing modes of interaction might allow them to select for different cellular Hsp90 complexes, resulting in wide-ranging biological effects.18,75
N-terminal ATP pocket binders
Geldanamycin, its ansamycin derivatives (17AAG, 17DMAG, IPI-504), and several of the newer synthetic inhibitors target the nucleotide-binding pocket in the N-terminal domain. Included among these are numerous inhibitors derived from various chemical scaffolds: resorcinol – radicicol, AUY922, AT13387, STA-9090/Ganetespib, KW-2478; purine class – PU3, PU-H71, MPC-3100; CUDC-305/Debio 0932, BIIB021; pyrazole – CCT018159; pyrimidine – HSP990.20,21,74,76–78
C-terminal binders
A second potential drug inhibitory site has been identified in the Hsp90 C-terminal domain that is targeted by coumarin-based antibiotics such as novobiocin79 (reviewed by Chiosis et al,80 and Chiosis81), which binds at the interface between two monomers.82 Improvements in the affinity of these compounds for Hsp90 have allowed them to induce apoptosis in cancer cells, and in some cases, demonstrate superior efficacy compared to 17AAG.83,84 Evidence suggests that coumarin antibiotic-based inhibitors interfere with dimerization of the Hsp90 C-terminal domain, signifying a novel mode of action through which they antagonize Hsp90 function by destabilizing the Hsp90 dimer.85 Novobiocin blocks the binding of TPR immunophilins as well as Cdc37 to Hsp9085 and therefore acts locally and allosterically to induce global conformational changes within Hsp90. Cisplatin, an antineoplastic reagent that arrests cell division resulting in apoptosis and inhibition of tumor growth,86,87 also binds within the Hsp90 C-terminal region and disrupts Hsp90 chaperone activity.88 In further studies, cisplatin inhibited the hormone-binding and transcriptional activities of both AR and GR and caused the proteasomal depletion of GR.89 Cisplatin binds to a different site to novobiocin in the Hsp90 C-terminal domain and most likely interacts covalently with a reactive cysteine (Cys596 in human Hsp90) important for Hsp90 function.89,90 Celastrol binds to the C-terminal domain and inhibits Hsp90 ATPase activity, but not ATP binding. This natural triterpenoid,91 which acts in a distinct manner to either geldanamycin or novobiocin, may represent a new class of inhibitors with the ability to capture a unique Hsp90 conformation, enabling it to allosterically regulate Hsp90 chaperone activity and disrupt Hsp90–Cdc37 complex formation.82,92 Recent evidence suggests that celastrol and the Hsp90 modulators, gedunin (also a natural product93) and gamendazole, a structurally dissimilar small molecule based on the indazole scaffold,94 share a common mechanism of action.82,95
N-terminal/middle domain binders
An analog of the natural product sansalvamide A, SM145, binds between the N-terminal and middle domains and allosterically inhibits the binding of Hsp90 to all TPR containing cochaperones tested, including FKBP51, FKBP52, CyP40, and Hop.96,97 SM145 likely impacts access to the Hsp90 MEEVD C-terminal peptide by inducing or stabilizing an Hsp90 conformation that obstructs access to the TPR-docking site. SM145 was shown to induce caspase 3-dependent apoptosis,96 interfered with the chaperoning ability of Hsp90 to refold proteins, and caused a dramatic depletion of GR in HeLa cells.97
Histone deacetylase inhibitors
Posttranslational phosphorylation of specific serine, threonine, and tyrosine residues in Hsp90 may affect its chaperone activity and sensitivity to Hsp90 inhibitors through influences on ATP binding and by impacting association with various cochaperones as well as formation of distinct client protein complexes98–101 (reviewed by Mollapour and Neckers102). Additionally, reversible acetylation has been implicated as a functionally important posttranslational modification of Hsp90, identifying the chaperone as a downstream target of histone deacetylase inhibitors, resulting in acetylation of Hsp90, inhibition of its ATP binding, and depletion of several Hsp90 client proteins103,104 (reviewed by Mollapour and Neckers102). Hsp90 was shown to be a substrate of histone deacetylase 6 (HDAC6) which modulates Hsp90 chaperone activity by regulating its level of acetylation.105 Silencing or pharmacological inhibition of HDAC6 caused Hsp90 hyperacetylation, its dissociation from p23, a cochaperone essential for the maturation of Hsp90-client protein complexes, and loss of its chaperone activity. The loss or inactivation of HDAC6 compromised the Hsp90-dependent maturation of both GR and the aryl hydrocarbon receptor (AhR), making them functionally defective in ligand binding and transcriptional activity.105,106
Peptidomimetics targeting Hsp90 client protein and cochaperone interactions
The targeting of Hsp90–client protein interaction as a strategy for disrupting Hsp90 function has been achieved for Hsp90–survivin complexes using the peptidomimetic, shepherdin, modeled on the minimal survivin peptide (Lys79–Leu87) required for Hsp90 binding.107 Survivin, an important regulator of cell proliferation and differentiation, has essential roles in mitotic control and apoptosis inhibition; its high expression levels in cancer cells versus normal tissues correlating with unfavorable outcomes.108 Hsp90 is crucial for survivin stability in vivo,109 with the binding interface for the client protein mapping to the N-terminal ATP-binding domain. Shepherdin was shown to make extensive contacts with the Hsp90 ATP pocket and cell-permeable variants abrogated Hsp90 function leading to the loss of multiple Hsp90 client proteins, including survivin and Akt. The reagent potently induced death of tumor cells and inhibited human tumor growth in mouse models.107 In a variation of this theme, Horibe et al110 have generated the cell-permeable peptidomimetic, Antp–TPR hybrid peptide, based on a 12-amino acid sequence from the Hsp90-binding TPR2A domain of Hop (301KAYARIGNSYFK312). The Antp–TPR hybrid peptide inhibited Hsp90 interaction with the Hop TPR2A domain, inducing death in a range of cancer cell lines. Notably, the Hsp90-targeting peptide displayed significant antitumor activity in a xenograft model of human pancreatic cancer in mice.110
Small molecule enhancers of Hsp90 ATPase activity
As already explained, several Hsp90 inhibitors have been isolated from natural products and others chemically synthesized as small molecules based on different chemotype scaffolds. A virtual screening approach has identified 4-hydroxytamoxifen (4-OHT), the active metabolite of the antiestrogenic compound, tamoxifen, as a putative Hsp90 ligand with ATPase-enhancing (2–4-fold) properties.111 4-OHT was shown to dock well to the Hsp90 ATP pocket; 4-OHT bound Hsp90 more strongly than ATP, but less avidly than ADP. A more recent Förster resonance energy transfer-based screen112 has identified novel modulators that can accelerate the Hsp90 conformation cycle in a mode similar to that of the Aha1 cochaperone, which activates the Hsp90 ATPase.113 While increasing the affinity of Hsp90 toward GR client protein in vivo, acceleration of the Hsp90 chaperone cycle by the modulatory compounds interfered with GR processing, potentially offering a strategy for therapeutic intervention.112
Hsp90 paralog-selective ligands
While the N-terminal Hsp90 inhibitors share a similar binding site and bind with comparable affinity to the four known Hsp90 paralogs, Hsp90α, Hsp90β, GRP94, and TRAP1, slight structural differences and conformational flexibility within the adenine-binding cavity has revealed adjacent subpockets that allow marginally distinct interaction modes with the major clinically relevant ligands, enabling the development of paralog-selective inhibitors114 (reviewed by Taldone et al74). Since each Hsp90 paralog governs the chaperoning of a unique set of client proteins, selective ligands may result in higher efficacy and therapeutic control and pave the way for better understanding the biological role of these paralogs, particularly in specific disease contexts. Thus, an 8-residue amino acid sequence present within the N-terminal binding pocket of Hsp90α and β, which differs in the first two amino acids of the corresponding sequence in both GRP94 and TRAP1, contributed to the stability of chemotype-induced conformations of the Hsp90α and β isoforms, resulting in a higher level of Hsp90α/β selectivity compared with GRP94 and TRAP1 (>1,000-fold).74,115,116 Similarly, insertion of several purine-scaffold class chemical compounds into a new allosteric pocket, arising from the conformational flexibility of GRP94, led to the development of the purine-based ligands, PU-H54 and PU-WS13, that were >100-fold more selective for GRP94 over Hsp90α/β and TRAP1.74,117 Another GRP94-specific ligand, NECA (5′-N-ethylcarboxamidoadenosine), is an adenine-based nucleotide analog that does not bind Hsp90.74,118 GRP94 accommodates NECA at two sites – the adenine moiety fits into the adenine cavity, while the 5′-N-ethylcarboxamido moiety occupies a novel and adjacent second pocket. This secondary site cannot be accessed in Hsp90.118 Altieri et al49 tethered geldanamycin to a mitochondria-targeting sequence to generate the reagent, gamitrinib, which, on transfer into mitochondria, engages with the resident Hsp90 paralog, TRAP1.48
Potential clinical applications of Hsp90 inhibitors in endocrine and other disorders
Most organs and physiological systems in higher organisms are sensitive to glucocorticoids, and these hormones are strongly linked to pathophysiology in humans with glucocorticoid-regulated components being evident in common behavioral disorders, such as anxiety and depression, obesity and the metabolic syndrome, atherosclerosis and osteoporosis, as well as autoimmune, inflammatory, and allergic disorders.119–121 The numerous effects of glucocorticoids are mediated by GR through its interaction with multiple cell signaling systems, and the pathogenesis of several of the aforementioned disorders includes target tissue hypersensitivity or resistance to these hormones. As noted, GR is a well characterized Hsp90 client protein that requires interaction of its unliganded LBD with Hsp90 and associated cochaperones to attain hormone-binding competency and subsequent hormone-activated transcriptional activity.6,7,9,10 Early pharmacological evidence using the Hsp90 inhibitor, geldanamycin, confirmed an essential role for Hsp90 function in both GR hormone-binding activity and receptor protein stability in vivo.122 Additional studies confirmed ERα and AR as targets for ansamycin type Hsp90 inhibitors; the destabilization and dramatic depletion of these receptors in breast and prostate cancer cells, respectively, strongly indicated a therapeutic utility for these agents in human cancers, generally.123–126
Both ERα and AR, in addition to the receptor tyrosine kinase HER2 (human epidermal growth factor receptor 2), which is amplified or overexpressed in up to 30% of breast cancers,127,128 are directly associated with the well-documented hallmarks of cancer,129 and all are clinically validated cancer targets.130 Given the association of ERα with Hsp90 and the fact that HER2 is one of the most Hsp90-dependent client proteins known, breast cancer is a prime target for Hsp90 inhibitors.125 Targeting Hsp90 in prostate cancer is a particularly attractive therapeutic strategy since AR, the key driver of prostate cancer progression, is a sensitive Hsp90 client protein.131 A major advantage of Hsp90 inhibitors is that they are able to simultaneously shut down several pathways necessary for cancer development, reducing the likelihood of drug-resistant cancers. We will now expand on the therapeutic applications of various Hsp90 inhibitors in the treatment of specific endocrine and other diseases.
Metabolic syndrome
An array of closely associated metabolic disorders, including abdominal obesity, hyperglycemia, dyslipidemia, hypertension, and insulin-resistance, constitute the metabolic syndrome. The underlying cause of hyperglycemia and associated fatty liver phenotype is glucocorticoid excess and the resistance to insulin action.119–121 Metabolic disruptions underlying these conditions arise from altered glucocorticoid activity in target organs, in particular, liver, adipose tissue, skeletal muscle, and pancreatic β-cell. In addition, adipocytes from omental (but not subcutaneous) fat express 11β-hydroxysteroid dehydrogenase type 1, converting cortisone to cortisol. Therefore, in central obesity, there is increased cortisol production as well as altered action/sensitivity.132 Furthermore, in addition to GR, the mineralocorticoid receptor (MR) has been shown to play a pivotal role in adipocyte biology and helps mediate both aldosterone and glucocorticoid effects on adipose tissue development.133 Peroxisome proliferator-activated receptor γ (PPARγ), also a member of the nuclear hormone receptor superfamily, is indispensable for adipocyte differentiation and adipose tissue function (reviewed by Lehrke and Lazar,134 Tontonoz and Spiegelman,135 and Christodoulides and Vidal-Puig136). All three nuclear receptors, GR, MR, and PPARγ, are client proteins of Hsp90. Biochemical and animal studies with geldanamycin analogs and novobiocin have revealed that these Hsp90 inhibitors simultaneously destabilize Hsp90 complexes with these receptors, curtailing their adipogenic transcriptional programs and causing a blockade of adipogenesis.137,138 The results identify Hsp90 as a potential therapeutic target in the control of obesity and its related metabolic complications.
Cushing’s syndrome
Cushing’s disease (pituitary-dependent Cushing’s syndrome) is a neuroendocrine condition caused by an adrenocorticotropic hormone (ACTH)-secreting adenoma, which leads to a hypercortisolemic state.139 The principal biochemical features are a resetting of hypothalamic–pituitary–adrenal feedback such that ACTH is secreted in the face of abnormally high levels of circulating cortisol. With the prolonged and inappropriate exposure to excessive glucocorticoids, patients may develop multiple metabolic problems including central obesity, hyperglycemia, hypertension, proximal myopathy, and osteoporosis.140 Although usually treated surgically, the syndrome is frequently a management challenge, especially in the case of recurrent disease, signifying unmet needs in the treatment of afflicted patients.141 In this regard, retinoic acid displayed specific inhibitory effects on ACTH biosynthesis in experimental Cushing’s syndrome.142 Recent studies have revealed the overexpression of Hsp90α and β isoforms in glucocorticoid-resistant corticotroph adenomas in comparison to normal pituitary.143 Pharmacological inhibition of Hsp90 with the N-terminal Hsp90 inhibitor, 17AAG, a derivative of geldanamycin and the C-terminal inhibitory reagents, novobiocin and silibinin, an analog of the natural product, silymarin,144 showed vastly different outcomes for GR activity when used at low concentrations.143 Thus, 17AAG promoted GR degradation in a corticotroph cell line, whereas novobiocin and silibinin, which act at a different stage of the Hsp90 chaperoning cycle, caused a release of mature, correctly folded GR from Hsp90 that was capable of hormone binding and transcriptional activity. In a mouse allograft model of Cushing’s disease, silibinin was shown to lower ACTH and corticosterone levels, thus partially overcoming glucocorticoid resistance and relieving the symptoms of Cushing’s disease.143 These findings reveal a new pharmacological mechanism of action for a select class of Hsp90 inhibitors that might be used to restore glucocorticoid sensitivity in ACTH-secreting adenomas in individuals with Cushing’s syndrome.
Multiple endocrine neoplasia type 2, sporadic medullary thyroid carcinoma – RET tyrosine kinase mutations
RET belongs to a subclass of receptor tyrosine kinases that binds growth factors of the glial-derived neurotropic factor family.145,146 Deregulated receptor tyrosine kinases can become potent oncoproteins. Thyroid cancer is the most prevalent endocrine malignancy and is frequently associated with oncogenic conversion of RET kinase.147 In medullary thyroid carcinomas (MTC), a malignancy arising from calcitonin-secreting C-cells of the thyroid, somatic mutations in RET are present in up to 50% of cases, whereas germline point mutations in RET result in inherited cancer syndromes, which include MTC multiple endocrine neoplasia type 2A (MEN2A), type 2B (MEN2B), and familial MTC. Papillary thyroid carcinomas (PTC), which arise from the follicular epithelial cells of the thyroid and account for 80%–90% of thyroid carcinomas, can result from recombination of the intracellular kinase-encoding RET domain with heterologous genes, leading to generation of RET/PTC fusion oncogenes.146 Small molecule tyrosine kinase inhibitors may be effective in treatment strategies for some RET-dependent cancers.147,148 For example, vandetanib, a tyrosine kinase inhibitor that selectively targets RET-, VEGFR-, and EGFR-dependent signaling, as well as other successful agents targeting VEGFR,149 may provide viable therapeutic options for patients with advanced MTC, a rare disease with no effective treatment.150 Similarly, the small molecule tyrosine kinase inhibitors lenvatinib and sorafenib improve progression-free survival in radioiodine-refractory differentiated thyroid cancer (encompassing papillary and follicular cancer).151–153 A previous report identified the RET/PTC1 protein as an Hsp90 client,154 and a more recent study has confirmed that RET, as well as MEN2-associated RET mutants are stabilized by Hsp90, require the chaperone for their function, and are depleted from human MTC-derived cell lines by the blockade of Hsp90 with 17AAG.155
Multiple myeloma
Multiple myeloma is a common hematological cancer characterized by the proliferation of malignant plasma cells within the bone marrow. Local secretion of cytokines (eg, interleukin-6 and receptor activator of NF-ºB ligand) from myeloma cells accumulating on bone surfaces stimulates osteoclast growth and activity, resulting in bone loss that requires the use of bisphosphonates to prevent and treat complications arising from bone damage and the risk of fractures.156 The canonical Wnt signaling pathway has an emerging role in multiple myeloma, in which the constitutively active β-catenin is relevant to tumor cell growth, survival, and migration.157,158 To exploit this tumor dependence on Wnt signaling, small molecule compounds and interfering RNA targeting β-catenin have shown promise as therapeutic approaches in the treatment of the disease.157,158 As already noted, GRP94 deficiency in human multiple myeloma cells resulted in apoptosis through inhibition of the Wnt–survivin pathway.70 Although the pan-Hsp90 inhibitors showed potential as therapeutic agents for multiple myeloma, the role of individual Hsp90 isoforms and family members in these treatments was not determined.159–161 The GRP94-specific inhibitor, PU-WS13, was shown to induce apoptosis and block the growth of multiple myeloma cells, yet did not induce death in pre-B leukemic cells, strongly indicating that GRP94 is a valid therapeutic target in myeloma.70
Autosomal dominant polycystic kidney disease
Autosomal dominant polycystic kidney disease (ADPKD) is the most common of the inherited renal cystic diseases, a group of disorders characterized by the development of renal cysts and a variety of extrarenal manifestations that may include hepatic and pancreatic cysts, cerebral aneurysm, and cardiac valve disease.162 The disease is a progressive genetic syndrome with an incidence of 1:700, with symptoms typically manifesting at middle age, with kidney function increasingly impaired by abnormally proliferating cysts. Many patients will progress to end-stage renal disease requiring dialysis. ADPKD arises from inactivating mutations in the PKD1 or PKD2 genes, disrupting numerous signaling pathways that regulate kidney homeostasis: PKD1, which encodes polycystin-1 (PC1, ~460 kDa), and PKD2, which encodes polycystin-2 (PC2, ~110 kDa), constitute a subfamily of transient receptor potential channels. Several putative pathways which are either up- or downregulated in polycystic kidney disease provide a possible rationale for treatment with a range of therapeutic agents, including vasopressin 2 receptor antagonists, somatostatin, triptolide, and tyrosine kinase inhibitors.162 Numerous parallels may be drawn between ADPKD and cancer with regard to signaling mechanisms controlling growth, apoptosis, and differentiation.163 Many signaling proteins hyperactivated in ADPKD (eg, HER2, Akt, Src, etc) are clients of Hsp90,4,162 suggesting that Hsp90 inhibitors might improve kidney function by simultaneously disrupting the activity of multiple proteins supporting progressive cyst growth. Use of STA-2842, a novel next-generation, highly specific Hsp90 inhibitor that is structurally similar to STA-9090/ganetespib, induced the degradation of several ADPKD-relevant Hsp90 client proteins in PKD1-deficient kidney cells and reduced the formation of renal cysts and kidney growth in a mouse model of the disease.164 These findings thus provide proof-of-concept for the use of Hsp90 inhibitors in treating ADPKD.
Male contraception
In studies of Hsp90α-deficient male mice, Grad et al33 demonstrated testicular atrophy and infertility caused by apoptosis of spermatocytes, indicating that the Hsp90α isoform is required for spermatogenesis. Subsequent conditional deletion of Hsp90α, in adult mice, showed that the chaperone is essential for germ cell development in mature adult testes.34 Extended exposure of pubertal male mice to high doses of the Hsp90α/β N-terminal domain inhibitor, pochoxime A, a new radicicol derivative with efficacy favorable for entry into brain and testes,28,165,166 caused testicular atrophy in some of the animals similar to the phenotype seen in mice with genetic disruption of Hsp90α.33 These results suggest that such drugs could be used to generate pharmacological infertility in male mice. In this context, it is of considerable interest that gamendazole, a new class of Hsp90 inhibitor selective for Hsp90β, which binds to the Hsp90 C-terminal region in a manner similar to celastrol and gedunin,82 causes infertility by blocking spermatogenesis in rats.94,167 Gamendazole is therefore a promising candidate for the development of a novel male contraceptive.
Conclusion
There are at least 13 Hsp90 inhibitors currently in clinical development for a broad range of tumors including breast, prostate, gastrointestinal, melanoma, and hematological malignancies, and in vitro studies continue to identify new and novel small molecule compounds that selectively target the Hsp90 molecular chaperone in its various paralog forms.74,168 Simultaneously, our increased understanding of Hsp90 biology and mechanisms of action when bound to different Hsp90 disruptors provide an expanding list of disease states that might benefit therapeutically through Hsp90 modulation. In addition to the disorders already detailed in this review, potential diseases in which therapeutic treatment with Hsp90 inhibitors is indicated are summarized in Table 1. Thus gamitrinib, a mitochondrial-directed small-molecule inhibitor selective for TRAP1 and mitochondrial Hsp90, was shown to suppress localized and metastatic prostate cancer growth in a genetic mouse model of the disease.49 In addition to their application to HER2-positive breast cancer,117,169 Hsp90 inhibitors also show potential in the treatment of triple-negative breast cancer (ie, tumors clinically defined by the negative expression of ERα, progesterone receptor, and HER2)170,171 and aromatase inhibitor-resistant subtypes.172 Recent studies have found an association between GR regulation and the response to traumatic stress.173,174 The HDAC6 inhibitor, ACY-738, was shown to increase Hsp90 acetylation in mice, promoting increased incorporation of the FKBP51 cochaperone into Hsp90–GR complexes and causing a suppression of GR function. This led to an enhanced adaptation to stress.174 Thus, through its modulating influences on Hsp90, HDAC6 has been confirmed as a viable therapeutic target for psychosocial stress and depression. The continuing evolution of Hsp90 inhibitory drugs and their relevance to specific diseases hold great promise of exciting developments in the future.
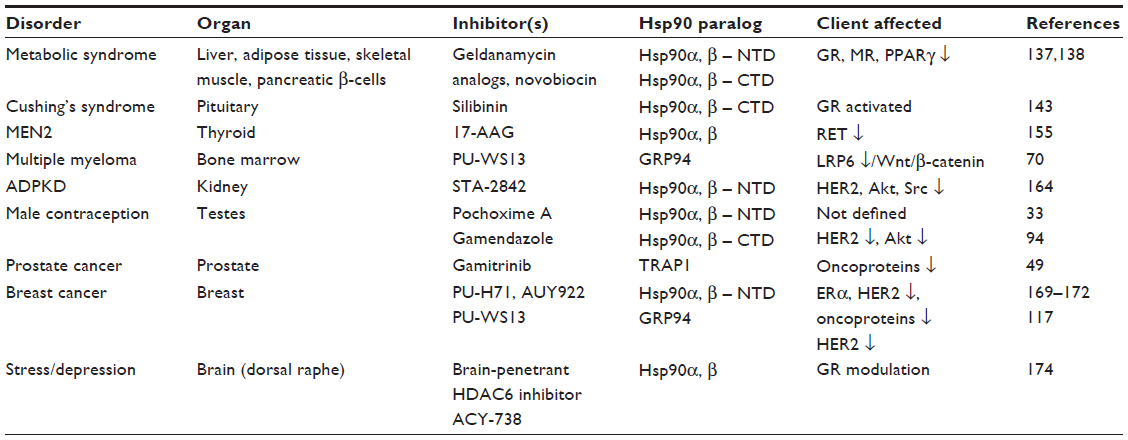 | Table 1 Regulation of endocrine and other disorders by Hsp90 modulators |
Acknowledgments
We acknowledge research support from the National Health and Medical Research Council of Australia, the National Breast Cancer Foundation, the Cancer Council of Western Australia, the Prostate Cancer Foundation of Australia, and the Sir Charles Gairdner Hospital Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. | |
Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. | |
Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One. 2011;6(10):e26044. | |
Taipale M, Jarosz DF, Lindquist S. Hsp90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. | |
Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987–1001. | |
Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18(3):306–360. | |
Smith DF. Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones. 2004;9(2):109–121. | |
Picard D. Chaperoning steroid hormone action. Trends Endocrinol Metab. 2006;17(6):229–236. | |
Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood). 2003;228(2):111–133. | |
Ratajczak T, Ward BK, Minchin RF. Immunophilin chaperones in steroid receptor signalling. Curr Top Med Chem. 2003;3(12):1348–1357. | |
Smith DF, Toft DO. Minireview. The intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22(10):2229–2240. | |
Röhl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci. 2013;38(5):253–262. | |
Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157(7):1685–1697. | |
Roe SM, Ali MM, Meyer P, et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(Cdc37). Cell. 2004;116(1):87–98. | |
Meyer P, Prodromou C, Hu B, et al. Structural and functional analysis of the middle segment of Hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell. 2003;11(3):647–658. | |
Li J, Richter K, Reinstein J, Buchner J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat Struct Mol Biol. 2013; 20(3):326–331. | |
Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic Hsp90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–549. | |
Moulick K, Ahn JH, Zong H, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–826. | |
Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. | |
Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–755. | |
Travers J, Sharp S, Workman P. Hsp90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today. 2012;17(5–6):242–252. | |
Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004; 562(1–3):11–15. | |
Kunisawa J, Shastri N. Hsp90α chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24(5):523–534. | |
Bouchier-Hayes L, Oberst A, McStay GP, et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Mol Cell. 2009;35(6):830–840. | |
Ichiyanagi T, Imai T, Kajiwara C, et al. Essential role of endogenous heat shock protein 90 of dendritic cells in antigen cross-presentation. J Immunol. 2010;185(5):2693–2700. | |
Imai T, Kato Y, Kajiwara C, et al. Heat shock protein 90 (Hsp90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci U S A. 2011;108(39):16363–16368. | |
Eustace BK, Sakurai T, Stewart JK, et al. Functional proteomic screens reveal an essential extracellular role for Hsp90α in cancer cell invasiveness. Nat Cell Biol. 2004;6(6):507–514. | |
Wang X, Song X, Zhuo W, et al. The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc Natl Acad Sci U S A. 2009;106(50):21288–21293. | |
Lei H, Romeo G, Kazlauskas A. Heat shock protein 90α-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ Res. 2004;94(7):902–909. | |
Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 α impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007;282(13):9364–9371. | |
Cortes-González C, Barrera-Chimal J, Ibarra-Sanchez M, et al. Opposite effect of Hsp90α and Hsp90β on eNOS ability to produce nitric oxide or superoxide anion in human embryonic kidney cells. Cell Physiol Biochem. 2010;26(4–5):657–668. | |
Metchat A, Akerfelt M, Bierkamp C, et al. Mammalian heat shock factor 1 is essential for oocyte meiosis and directly regulates Hsp90α expression. J Biol Chem. 2009;284(14):9521–9528. | |
Grad I, Cederroth CR, Walicki J, et al. The molecular chaperone Hsp90α is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS One. 2010;5(12):e15770. | |
Kajiwara C, Kondo S, Uda S, et al. Spermatogenesis arrest caused by conditional deletion of Hsp90α in adult mice. Biol Open. 2012; 1(10):977–982. | |
Voss AK, Thomas T, Gruss P. Mice lacking Hsp90β fail to develop a placental labyrinth. Development. 2000;127(1):1–11. | |
Millson SH, Truman AW, Racz A, et al. Expressed as the sole Hsp90 of yeast, the α and β isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90β generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 2007;274(17):4453–4463. | |
Chadli A, Graham JD, Abel MG, et al. GCUNC45 is a novel regulator for the progesterone receptor/Hsp90 chaperoning pathway. Mol Cell Biol. 2006;26(5):1722–1730. | |
Chadli A, Felts SJ, Toft DO. GCUNC45 is the first Hsp90 co-chaperone to show α/β isoform specificity. J Biol Chem. 2008;283(15):9509–9512. | |
Correia AL, Mori H, Chen EI, Schmitt FC, Bissell MJ. The hemopexin domain of MMP3 is responsible for mammary epithelial invasion and morphogenesis through extracellular interaction with Hsp90β. Genes Dev. 2013;27(7):805–817. | |
Sternlicht MD, Lochter A, Sympson CJ, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98(2):137–146. | |
Pires ES, Khole VV. A block in the road to fertility: autoantibodies to heat-shock protein 90-β in human ovarian autoimmunity. Fertil Steril. 2009;92(4):1395–1409. | |
Choudhury A, Khole VV. Hsp90 antibodies: a detrimental factor responsible for ovarian dysfunction. Am J Reprod Immunol. 2013; 70(5):372–385. | |
Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The Hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275(5):3305–3312. | |
Leskovar A, Wegele H, Werbeck ND, Buchner J, Reinstein J. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem. 2008;283(17):11677–11688. | |
Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol Cell. 2014;53(2):330–343. | |
Rasola A, Neckers L, Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014;24(8):455–463. | |
Cechetto JD, Gupta RS. Immunoelectron microscopy provides evidence that tumor necrosis factor receptor-associated protein 1 (TRAP-1) is a mitochondrial protein which also localizes at specific extramitochondrial sites. Exp Cell Res. 2000;260(1):30–39. | |
Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131(2):257–270. | |
Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2012;1823(3):767–773. | |
Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5(7):e172. | |
Landriscina M, Laudiero G, Maddalena F, et al. Mitochondrial chaperone Trap1 and the calcium binding protein Sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010;70(16):6577–6586. | |
Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31(14):3038–3062. | |
Corti O, Brice A. Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson’s disease. Curr Opin Neurobiol. 2013;23(1):100–108. | |
Saisawat P, Kohl S, Hilger AC, et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 2014;85(6):1310–1317. | |
Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol. 2010;21(5):479–485. | |
Marzec M, Eletto D, Argon Y. GRP94: An Hsp90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012;1823(3):774–787. | |
Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14(4):263–276. | |
Krukenberg KA, Bottcher UM, Southworth DR, Agard DA. Grp94, the endoplasmic reticulum Hsp90, has a similar solution conformation to cytosolic Hsp90 in the absence of nucleotide. Protein Sci. 2009;18(9):1815–1827. | |
Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the Hsp90 chaperones. Mol Cell. 2007;28(1):41–56. | |
Lee AS. Mammalian stress response: induction of the glucose-regulated protein family. Curr Opin Cell Biol. 1992;4(2):267–273. | |
Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101(5):451–454. | |
Ostrovsky O, Makarewich CA, Snapp EL, Argon Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc Natl Acad Sci U S A. 2009;106(28):11600–11605. | |
Weekes MP, Antrobus R, Talbot S, et al. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J Proteome Res. 2012;11(3):1475–1484. | |
Ostrovsky O, Ahmed NT, Argon Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell. 2009;20(6):1855–1864. | |
Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10(3):272–282. | |
Satoh T, Chen Y, Hu D, Hanashima S, Yamamoto K, Yamaguchi Y. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol Cell. 2010;40(6):905–916. | |
Seidler PM, Shinsky SA, Hong F, Li Z, Cosgrove MS, Gewirth DT. Characterization of the Grp94/OS-9 chaperone-lectin complex. J Mol Biol. 2014;426(21):3590–3605. | |
Mao C, Wang M, Luo B, et al. Targeted mutation of the mouse GRP94 gene disrupts development and perturbs endoplasmic reticulum stress signaling. PLoS One. 2010;5(5):e10852. | |
Liu B, Staron M, Hong F, et al. Essential roles of GRP94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc Natl Acad Sci U S A. 2013;110(17):6877–6882. | |
Hua Y, White-Gilbertson S, Kellner J, et al. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res. 2013;19(22):6242–6251. | |
Morales C, Rachidi S, Hong F, et al. Immune chaperone gp96 drives the contributions of macrophages to inflammatory colon tumorigenesis. Cancer Res. 2014;74(2):446–459. | |
Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–410. | |
Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10(6):283–290. | |
Taldone T, Ochiana SO, Patel PD, Chiosis G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci. 2014;35(11):592–603. | |
Beebe K, Mollapour M, Scroggins B, et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4(7):1065–1074. | |
Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin Drug Discov. 2011;6(5):559–587. | |
Menezes DL, Taverna P, Jensen MR, et al. The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol Cancer Ther. 2012;11(3):730–739. | |
Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332(2):275–285. | |
Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92(3):242–248. | |
Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today. 2004;9(20):881–888. | |
Chiosis G. Targeting chaperones in transformed systems – a focus on Hsp90 and cancer. Expert Opin Ther Targets. 2006;10(1):37–50. | |
Matts RL, Dixit A, Peterson LB, et al. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem Biol. 2011;6(8):800–807. | |
Matthews SB, Vielhauer GA, Manthe CA, et al. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate. 2010;70(1):27–36. | |
Kusuma BR, Peterson LB, Zhao H, Vielhauer G, Holzbeierlein J, Blagg BS. Targeting the heat shock protein 90 dimer with dimeric inhibitors. J Med Chem. 2011;54(18):6234–6253. | |
Allan RK, Mok D, Ward BK, Ratajczak T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J Biol Chem. 2006;281(11):7161–7171. | |
Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J Biol Chem. 1994;269(2):787–790. | |
Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59(4):657–663. | |
Itoh H, Ogura M, Komatsuda A, Wakui H, Miura AB, Tashima Y. A novel chaperone-activity-reducing mechanism of the 90-kDa molecular chaperone Hsp90. Biochem J. 1999;343(Pt 3):697–703. | |
Rosenhagen MC, Soti C, Schmidt U, et al. The heat shock protein 90-targeting drug cisplatin selectively inhibits steroid receptor activation. Mol Endocrinol. 2003;17(10):1991–2001. | |
Nardai G, Sass B, Eber J, Orosz G, Csermely P. Reactive cysteines of the 90-kDa heat shock protein, Hsp90. Arch Biochem Biophys. 2000;384(1):59–67. | |
Ngassapa O, Soejarto DD, Pezzuto JM, Farnsworth NR. Quinone-methide triterpenes and salaspermic acid from Kokoona ochracea. J Nat Prod. 1994;57(1):1–8. | |
Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit Hsp90 and Cdc37 interaction. J Biol Chem. 2009;284(51):35381–35389. | |
Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. | |
Tash JS, Chakrasali R, Jakkaraj SR, et al. Gamendazole, an orally active indazole carboxylic acid male contraceptive agent, targets HSP90AB1 (HSP90β) and EEF1A1 (eEF1A), and stimulates Il1a transcription in rat Sertoli cells. Biol Reprod. 2008;78(6):1139–1152. | |
Matts RL, Brandt GE, Lu Y, et al. A systematic protocol for the characterization of Hsp90 modulators. Bioorg Med Chem. 2011;19(1):684–692. | |
Ardi VC, Alexander LD, Johnson VA, McAlpine SR. Macrocycles that inhibit the binding between heat shock protein 90 and TPR-containing proteins. ACS Chem Biol. 2011;6(12):1357–1366. | |
McConnell JR, Alexander LA, McAlpine SR. A heat shock protein 90 inhibitor that modulates the immunophilins and regulates hormone receptors without inducing the heat shock response. Bioorg Med Chem Lett. 2014;24(2):661–666. | |
Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006;25(2):367–376. | |
Mollapour M, Tsutsumi S, Donnelly AC, et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010;37(3):333–343. | |
Mollapour M, Tsutsumi S, Kim YS, Trepel J, Neckers L. Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity. Oncotarget. 2011;2(5):407–417. | |
Wang X, Lu XA, Song X, et al. Thr90 phosphorylation of Hsp90α by protein kinase A regulates its chaperone machinery. Biochem J. 2012;441(1):387–397. | |
Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823(3):648–655. | |
Yu X, Guo ZS, Marcu MG, et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94(7):504–513. | |
Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13(16):4882–4890. | |
Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. | |
Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284(12):7436–7445. | |
Plescia J, Salz W, Xia F, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell. 2005;7(5):457–468. | |
Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3(1):46–54. | |
Fortugno P, Beltrami E, Plescia J, et al. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A. 2003;100(24):13791–13796. | |
Horibe T, Kohno M, Haramoto M, Ohara K, Kawakami K. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J Transl Med. 2011;9:8. | |
Zhao R, Leung E, Gruner S, Schapira M, Houry WA. Tamoxifen enhances the Hsp90 molecular chaperone ATPase activity. PLoS One. 2010;5(4):e9934. | |
Zierer BK, Weiwad M, Rubbelke M, et al. Artificial accelerators of the molecular chaperone Hsp90 facilitate rate-limiting conformational transitions. Angew Chem Int Ed Engl. 2014;53(45):12257–12262. | |
Panaretou B, Siligardi G, Meyer P, et al. Activation of the ATPase activity of Hsp90 by the stress-regulated cochaperone Aha1. Mol Cell. 2002;10(6):1307–1318. | |
Taldone T, Patel PD, Patel M, et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013;56(17):6803–6818. | |
Ernst JT, Liu M, Zuccola H, et al. Correlation between chemotype-dependent binding conformations of Hsp90α/β and isoform selectivity-implications for the structure-based design of Hsp90α/β selective inhibitors for treating neurodegenerative diseases. Bioorg Med Chem Lett. 2014;24(1):204–208. | |
Ernst JT, Neubert T, Liu M, et al. Identification of novel Hsp90α/β isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington’s disease. J Med Chem. 2014;57(8):3382–3400. | |
Patel PD, Yan P, Seidler PM, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9(11):677–684. | |
Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278(48):48330–48338. | |
Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21(1):55–89. | |
Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275(1–2):43–61. | |
Chrousos GP, Kino T. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009;1179:153–166. | |
Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10(6):705–712. | |
Fliss AE, Benzeno S, Rao J, Caplan AJ. Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol. 2000; 72(5):223–230. | |
Vanaja DK, Mitchell SH, Toft DO, Young CY. Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones. 2002;7(1):55–64. | |
Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs. 2004;15(7):651–662. | |
Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3(8):1021–1030. | |
Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353(16):1652–1654. | |
Mitri Z, Constantine T, O’Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. Heat shock protein 90: inhibitors in clinical trials. J Med Chem. 2010;53(1):3–17. | |
Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8(4):440–448. | |
Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet. 1997;349(9060):1210–1213. | |
Caprio M, Feve B, Claes A, Viengchareun S, Lombes M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007;21(9):2185–2194. | |
Lehrke M, Lazar MA. The many faces of PPARγ. Cell. 2005;123(6):993–999. | |
Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem. 2008;77:289–312. | |
Christodoulides C, Vidal-Puig A. PPARs and adipocyte function. Mol Cell Endocrinol. 2010;318(1–2):61–68. | |
Nguyen MT, Csermely P, Soti C. Hsp90 chaperones PPARγ and regulates differentiation and survival of 3T3-L1 adipocytes. Cell Death Differ. 2013;20(12):1654–1663. | |
Desarzens S, Liao WH, Mammi C, Caprio M, Faresse N. Hsp90 blockers inhibit adipocyte differentiation and fat mass accumulation. PLoS One. 2014;9(4):e94127. | |
Dahia PL, Grossman AB. The molecular pathogenesis of corticotroph tumors. Endocr Rev. 1999;20(2):136–155. | |
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367(9522):1605–1617. | |
Tritos NA, Biller BM, Swearingen B. Management of Cushing disease. Nat Rev Endocrinol. 2011;7(5):279–289. | |
Paez-Pereda M, Kovalovsky D, Hopfner U, et al. Retinoic acid prevents experimental Cushing syndrome. J Clin Invest. 2001;108(8):1123–1131. | |
Riebold M, Kozany C, Freiburger L, et al. A C-terminal Hsp90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat Med. 2015;21(3):276–280. | |
Zhao H, Brandt GE, Galam L, Matts RL, Blagg BS. Identification and initial SAR of silybin: an Hsp90 inhibitor. Bioorg Med Chem Lett. 2011;21(9):2659–2664. | |
Manié S, Santoro M, Fusco A, Billaud M. The RET receptor: function in development and dysfunction in congenital malformation. Trends Genet. 2001;17(10):580–589. | |
Santoro M, Melillo RM, Carlomagno F, Vecchio G, Fusco A. Minireview: RET: normal and abnormal functions. Endocrinology. 2004;145(12):5448–5451. | |
Santoro M, Carlomagno F. Drug insight: small-molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2(1):42–52. | |
Vidal M, Wells S, Ryan A, Cagan R. ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res. 2005;65(9):3538–3541. | |
Sherman SI. Advances in chemotherapy of differentiated epithelial and medullary thyroid cancers. J Clin Endocrinol Metab. 2009;94(5):1493–1499. | |
Wells SA Jr, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–772. | |
Brose MS, Nutting CM, Jarzab B, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319–328. | |
Schlumberger M, Tahara M, Wirth LJ. Lenvatinib in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(19):1868. | |
Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372(7):621–630. | |
Marsee DK, Venkateswaran A, Tao H, et al. Inhibition of heat shock protein 90, a novel RET/PTC1-associated protein, increases radioiodide accumulation in thyroid cells. J Biol Chem. 2004;279(42):43990–43997. | |
Alfano L, Guida T, Provitera L, et al. RET is a heat shock protein 90 (Hsp90) client protein and is knocked down upon Hsp90 pharmacological block. J Clin Endocrinol Metab. 2010;95(7):3552–3557. | |
Roodman GD. Biology of osteoclast activation in cancer. J Clin Oncol. 2001;19(15):3562–3571. | |
Sukhdeo K, Mani M, Zhang Y, et al. Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc Natl Acad Sci U S A. 2007;104(18):7516–7521. | |
Ashihara E, Kawata E, Nakagawa Y, et al. β-Catenin small interfering RNA successfully suppressed progression of multiple myeloma in a mouse model. Clin Cancer Res. 2009;15(8):2731–2738. | |
Usmani SZ, Bona RD, Chiosis G, Li Z. The anti-myeloma activity of a novel purine scaffold Hsp90 inhibitor PU-H71 is via inhibition of both Hsp90a and Hsp90b1. J Hematol Oncol. 2010;3:40. | |
Ishii T, Seike T, Nakashima T, et al. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J. 2012;2(4):e68. | |
Stuhmer T, Iskandarov K, Gao Z, et al. Preclinical activity of the novel orally bioavailable Hsp90 inhibitor NVP-HSP990 against multiple myeloma cells. Anticancer Res. 2012;32(2):453–462. | |
Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149–168. | |
Grantham JJ. Polycystic kidney disease: neoplasia in disguise. Am J Kidney Dis. 1990;15(2):110–116. | |
Seeger-Nukpezah T, Proia DA, Egleston BL, et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci U S A. 2013;110(31):12786–12791. | |
Barluenga S, Wang C, Fontaine JG, et al. Divergent synthesis of a pochonin library targeting Hsp90 and in vivo efficacy of an identified inhibitor. Angew Chem Int Ed Engl. 2008;47(23):4432–4435. | |
Barluenga S, Fontaine JG, Wang C, et al. Inhibition of Hsp90 with pochoximes: SAR and structure-based insights. Chembiochem. 2009;10(17):2753–2759. | |
Tash JS, Attardi B, Hild SA, Chakrasali R, Jakkaraj SR, Georg GI. A novel potent indazole carboxylic acid derivative blocks spermatogenesis and is contraceptive in rats after a single oral dose. Biol Reprod. 2008;78(6):1127–1138. | |
Gomez-Monterrey I, Sala M, Musella S, Campiglia P. Heat shock protein 90 inhibitors as therapeutic agents. Recent Pat Anticancer Drug Discov. 2012;7(3):313–336. | |
De Mattos-Arruda L, Cortes J. Breast cancer and Hsp90 inhibitors: is there a role beyond the HER2-positive subtype? Breast. 2012;21(4):604–607. | |
Caldas-Lopes E, Cerchietti L, Ahn JH, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106(20):8368–8373. | |
Mehta PP, Whalen P, Baxi SM, Kung PP, Yamazaki S, Yin MJ. Effective targeting of triple-negative breast cancer cells by PF-4942847, a novel oral inhibitor of Hsp90. Clin Cancer Res. 2011;17(16):5432–5442. | |
Wong C, Chen S. Heat shock protein 90 inhibitors: new mode of therapy to overcome endocrine resistance. Cancer Res. 2009;69(22):8670–8677. | |
Yehuda R, Flory JD, Bierer LM, et al. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatr. 2015;77(4):356–364. | |
Jochems J, Teegarden SL, Chen Y, et al. Enhancement of stress resilience through histone deacetylase 6-mediated regulation of glucocorticoid receptor chaperone dynamics. Biol Psychiatr. 2015;77(4):345–355. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.