Back to Journals » International Journal of Nanomedicine » Volume 21
How PLGA Microspheres are Emerging as a Key Drug Delivery System
Authors Qian S, Ma L, Liu C, Lin Y, Wu J, Wang X
Received 30 January 2026
Accepted for publication 30 May 2026
Published 15 June 2026 Volume 2026:21 600160
DOI https://doi.org/10.2147/IJN.S600160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Farooq A. Shiekh
Sai Qian,1– 3 Lan Ma,1,2 Chenlu Liu,1,2 Yuxi Lin,1,2 Jiangfeng Wu,1,2 Xiaolian Wang1,2
1Hubei Key Laboratory of Tumor Microenvironment and Immunotherapy, China Three Gorges University, Yichang, Hubei, 443000, People’s Republic of China; 2College of Basic Medical Sciences, China Three Gorges University, Yichang, Hubei, 443000, People’s Republic of China; 3College of Medicine and Health Sciences, China Three Gorges University, Yichang, Hubei, 443000, People’s Republic of China
Correspondence: Xiaolian Wang, Email [email protected] Jiangfeng Wu, Email [email protected]
Abstract: Poly(lactic-co-glycolic acid) (PLGA) microspheres are a clinically established platform for sustained and controlled drug delivery, offering tunable degradation and reduced dosing frequency. This review critically appraises PLGA microsphere technology, linking materials science with clinical translation. We examine how molecular parameters—molecular weight, lactide-to-glycolide ratio, and terminal group chemistry—affect degradation and release kinetics. Key fabrication methods (emulsion-solvent evaporation, spray drying, membrane emulsification, and microfluidics) are compared for their control over encapsulation efficiency, particle size, and scalability. Release mechanisms, including diffusion, swelling, and erosion, are discussed alongside strategies to mitigate burst release. The review also addresses in vivo pharmacokinetics, recent clinical progress in oncology and vaccine delivery, regulatory challenges, manufacturing hurdles, and future directions such as stimuli-responsive microspheres and AI-guided formulation design.
Keywords: PLGA microspheres, drug delivery systems, sustained release kinetics, clinical translation
Introduction
PLGA microspheres have transitioned from an experimental excipient to a clinically validated platform in controlled drug delivery, representing one of the most significant advances in long-acting injectable formulations over the past two decades. The sheer pace of progress is reflected in the steady upward trajectory of both published research articles and global patent activity (Figures 1 and 2), signals that underscore the strong commitment of both academia and industry to turning PLGA-based technologies into clinically viable therapies. Conventional drug administration suffers from rapid systemic elimination, fluctuating plasma levels, and poor patient compliance. These drawbacks have long limited the therapeutic potential of many small-molecule drugs and biologics. Against this backdrop, PLGA sustained-release microspheres have stepped forward as a compelling answer: they widen the therapeutic window, lower the frequency of dosing, and help keep off-target side effects in check.1
 |
Figure 1 Trends in PLGA microsphere research publications (2006–2025). Notes: Publication data were retrieved from Web of Science Core Collection on [2026.05.08], using the search query: TS=((“PLGA” OR “poly(lactic-co-glycolic acid)”) AND (“microsphere*” OR “microparticle*”)). Data for 2024–2025 may be incomplete due to database indexing lag. |
 |
Figure 2 Trends in PLGA microsphere patent filings (2006–2025). Notes: Patent data were retrieved from Lens.org on [2026.05.08], using the same keyword strategy: (“PLGA” OR “poly(lactic-co-glycolic acid)”) AND (“microsphere*” OR “microparticle*”). Data for 2024–2025 may be incomplete due to database indexing lag. |
The widespread adoption of PLGA is primarily attributable to its outstanding safety record and the high degree of tunability of its physicochemical properties. Because the copolymer has received clearance for parenteral use from both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), it can be administered with confidence; once inside the body, it degrades via simple hydrolysis into lactic acid and glycolic acid.2 These endogenous compounds then enter the tricarboxylic acid (TCA) cycle and are ultimately excreted as carbon dioxide and water. This built-in biocompatibility allows PLGA implants to avoid the immune rejection and persistent inflammation that frequently accompany non-degradable polymeric devices. What makes PLGA chemistry especially powerful is its adjustability: by varying the molecular weight distribution, the lactide-to-glycolide (LA/GA) molar ratio, and the chemical nature of the terminal groups, scientists can precisely control the rate at which the polymer breaks down, and therefore can engineer drug release over timeframes ranging from a few weeks to several months.3
More than a dozen PLGA microsphere products—Lupron Depot®, Sandostatin LAR®, and Risperdal Consta® among them—have reached the market and are used routinely in the clinic.4 Despite this commercial success, the rational design of generic and novel PLGA formulations remains fraught with substantial scientific and engineering hurdles. During process scale‑up, it remains difficult to keep the particle size distribution tight and reproducible; the well‑known problem of a large initial burst release, which can put patient safety at risk, still awaits a fully satisfactory solution; and preserving the delicate structure of biologic payloads throughout the manufacturing process is a persistent concern.5 On top of these issues, building a reliable in vitro–in vivo correlation (IVIVC) for PLGA release is very much a work in progress—it demands a far clearer picture of how the polymer erodes, how the pore network inside the microspheres evolves over time, and how drug molecules diffuse through the ever‑changing physiological microenvironment.6
This review provides a concise yet wide-ranging look at where PLGA microsphere technology stands today. It picks apart how the choices made at the level of materials, fabrication, and PLGA’s own molecular architecture collectively shape the way a microsphere releases its cargo. Responding to the growing emphasis on translational relevance, we also widen the lens to include factors that matter in the clinic: in vivo pharmacokinetic behavior, the regulatory considerations that come with scaling up a manufacturing process, and the expanding field of intelligent microspheres that can respond to biological or external stimuli.7 The review rests on a combination of established scientific foundations and very recent literature, aiming to give both researchers and clinicians a practical, forward-looking guide for moving the next generation of PLGA-based delivery systems closer to clinical use.
Material Selection: PLGA, Solvents, and Stabilizers
Designing PLGA microspheres rationally begins with three key decisions: which polymer matrix to use, what organic solvent system to employ, and which emulsion stabilizer to add. Each of these choices shapes the final product’s critical quality attributes—encapsulation efficiency, particle size distribution, surface morphology, residual solvent levels—and ultimately governs drug release in vivo.1
PLGA as the Core Matrix Material
PLGA owes its standing as the premier biodegradable polymer for long-acting injectable microspheres to the regulatory clearance it has received from both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for parenteral use.8 Its widespread adoption can be traced to its ability to offer superb biocompatibility, easily adjustable mechanical properties, and highly foreseeable degradation behavior. When implanted, the polymer backbone spontaneously breaks down through non-enzymatic hydrolysis, liberating lactic acid (LA) and glycolic acid (GA). Because these monomeric products are natural to the body, they are routed through the tricarboxylic acid (TCA) cycle and eventually excreted as carbon dioxide and water, essentially eliminating any long-term safety worries.3,9
The properties of the resulting PLGA polymer are intimately linked to the synthesis route employed. The two principal synthetic routes are direct polycondensation and ring-opening polymerization (ROP).10
Ring-opening polymerization, or ROP for short, starts from the cyclic dimers of lactide and glycolide and has become the method of choice on an industrial scale. The reason is straightforward: ROP can reliably deliver high‑molecular‑weight PLGA whose chain architecture is far better controlled. The most widely used catalysts are tin‑based—stannous octoate being the classic example—and, while they are certainly efficient at opening the rings, they produce a largely random arrangement of the two monomers along the chain.11
Direct polycondensation, which essentially involves simply condensing LA and GA monomers together, is the simpler of the two routes and costs less to run. What you get, however, is PLGA whose molecular weight stays low, whose chains span a wide range of lengths, and whose monomer sequence along the backbone cannot be dictated with any real precision. Put another way, the degradation trajectory that a direct‑polycondensation polymer will follow is, much of the time, frustratingly difficult to forecast.12,13
A key recent advance is the ability to precisely control PLGA microstructure using next‑generation catalysts. In particular, regio‑ and stereoselective ring‑opening polymerization enables the synthesis of isotactic, alternating PLGA, where LA and GA units repeat in strict order.14 This ordered sequence alters polymer crystallinity, water affinity, and hydrolytic stability, leading to degradation profiles distinct from random copolymers. As a result, alternating PLGA offers more predictable drug release and represents a frontier in rational polymer design.
Organic Solvent Selection and Its Ramifications
The choice of organic solvent in the emulsion-solvent evaporation method is a critical determinant of microsphere morphology, drug loading, and process safety. Dichloromethane (DCM) and ethyl acetate (EtAc) are the most frequently employed solvents, each presenting a distinct profile of advantages and drawbacks.
Dichloromethane (DCM): DCM remains the gold-standard solvent due to its excellent solvency for PLGA, low boiling point (facilitating rapid evaporation), and low aqueous solubility, which collectively promote high encapsulation efficiency and uniform particle size.15 However, microspheres prepared with DCM often exhibit a porous or rough surface morphology. Such a porous surface tends to promote a more pronounced burst release of the drug during the initial phase. More critically, DCM is classified as a Class 2 residual solvent by the ICH, and its potential toxicity necessitates rigorous control of residual levels, a major challenge during the scale-up of manufacturing processes.5,6
Ethyl Acetate (EtAc): EtAc is a Class 3 solvent, which means it carries a better safety profile, and it has received attention as a greener replacement for DCM. On the other hand, it dissolves more readily in water and evaporates more slowly than DCM, and these two properties produce microspheres that look very different. The literature shows that EtAc usually gives particles that are largely free of pores, often taking on a hollow or “doughnut‑like” architecture. That architecture does keep the early burst in check, but it comes with a trade‑off: drug loading tends to drop, especially for hydrophilic compounds, because rapid-phase separation pushes a large fraction of the drug into the surrounding aqueous phase before the microspheres have a chance to harden.16 (A comparative analysis of DCM and EtAc is provided in Table 1)
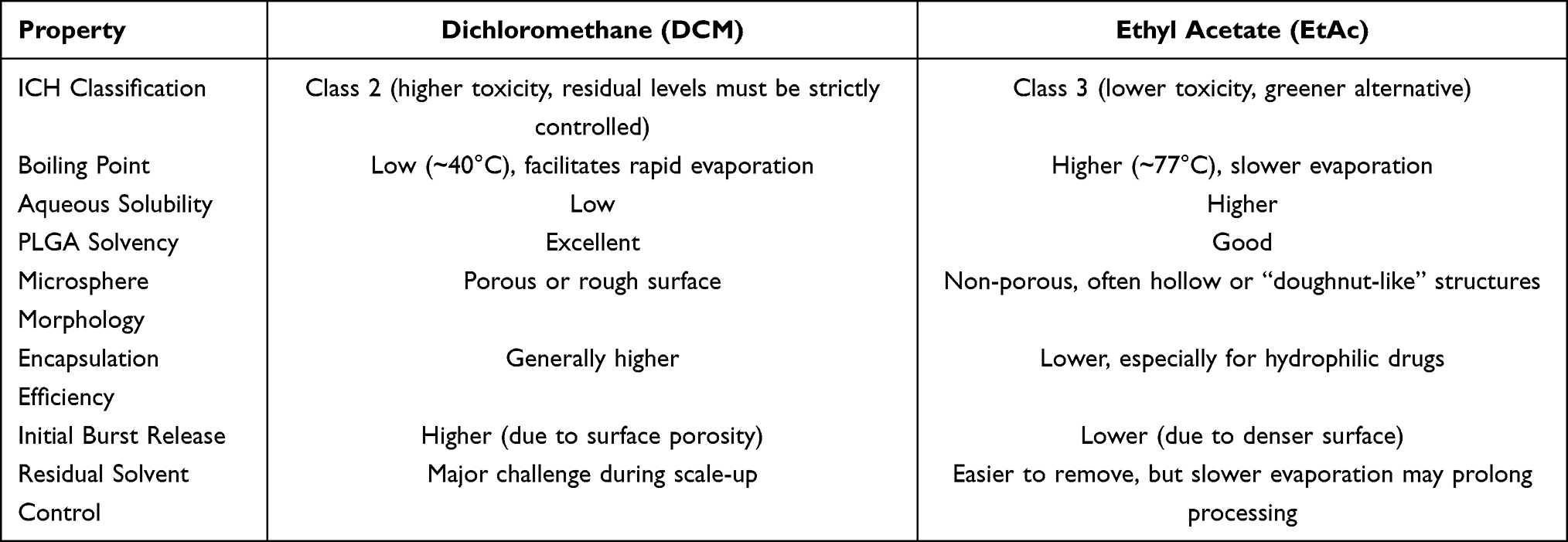 |
Table 1 Comparative Analysis of Commonly Used Organic Solvents for PLGA Microsphere fabrication5,6,15,16 |
The Indispensable Role of Emulsifiers and Stabilizers
During emulsification, the creation of separate, well-defined spherical microspheres depends on a stabilizer that can reduce interfacial tension and keep the scattered organic-phase droplets from merging into unwanted larger masses. In the context of PLGA microsphere production, the emulsifier that finds the widest application remains poly(vinyl alcohol) (PVA). Its molecules accumulate at the interface of the freshly created droplets, forming a protective steric shell that prevents the droplets from coalescing.17 PVA enjoys a long history of safe use in pharmaceutical products and has been officially classified by the FDA as a Generally Recognized as Safe (GRAS) excipient for a variety of administration routes. It is this excellent record of biocompatibility that has made PVA the stabilizer of first choice for microsphere products destined for parenteral use.7
PVA is highly effective, but it does leave behind residual polymer on the microsphere surface, and that residue can alter both the kinetics of drug release and how the particles interact with biological systems once they are administered. Emulsion stability and the size distribution of the finished microspheres are also strongly shaped by two key properties of the PVA itself—its molecular weight and the extent of hydrolysis. When ultra‑high purity is a must, researchers have looked at alternative stabilizers, poloxamers (Pluronic F‑127 being one well‑known example) and polysorbates such as Tween 20/80 among them; these alternatives, however, do not always match the stabilizing power of PVA.18,19
Preparation Methods for PLGA Microspheres
How a PLGA microsphere is made leaves a clear footprint on the quality attributes that matter most—measured by how broad the size distribution turns out, what the surface looks like, how much of the drug actually ends up encapsulated, and the way the drug is released over time. Most of the fabrication technologies in use today can be sorted under four headings: emulsion‑solvent evaporation, spray drying, membrane emulsification, and microfluidics. What distinguishes one from another is a characteristic mix of strengths and limitations, especially with regard to amenability to scale‑up, batch‑to‑batch consistency, and the range of therapeutic payloads they can handle.20
Emulsion-Solvent Evaporation Method
For PLGA microspheres, the emulsion‑solvent evaporation method remains by far the most widely used and industrially proven manufacturing route. In short, the process works by first dispersing an organic phase containing the polymer—and, where applicable, the drug—into an aqueous phase, then removing the volatile solvent so that hardened particles are left behind. Which variant of the method is chosen depends on the type of emulsion that the formulation is built around, and there are essentially two options: single emulsion and double emulsion procedures. Single emulsion variants can be run in either oil‑in‑water (O/W) or water‑in‑oil (W/O) mode. Double emulsion variants almost always rely on a water‑in‑oil‑in‑water (W/O/W) design. When the drug is hydrophobic, the O/W configuration is the better fit, whereas the W/O and W/O/W configurations are the routes to reach for when the therapeutic payload is hydrophilic.21
In the single emulsion (O/W) process, the starting point is an organic solvent that does not mix with water, into which both the PLGA polymer and the hydrophobic drug are dissolved together. The resulting organic mixture is then dispersed as fine droplets into a water-based phase to which a stabilizer—most often poly(vinyl alcohol) (PVA)—has been added. With continued stirring or the application of reduced pressure, the organic solvent is gradually driven off; as this happens, the polymer-rich phase solidifies, locking the drug inside the freshly created microspheres. Hydrophilic drugs are more commonly processed via the double emulsion (W/O/W) route. Here, an aqueous solution of the drug is first emulsified with the PLGA-containing organic solvent, producing a primary water-in-oil (W/O) emulsion. This primary emulsion is next introduced into a larger volume of water that already contains PVA, generating a short-lived water-in-oil-in-water (W/O/W) double emulsion. Stripping away the volatile organic solvent causes the microspheres to solidify, and by that point the hydrophilic cargo remains trapped within the internal water pockets of the hardened particles.
Researchers at Shandong Luye Pharmaceutical Co., Ltd., turned to a combination of the W/O/W emulsion‑solvent evaporation method and Box‑Behnken response surface methodology as a way of systematically optimizing leuprolide acetate‑loaded PLGA microspheres. Once the formulation had been optimized—a theoretical drug loading of 7.59%, a PVA concentration of 2%, and a water‑to‑oil ratio of 200:1—the particles came out with a mean diameter of 20.81 ± 1.34 µm and an encapsulation efficiency that reached 95.88 ± 1.56%.22 Under the scanning electron microscope, the particles looked spherical, with smooth, rounded surfaces that were dotted with plentiful pores. When the same microspheres were put through accelerated in vitro release tests, the drug came out following a first‑order kinetic model. In short, this piece of work drives home how much can be gained by applying a systematic experimental design to fine‑tune the quality of PLGA microspheres made by the emulsion route.
The emulsion‑solvent evaporation method remains the industrial standard due to its straightforward equipment needs, simple operation, and decades of reliable production history. Few other platform technologies can match its versatility—the same basic workflow handles small‑molecule drugs and biologics quite comfortably. Where the method runs into trouble, though, is in the consistency of the product it delivers. The microspheres that emerge are apt to show a wide, often multi‑modal size distribution, and that breadth brings with it uneven drug loading, release kinetics that shift from batch to batch, and reproducibility that is simply not tight enough for a parenteral dosage form. A further practical headache is the sheer volume of organic solvent and water that the process consumes; at production scale this raises both environmental and cost concerns. And for hydrophilic drugs, high entrapment efficiency has remained something of a holy grail—these compounds tend to escape into the external aqueous phase before the droplets have a chance to harden, a difficulty that has frustrated formulation scientists for years.23–25
Spray Drying Method
In spray drying, a liquid feed—which can be a solution, a suspension, or an emulsion that already contains both the PLGA polymer and the therapeutic agent—is forced through a nozzle and broken into fine droplets that are immediately contacted with a hot drying gas, usually air or nitrogen. Because the organic solvent in those droplets is volatile, it flashes off almost instantly under the high temperature; what remains behind solidifies into discrete, dry microparticles. What makes spray drying attractive is that it is a genuinely continuous, single‑step operation, and it can be taken from the laboratory bench to pilot scale without having to rethink the entire manufacturing approach. It also gives the formulator an unusual degree of control: by adjusting the inlet temperature, the rate at which the feed is pumped, or the pressure used to atomise the liquid, and by selecting the molecular weight of the PLGA or the concentration of polymer in the feed, one can steer the size, morphology, drug loading, encapsulation efficiency, and release kinetics of the microparticles with considerable precision.26
For all its benefits, spray drying also introduces some real problems that are hard to ignore. High on that list is a pronounced early burst: a substantial fraction of the drug payload can escape from the microspheres almost immediately, long before it is supposed to. Mechanistic investigations into the kinetics of this burst have begun to explain why. In experiments that use bovine serum albumin (BSA) as a model protein, surface‑sensitive tools—X‑ray photoelectron spectroscopy (XPS) and time‑of‑flight secondary ion mass spectrometry (ToF‑SIMS)—paint a consistent picture: microspheres produced with a two‑fluid nozzle tend to develop a PLGA‑rich outer skin, beneath which the BSA sits trapped. The outer skin does not hold up well; it is prone to degrade and to form pores, and once that happens the encapsulated protein diffuses outward more quickly, feeding the burst release that is so commonly observed.27
Despite its practical advantages, spray drying exposes sensitive biological payloads to heat and shear during atomization and drying, which poses a substantial risk of protein unfolding and loss of biological activity. The rapid cooling that accompanies solvent evaporation does afford some protection against thermal damage, but it would be misleading to suggest that it eliminates the problem; spray drying a biologic that cannot tolerate high temperatures is rarely straightforward—it takes careful, case‑by‑case adjustment of the process parameters to get it right. Moving the process from a bench‑top dryer to a production‑scale unit introduces another layer of difficulty: keeping the particle size distribution and the morphology of the microspheres constant from one batch to the next at production scale remains a tough engineering task.28 Even with these limitations, spray drying keeps its place as an attractive manufacturing route for PLGA microspheres, provided the drug is a stable small molecule. Where speed, continuous operation, and efficient use of solvent are priorities, the benefits tend to outweigh the risks.
Membrane Emulsification Method
Membrane emulsification has emerged as a comparatively new and elegant strategy for producing uniform, monodisperse microspheres. The technique can be broadly split into two modes: direct membrane emulsification and indirect (or premix) membrane emulsification. In the direct mode, the dispersed phase—commonly an organic PLGA solution—is forced through the evenly sized pores of a microporous membrane and directly enters the continuous phase; there, droplets are formed and detach under controlled shear. In the indirect mode, a coarse pre-emulsion is first generated by ordinary stirring and is then extruded through the membrane under pressure, which reduces and homogenizes the droplet size. As the pre-emulsion is squeezed through the pores, the larger droplets break apart, giving rise to final emulsion droplets that are typically only one-third to one-half the diameter of the original membrane pores. Consequently, the indirect route can produce microspheres that are significantly smaller than those obtained by the direct route.29
The capabilities of the technique are perhaps best illustrated by Shirasu Porous Glass (SPG) membrane emulsification. When researchers set out to encapsulate the hydrophilic model compound blue dextran (BLD), they coupled the solvent evaporation method with SPG membrane emulsification and obtained monodisperse PLGA microspheres.30 The investigation checked how stable and productive the primary W/O emulsion was and showed that, simply by choosing SPG membranes with different pore sizes, it was possible to generate microspheres with sizes anywhere from 2.0 to 10 µm. What stood out was the remarkably narrow size distribution, and the initial release rate of the drug could be adjusted simply by adding polyethylene glycol (PEG) to the aqueous phase.
Membrane emulsification has several practical strengths. Microspheres can be prepared quite fast, the particles are remarkably monodisperse, and the process tends to give very similar results from one batch to the next. Because it works under mild conditions—there is little mechanical force and no harsh stirring—it is one of the few techniques that can handle delicate biomacromolecules without destroying their structure. The main drawback is throughput: compared with the workhorses of industrial production, membrane emulsification runs more slowly. Membranes also foul over time, and if the organic phase wets the pores, performance drifts in ways that are hard to predict. A successful process therefore starts with choosing a membrane whose wettability and chemical resistance match the solvent system being used. Incorrect selection of membrane properties often leads to unreliable process performance.
Microfluidic Technology
Microfluidic technology has emerged as a highly precise and reliable method for producing PLGA microspheres, offering exceptional control over particle size, morphology, and internal architecture.31 In a typical protocol, the drug and the PLGA carrier are first co‑dissolved in an organic solvent to generate the organic phase; the aqueous Phase Is simply a solution of a surfactant—PVA is the most common choice. That aqueous phase is fed into the tiny channels of a microfluidic chip under flow conditions that can be adjusted with great accuracy, and it is the interplay of pressure and shear where the two immiscible streams meet that gives rise to extremely uniform droplets. Once the organic solvent has been stripped away, what remains is a population of monodisperse microspheres whose uniformity is striking.32 Because the channels are microscale and the flow parameters can be set digitally, microfluidic technology delivers microspheres that are not only highly uniform but also remarkably reproducible from batch to batch.
Microfluidic technology has seen a number of advances in the recent past that have extended its reach and helped to overcome some of the hurdles that have held it back. One particularly interesting development is centrifugal microfluidics—an approach that uses centrifugal force to pump fluids and to pinch off droplets inside a spinning microchannel. In a recent demonstration, the method was applied to risperidone‑loaded PLGA microspheres (RIS‑MS).33 Key variables—PLGA type and concentration, the internal diameter of the microchannel, and the centrifugal speed—were systematically tuned, and the group found that when the PLGA content was kept at or above 15%, they could operate microchannels with internal diameters of 170–210 µm at a speed of 400 rpm and still obtain well‑formed particles. Pharmacokinetic evaluation in rabbits showed that the RIS‑MS maintained steady plasma levels and released the drug over a period of 42 days—a result that underlines just how promising this technology is for long‑acting injectable therapies.
The push toward automation has also produced integrated microfluidic platforms that are both scalable and programmable. In one example, long-acting hydromorphone-loaded PLGA (HM-PLGA) microspheres were fabricated on a system that pulled together a novel micro-mixer for particle generation, syringe and HPLC pumps that kept the feed flowing continuously, a Raman spectrometer that served as a process analytical technology (PAT) tool, and a programmable automation unit that coordinated the entire operation.34 Because the platform linked all of these elements, researchers could systematically scan a wide formulation space instead of having to vary parameters one at a time by hand. The microspheres that emerged from the process could be steered to the desired size, reached a maximum drug loading of 7.71%, and gave an encapsulation efficiency of 69.40%. When the same particles were tested in beagle dogs, drug release was sustained for more than 11 days—concrete evidence that continuous manufacturing of size-controlled, drug-loaded microspheres on an automated microfluidic platform is a practical proposition.
A further demonstration of the versatility of microfluidics comes from work on core‑shell PLGA microspheres, a geometry that is particularly helpful when the goal is to protect water‑soluble peptides and proteins and to release them gradually. In that study, a glass capillary microfluidic device was operated on the water‑in‑oil‑in‑water (W/O/W) emulsion principle to prepare leuprolide acetate‑loaded PLGA microspheres. By adding gelatin to the internal aqueous phase and carefully tuning the pH of the collection solution, the group was able to obtain highly monodisperse microspheres that were about 80 µm in diameter and displayed a distinct core‑shell architecture. The encapsulation efficiency attained was 80.28%, and in vitro drug release was sustained over roughly 28 days.35
Despite these impressive capabilities, microfluidic technology is not without its challenges. Throughput remains the most stubborn limitation: a single channel can only generate a limited number of microspheres per hour, and this output is typically far too low to meet industrial manufacturing volumes. This is why a great deal of current research is focused on running many channels in parallel and on designing droplet generators that can operate at much higher frequencies. A less obvious but equally important hurdle is the cost of entry—the instrumentation itself requires a sizeable initial investment—and the level of technical know‑how needed to build the chips and to keep the systems running reliably. In some contexts, the combination of high capital cost and demanding operational expertise can be enough to discourage adoption.28
Summary and Comparative Assessment
No single fabrication route is ideal for every application; each one brings a distinctive mix of strengths and weaknesses, and choosing among them means weighing what a particular drug delivery problem actually demands. Emulsion‑solvent evaporation has long been the industrial backbone because it is simple, flexible, and built on decades of manufacturing experience, even if it tends to give wider particle‑size distributions and less consistent batch‑to‑batch performance. Spray drying, by contrast, can be run continuously and is comparatively easy to scale, but the risks of heat damage and a large early burst cannot be dismissed lightly.26,36 Membrane emulsification produces exceedingly uniform particles under very gentle conditions—an obvious advantage when the cargo is a fragile biologic—though the low throughput and the practical headaches of membrane fouling and pore wetting continue to limit its reach. Microfluidic technology has moved to the forefront of precision particle engineering, largely because it allows the user to dictate size, shape, and internal structure to a degree that is difficult to match. What has made microfluidics even more compelling is the recent arrival of automated, scalable platforms that begin to close the gap between what can be done at the laboratory bench and what must be achieved on a production floor.32–35 Because the properties of a PLGA sustained‑release microsphere are so strongly influenced by how it is made, selecting the right fabrication route—driven by the specific application—can tune particle size, encapsulation efficiency, drug loading, and release behaviour so that the final product meets the stringent demands of clinical use. (Table 2 places these four methods side by side, highlighting how they compare in terms of particle‑size control, scalability, encapsulation efficiency, and their key limitations)
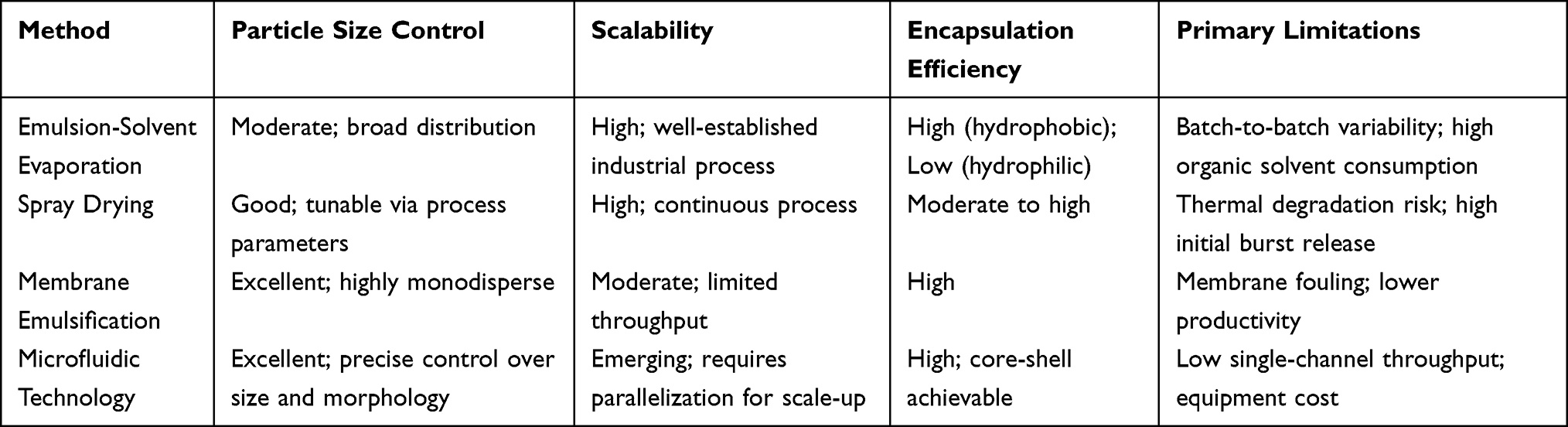 |
Table 2 Comparison of Preparation Methods for PLGA microspheres24–35 |
Molecular Properties of PLGA
How much drug a PLGA microsphere can carry, how efficiently it traps that drug inside, how fast the polymer matrix breaks down, and the release pattern that eventually unfolds—all of these performance characteristics trace back, in one way or another, to the way the polymer itself is put together at the molecular level. Coming to grips with these structure–property relationships is not an academic exercise; it is what makes the rational design of sustained-release formulations possible in the first place. The molecular features that matter most are the relative molecular weight and how broadly the chain lengths are spread, the lactide-to-glycolide (LA/GA) ratio, the chemistry of the chain ends, whether the polymer is linear or branched, its glass transition temperature (Tg), and its crystallinity.37
Relative Molecular Weight and Molecular Weight Distribution
As the PLGA chain is extended by the incorporation of more LA and GA monomers, the degree of polymerization rises, and this directly sets the molecular weight of the polymer—quantities that are routinely quoted in their weight-average (Mw) and number-average (Mn) forms. The reason why these numbers matter is that they profoundly shape the properties of the resulting microspheres through a series of interlinked physical and chemical pathways.
As the molecular weight of PLGA increases, the polymer chains become longer and entangle more frequently, which raises the viscosity of the organic phase during fabrication. Consequently, dissolved drug molecules find it harder to escape into the surrounding aqueous medium, so the encapsulation efficiency tends to rise and the early burst tends to shrink. High molecular weight also changes how the polymer degrades. PLGA breaks down through random scission of ester linkages along its backbone, and a longer chain simply has more bonds that need to be cut before any of the fragments become small enough to dissolve in water and diffuse out of the matrix. The overall effect is that degradation slows down considerably, and drug release can be stretched over a much longer timeframe. At the same time, high‑molecular‑weight PLGA contains fewer free carboxyl end groups—the very groups that speed up hydrolysis through an autocatalytic cycle—so having fewer of them adds to the extended degradation timeline.38
Looking beyond the average molecular weight, the breadth of the molecular weight distribution—usually expressed as the polydispersity index, or PDI—has an important role that is sometimes overlooked. The commercial PLGA used in practice is rarely monodisperse; it contains chains of many different lengths, and data from the last few years make it clear that this spread in molecular weight directly shapes how the release profile evolves from one phase to the next. One systematic review that catalogued the factors responsible for burst release placed molecular weight among the twelve most influential parameters, which reinforces how crucial it is to control molecular weight when designing a formulation.39 What the literature does show, quite consistently, is that microspheres prepared from lower‑Mw PLGA tend to follow a biphasic-release pattern—an early burst, then a prolonged phase governed mainly by diffusion—whereas those formulated with higher‑Mw PLGA more frequently display three distinct phases: the initial burst, a lag interval during which very little drug is released, and a final stage of rapid release that is brought on by bulk erosion. The shift from two phases to three is, in essence, a kinetic consequence: higher‑Mw chains require many more hydrolysis events before the matrix loses enough integrity for erosion to overtake diffusion, so the terminal, erosion‑driven stage simply takes longer to arrive.
Lactide-to-Glycolide Ratio (LA/GA)
PLGA is built from two monomers—lactide (LA) and glycolide (GA)—and the particular character of the copolymer comes from the fact that these two building blocks bring very different properties to the chain. The LA unit carries an extra methyl group that hangs off the side; that small appendage makes the unit more hydrophobic and, just as importantly, shields the nearby ester bond from water. Because water has a harder time reaching that bond, the LA-rich regions of the polymer degrade more slowly. The GA unit, by contrast, has no such methyl appendage. Without that steric shield, water can approach the ester linkage far more easily, and as a result the GA-rich segments break down considerably faster.
The hydrophilic–hydrophobic balance of PLGA can be tuned across a wide window simply by adjusting the LA/GA ratio, giving researchers a powerful handle on how quickly the polymer absorbs water, degrades, and releases its drug cargo. When the LA/GA ratio is raised, the copolymer becomes more hydrophobic, takes up less water, and undergoes slower hydrolytic degradation—characteristics that make it well suited for long-acting, sustained-release therapies that need to operate over several months. When the copolymer is richer in GA, by contrast, it is more hydrophilic, water penetrates more rapidly, and the matrix degrades faster, favoring short-duration treatments or situations that demand a quick therapeutic onset. In terms of relative degradation speed, the trend runs as follows: PLGA 50:50 breaks down the fastest, PLGA 65:35 degrades slightly more slowly, PLGA 75:25 is slower still, and PLGA 85:15 persists the longest.
The LA/GA ratio does more than set the pace of bulk degradation—it also leaves a distinct fingerprint on the internal architecture of the microsphere and on the routes through which drug molecules diffuse out. Because GA‑rich polymers are more hydrophilic, water is drawn into the core of the particle more quickly. This faster hydration encourages the formation of a three‑dimensional network of water‑filled pores, and once those pores are connected, they provide ready channels for drug diffusion and, at the same time, greatly increase the area of polymer that is exposed to hydrolytic attack. What also matters is that the LA/GA ratio does not act alone; it works in concert with the way the two monomers are arranged along the chain. Glycolic blockiness and the average length of the lactic blocks, for instance, shape the local chemical environment inside the degrading microsphere and help determine how the pore network grows and rearranges over time.40
When one looks at the PLGA sustained-release microsphere products that have actually reached the market, two LA/GA ratios dominate: 50:50 and 75:25. The former is typically chosen when an intermediate duration of release is needed, whereas the latter is preferred when the goal is to extend drug release over a longer period. In other words, the LA/GA ratio has served as one of the key design handles that formulation scientists have used to match the copolymer to the physicochemical character of the drug and to the clinical window that needs to be covered.41,42
Terminal Functional Groups
PLGA polymers are typically categorized according to their terminal functional groups, with the three principal types being ester-terminated (capped), carboxyl-terminated (uncapped, –COOH), and hydroxyl-terminated (–OH). These terminal moieties exert a disproportionately large influence on encapsulation efficiency, drug–polymer interactions, and release kinetics, despite constituting only a minute fraction of the polymer mass.43
PLGA chains that end in carboxyl or hydroxyl groups are noticeably more hydrophilic than their ester‑capped analogues, and this difference becomes important during the emulsion‑based fabrication of microspheres. The hydrophilic chain ends tend to orient toward the boundary between the organic and aqueous phases, and as they do so, they create a surface that is riddled with pores. Those pores, in turn, give the drug an easy way out into the surrounding aqueous medium, which is why encapsulation efficiencies often suffer—especially when the drug being encapsulated is itself hydrophilic. Ester‑terminated PLGA, by contrast, does not have the same driving force to segregate to the interface; it produces microspheres with a denser, less open architecture, and consequently holds onto the encapsulated cargo more effectively.44
The chemistry of the chain ends also steers how strongly a drug interacts with the polymer. Ester‑terminated PLGA, for instance, creates water‑poor pockets that are a natural fit for hydrophobic drugs—the drug partitions preferentially into these regions, which raises the loading capacity and helps distribute the drug more evenly through the matrix. With carboxyl‑terminated PLGA, the picture changes: at physiological pH, the chain ends carry a negative charge, and that charge can act as an electrostatic bridge to cationic drugs and peptides. What this means in practice is that encapsulation efficiency can get a boost from those ionic interactions; the trade‑off, though, is that if the binding is too strong, it can slow down the release. Hydroxyl‑terminated PLGA sits somewhere in between, with a polarity that is moderate enough to accommodate a broader range of cargo, including drugs that can be chemically conjugated or that need a balanced hydrophobic–hydrophilic environment.1,43,45
How a microsphere releases its cargo depends very much on which terminal groups are present. Ester‑terminated PLGA microspheres tend to release drug by simple diffusion through the intact polymer matrix; this gives a comparatively slow release and keeps the initial burst under control—both features that lend themselves well to long‑acting, sustained‑delivery applications. Carboxyl‑terminated PLGA behaves quite differently: the polymer is more hydrophilic, and the carboxyl ends catalyse the hydrolysis of the very chains they are attached to. The result is a matrix that degrades faster and a release profile that is driven more by erosion, which typically means a larger early burst. Hydroxyl‑terminated PLGA falls somewhere between these two extremes. Early on, diffusion is the main driver of release; as hydrolysis gradually takes hold, the mechanism shifts toward erosion‑controlled liberation.46 Because the choice of end group leaves such a clear signature on the release pattern, it is one of the most effective tools available for tailoring a PLGA formulation to a particular therapeutic need.
Molecular Architecture: Linear versus Branched PLGA
The three‑dimensional shape of a PLGA molecule—whether it is a straightforward linear chain, a multi‑arm star, a highly branched dendrimer, or some other configuration—has a direct bearing on how fast the polymer degrades and, consequently, on how long it can sustain drug release.47 In commercial microsphere products, linear PLGA has remained by far the dominant choice, simply because its properties have been studied more thoroughly and the manufacturing infrastructure for producing it is already well established.
Branched PLGA—star-shaped, dendritic, and the like—differs from its linear counterpart in one key respect: per unit mass, it packs a noticeably higher density of terminal hydroxyl and carboxyl groups. Having more chain ends means that more sites are available for hydrolysis to begin, and that autocatalytic head start translates into faster overall degradation; in other words, branched architectures take less time to degrade. One promising demonstration comes from work on lipoyl ester‑terminated star PLGA loaded with risperidone. The star geometry helped hold the early burst in check, and at the same time allowed drug release to continue in a sustained fashion over an extended period—by day 45, 99.8% of the drug had been released, which is about as close to complete delivery as one can expect from a controlled‑release system.48
Beyond that, the branched architecture affects the way the polymer chains pack together and how the free volume is spread through the microsphere matrix—factors that directly influence how quickly water is taken up and how the internal pore network develops as degradation proceeds. Having this extra structural variable at one’s disposal gives the formulator an additional set of levers to pull when trying to shape a release profile, going beyond what can be achieved simply by adjusting the molecular weight or the LA/GA ratio.48
Glass Transition Temperature (Tg)
For any amorphous polymer, there is a characteristic temperature—the glass transition temperature, or Tg—at which it moves from a hard, brittle, glass‑like state into a more pliable, rubbery condition. PLGA is, by its very nature, an amorphous polymer, and its Tg usually falls somewhere between 40°C and 60°C. This range is worth noting because it sits quite close to the physiological temperature of 37 °C, and that near‑overlap turns out to have a real bearing on how the polymer behaves inside the body and, in particular, on the kinetics of drug release in vivo.
If one wants to understand what truly controls the pace at which a drug leaves a PLGA microsphere, the connection between release kinetics and the Tg of the formulation is one of the first places to look. When the temperature is kept below Tg, the polymer matrix is essentially frozen into a rigid, glassy state: the chains are locked in place, the material is stiff, and it acts as a fairly effective barrier. Diffusion is slow under these conditions, and drug release is correspondingly sluggish. Warm the system past its Tg, however, and the situation flips. The matrix sheds its rigidity, the polymer chains begin to move much more freely, and the free volume inside the material expands considerably. That extra space gives the embedded drug molecules enough room to wiggle out of their entanglements, and the overall release rate climbs noticeably.49
Within PLGA, Tg tends to rise with increasing LA content in the copolymer, higher polymer molecular weight, and greater chain-end crystallinity; conversely, it tends to drop as terminal group density or residual moisture content increases. Furthermore, residual surfactant remaining from the microsphere preparation process can exert a plasticizing effect, depressing Tg and altering-release behavior.50
What matters, in the end, is where a given PLGA formulation sits relative to the body’s own temperature of 37 °C. If the Tg of the polymer is well above that mark, the microspheres will still be in a rigid, glassy condition once they are placed in the body, and release will proceed largely by diffusion, with the initial burst kept relatively modest. If, instead, the Tg lies close to—or even below—37 °C, the matrix will have already passed into a softer, rubbery state soon after administration. Water can then rush in faster, the chains have more freedom to move, and the drug comes out more quickly, often with a larger burst. Whether the polymer finds itself on the glassy side or the rubbery side of its Tg after implantation is, therefore, far from a trivial detail; it directly shapes the in vivo performance and has to be taken into account from the very beginning of formulation design.1,51
Crystallinity
In a polymer such as PLGA, crystallinity tells us how much of the material is packed into regularly ordered domains and how much sits in a random, amorphous arrangement. Under the processing conditions that are normally used, PLGA stays predominantly in a disordered, amorphous state; yet the sequence in which lactide and glycolide repeat along the backbone is not entirely random, and the regularity it does possess can encourage short segments of the chains to organise themselves. It is this local ordering that influences the overall crystallinity of the sample.
Because the crystalline domains in PLGA are densely packed, they act as obstacles that resist the entry of water; therefore, moisture penetrates the polymer matrix far more easily through the disordered, amorphous zones.40 As a result, how fast and how far hydrolytic degradation progresses is largely determined by the fraction of the polymer that remains in the amorphous state. When crystallinity increases, the volume of amorphous material that is open to water uptake and hydrolysis shrinks, which decelerates the entire degradation process and prolongs the period over which the drug is released. The effect is even more striking in PLGA of lower molecular weight, simply because a larger share of the total polymer mass consists of crystalline regions.52
Crystallinity in PLGA is not a fixed attribute—it depends very much on how the two monomers are distributed along the chain. When the LA and GA units follow a more ordered, repetitive pattern, something that can be encouraged by tightening control over the synthesis, the polymer becomes noticeably more crystalline, and that shift in crystallinity, in turn, alters how the material degrades; the kinetics are not faster or slower in a simple sense, but follow a different trajectory.53 That said, a wholly crystalline PLGA is not physically attainable. Because the copolymer sequence is inherently mixed, a sizeable fraction of the matrix is always in an amorphous state, and it is precisely this amorphous fraction that acts as the main gateway through which water can enter and hydrolysis can proceed.54 (A comprehensive overview of the influence of PLGA molecular properties on microsphere performance is provided in Table 3)
 |
Table 3 Influence of PLGA Molecular Properties on Microsphere performance38–54 |
Release Mechanism of PLGA Microspheres
What makes PLGA microspheres clinically useful is, at heart, their capacity to keep drug concentrations within a therapeutic window for weeks or months at a stretch. They do this while also shielding the drug from degradation and helping more of it reach the systemic circulation in an active form. The way drug actually gets out of these microspheres is seldom a single, tidy mechanism. Instead, what one sees is a constantly shifting balance among several interlinked processes—water slowly seeping into the matrix, the polymer chains gradually breaking apart, and the whole structure losing its integrity over time—that together determine the rate and pattern of release.55
PLGA Degradation: The Foundational Process
Drug release from a PLGA microsphere is, at its core, driven by degradation of the polymer itself.56 The breakdown occurs through hydrolysis and follows a sequence of four recognisable stages:39
In the first stage, water molecules penetrate the loose, unstructured (amorphous) zones of the PLGA matrix, where they disturb the weak forces holding the polymer chains together. The absorbed water then softens the polymer and, as a result, the glass transition temperature, Tg, drops.
After hydration has begun, the ester bonds located along the polymer backbone start to break at scattered, random locations. As these bonds are cut, the average length of the chains decreases, the overall molecular weight falls, and the microsphere gradually loses the structural strength it originally possessed.
As time goes on, the breakage of bonds within the main chain becomes widespread. The long polymer strands are chopped into smaller oligomeric fragments, though these fragments are still physically stuck within the degrading structure around them.
The process does not stop at oligomers. These fragments continue to react with water and eventually split into their original building blocks—lactic acid and glycolic acid. Both of these small monomeric molecules dissolve easily in water, which allows them to leave the disintegrating matrix, diffuse into the surrounding tissue, and enter the body’s normal metabolic network. They are subsequently funnelled into the tricarboxylic acid (TCA) cycle, where they are ultimately turned into carbon dioxide and water and safely expelled from the body.
Mechanistic Pathways Governing Drug Release
Drug release from PLGA microspheres is not a one‑note process; instead, several distinct mechanisms operate side by side, often reinforcing one another:1,57–59
The first of these is diffusion through water‑filled pores. As water seeps into the polymer, it carves out a network of interconnected, water‑filled channels. Drug molecules that have dissolved in the aqueous phase then travel along their concentration gradient through these channels toward the external medium. This route tends to dominate during the early and middle stages of release, especially for drugs of lower molecular weight.
For hydrophobic drugs that prefer the polymer phase over water, a different pathway—diffusion through the PLGA network itself—comes into play. Here, the drug moves directly through the intact polymer, bypassing any pores. How fast this happens is governed by the drug’s partition coefficient and its diffusivity within the polymer, and both of these parameters are, in turn, sensitive to how easily the polymer chains can move and how much free volume is available.
As the polymer continues to take up water, it begins to swell. This swelling‑induced transport enlarges the pores that already exist and, just as importantly, opens up new ones. The expanding network of diffusion paths does two things: it gives drug molecules greater mobility and it helps push the release profile from the relatively quiet lag phase into the more rapid, erosion‑driven phase that follows.
Under some circumstances, degradation is confined largely to the outer surface of the microsphere. This surface erosion scenario is most likely when water penetration is slow compared with the rate at which the polymer hydrolyses. Material is gradually shed from the periphery, and drug that was near the surface is released as the outer layers peel away.
More commonly, however, PLGA undergoes bulk erosion: water enters the matrix faster than the chains can be cleaved, so hydrolysis is, at least initially, fairly uniform throughout the particle. But the process feeds on itself. The lactic acid and glycolic acid liberated by hydrolysis accumulate inside the core, lowering the local pH. Because acidic conditions catalyse further ester cleavage, a runaway, autocatalytic cycle sets in that causes the interior to degrade markedly faster than the surface. The result is a strikingly non‑uniform degradation pattern—and, in the later stages, a rush of drug release as the core disintegrates.40
These mechanisms do not operate in isolation but rather co-exist and dominate at different stages of the release timeline.
Temporal Evolution of Release: The Triphasic Profile
When one plots the amount of drug released from a PLGA microsphere against time, the resulting curve very often breaks into three clear segments that together reflect the shifting balance of the mechanisms described above.6 Not every formulation follows this triphasic pattern; as already noted in Relative Molecular Weight and Molecular Weight Distribution, microspheres made with lower‑molecular‑weight PLGA, or those with particular branched architectures, sometimes display a simpler biphasic profile that lacks a distinct lag period. Still, the triphasic sequence is the one most commonly encountered and the one that carries the greatest clinical relevance (Figure 3).
 |
Figure 3 Triphasic drug release profile and morphological evolution of PLGA microspheres during the release process. Notes: The upper panel illustrates the characteristic triphasic drug release curve from PLGA microspheres: Phase I (initial burst release, diffusion-controlled), driven by rapid dissolution of surface-associated drug; Phase II (lag phase), characterized by slow diffusion and gradual pore formation within the hydrated polymer matrix; and Phase III (accelerated release, erosion-controlled), triggered by bulk erosion and autocatalytic degradation. The lower panel schematically depicts the corresponding morphological evolution of a PLGA microsphere across these three phases: from an intact sphere with surface pores and initial drug egress (Phase I, left), through progressive internal pore enlargement and water penetration (Phase II, center), to extensive matrix degradation with interconnected pore networks enabling complete drug release (Phase III, right). |
The first segment—Phase I, the initial burst—begins virtually at the moment of administration. As soon as the microspheres come into contact with physiological fluid, a fraction of the drug is released almost immediately. What is happening here is straightforward: drug molecules that happen to be on or very near the particle surface, or that are already sitting inside open pores at the surface, simply dissolve and diffuse away. The size of this early burst is largely set by a handful of formulation variables: the total amount of drug loaded into the microspheres, the extent of surface porosity, and the relative preference of the drug for the polymer versus the aqueous surroundings. A certain level of burst can actually be useful—it helps reach an effective plasma concentration without delay. But if the burst is too large, it can push the plasma level into a range where toxicity becomes a concern, and it also wastes drug that was supposed to be released later during the sustained‑release phase.
After the burst subsides, the release profile flattens out into a Phase II lag period. The polymer is still taking up water and swelling, and ester bonds throughout the bulk of the microsphere are steadily being cut. Even so, the matrix retains enough integrity—the chains are still long enough and tangled enough—that drug molecules trapped deep inside cannot easily find a way out. What governs the release rate during this quiet phase is the slow, steady diffusion of drug through an increasingly hydrated polymer network, accompanied by the gradual creation and enlargement of pores. Because release is so restrained during Phase II, the length of this lag period largely dictates the total duration of drug delivery. And that length is exquisitely sensitive to the characteristics of the PLGA: a higher molecular weight, a lower GA content, or a more hydrophobic drug all tend to prolong the lag.
Phase II does not last indefinitely. At some point, the cumulative damage to the polymer chains reaches a tipping point, and the release profile takes off again—this is Phase III, the accelerated release phase. The trigger is the onset of bulk erosion. As more and more ester bonds are cleaved, the polymer fragments into water‑soluble oligomers and monomers, and these acidic breakdown products begin to accumulate inside the core. The resulting drop in internal pH catalyses still faster hydrolysis, creating a self‑amplifying, autocatalytic front that eats its way from the centre of the microsphere toward the outside. The structural integrity of the matrix collapses, and at the same time a massive, interconnected network of pores and channels opens up throughout the particle. Together, these two changes allow the remaining drug to diffuse out rapidly—and, in the final stages, to be carried out by convective flow. By the time Phase III has run its course, the drug reservoir is essentially exhausted, and delivery is complete.60
Modulating Release Kinetics Through Molecular Design
The rate at which a drug is released from PLGA microspheres is not fixed; it can be tuned with considerable precision by selecting the right polymer starting materials and by choosing an appropriate manufacturing route. Earlier sections of this review have already discussed, at length, how the molecular design of PLGA—its average molecular weight and the breadth of the distribution, the molar balance between lactide and glycolide, the chemistry of the chain ends, and the glass transition temperature—exerts a strong influence on the degradation rate, and consequently on the release kinetics. What the polymer chemist controls at the molecular scale is only half the story. The fabrication method itself—emulsion‑solvent evaporation produces microspheres that are structurally quite different from those made by microfluidics—sets key microstructural features such as particle size, pore architecture, and internal morphology. These features, in turn, determine whether the release profile is dominated by diffusion or by erosion. By altering these interconnected variables in a deliberate, systematic fashion, one can tune the release behavior to fit a desired time window, be it weeks or months. It is precisely this ability to dial in the release profile that allows PLGA‑based sustained‑release microspheres to achieve their full clinical potential.
Factors Affecting Microsphere Release
How a PLGA microsphere actually behaves once it is injected into the body depends on far more than the formulation itself; the local physiological environment—enzyme activities, pH gradients, interactions with lipids—also leaves its mark on the release profile. Testing every formulation iteration through exhaustive animal studies and clinical trials is both expensive and slow, which is why a robust in vitro–in vivo correlation, or IVIVC, has become an indispensable part of formulation development.6 When an IVIVC is well validated, it allows one to make reasoned predictions of in vivo performance directly from in vitro release data, and that in turn accelerates the iterative cycle of formulation optimization toward the target product profile.61 Building a truly dependable IVIVC, however, rests on a thorough understanding of the factors that shape the in vitro release behavior itself; without that foundation, reliable clinical performance cannot be expected.1
Extending from the molecular basis of PLGA addressed in Molecular Properties of PLGA, the release behavior of microspheres is also strongly shaped by formulation-dependent parameters that determine the particle’s physical structure and the condition of the encapsulated drug. Unlike the intrinsic polymer properties, which primarily control degradation kinetics, these factors influence the diffusion path length, the evolution of porosity, and the drug’s spatial distribution within the matrix. Accordingly, this section addresses three groups of factors: formulation variables (Formulation Factors), environmental conditions (Environmental Factors), and approaches for modulating release (Strategies for Modulating Microsphere Release Behavior).
Formulation Factors
Influence of Microsphere Particle Size
Among the formulation variables that can be adjusted, particle size stands out as a particularly powerful one because it shapes drug release through its own set of mechanisms—mechanisms that are quite separate from those linked to PLGA molecular weight, a topic that has been covered in Relative Molecular Weight and Molecular Weight Distribution. The main routes by which particle size exerts its influence are straightforward: it sets the length of the path that a drug molecule must travel to leave the particle, it affects how steep the pH gradient becomes inside the microsphere as acidic degradation products accumulate, and it determines the ratio of surface area to volume, which dictates how much of the drug is held near the surface versus buried deep in the core.
Smaller microspheres, by virtue of their abbreviated diffusion path length, facilitate rapid drug egress from the polymer matrix. In this situation, drug liberation remains largely governed by diffusion, and polymer erosion contributes minimally to the overall release behavior. In contrast, larger microspheres impose an extended diffusion distance that retards both the ingress of buffering species from the external release medium and the egress of acidic degradation products. This mass transport limitation results in a more pronounced intraparticulate pH drop within the core of larger microspheres, which, via the autocatalytic nature of PLGA hydrolysis, establishes a heterogeneous degradation profile wherein the core degrades more rapidly than the periphery.40,62
Beyond the average size, the breadth of the size distribution matters enormously. This spread is usually captured by a single number—the span, calculated as (D90 − D10) / D50—and it has a direct bearing on how uniformly the microspheres release their payload and on how consistently different batches perform. When the span is large, the microsphere population is a mixed bag: different particles degrade and release drug at very different rates. Consequently, the overall release profile becomes less predictable, and the initial burst tends to be larger and more variable. Tight manufacturing control that keeps the span narrow is therefore not a cosmetic detail; it is what ultimately delivers the kind of predictable in‑vivo performance that regulators and clinicians expect.55
It is also worth noting that the way particle size shapes the release profile can itself be shaped by the choice of manufacturing method. One particularly instructive demonstration comes from work published in 2025 on the continuous fabrication of long‑acting PLGA microspheres using an automated microfluidic platform. In that study, microspheres that had been produced with an exceptionally tight particle‑size distribution gave release profiles that were strikingly reproducible from batch to batch—a clear indication that precision in manufacturing and uniformity in particle size are two sides of the same coin when the goal is consistent therapeutic performance.34
Influence of Microsphere Porosity
Section 4.2 already described porosity as a conduit for drug diffusion, but porosity is much more than a passive structural feature—it is a formulation variable that can be deliberately tuned. Through the choice of solvent, the adjustment of processing parameters, or the deliberate addition of porogens, one can steer the pore architecture of a microsphere in a direction that produces a particular, pre‑defined release pattern.
On the face of it, a more porous microsphere will let water in faster and give drug molecules more surface area from which to diffuse, so the release speeds up and the early burst becomes larger. That may sound like a drawback, but porosity is very much a double‑edged sword. An interconnected pore network keeps the drug moving through the matrix without pause—essentially erasing the lag phase. It can also raise the total amount of drug that can be loaded, and because the acidic waste products of hydrolysis are able to escape through the same channels, the autocatalytic acceleration that plagues many PLGA systems is partly relieved. When the pores are made large enough to allow cells to migrate inside, the same design principles open the door to tissue‑engineering applications.
The key to the formulation of scientists is to balance these effects. For instance, whereas spray-dried particles derived from two-fluid nozzles inherently develop a surface-enriched PLGA layer prone to pore formation (Spray Drying Method), careful optimization of the spraying conditions can steer the morphology away from a burst-dominated profile if required. This tunability makes porosity one of the most powerful levers for tailoring release kinetics without altering the PLGA chemistry itself.27,55
Influence of Encapsulated Drug Properties
The nature of the drug being encapsulated—its preference for water or oil, and its ionisation behaviour—leaves a clear imprint on both the architecture of the microspheres and the way they release their cargo. Hydrophilic compounds are almost always processed through a water‑in‑oil‑in‑water (W/O/W) double‑emulsion route, and during the period when the droplets are hardening, these drugs have a strong tendency to escape from the organic phase into the surrounding aqueous medium. As a result, less of the drug is retained, loading efficiency drops, and the initial burst tends to be proportionally larger. Lipophilic drugs, by contrast, are typically handled by a simpler oil‑in‑water (O/W) single‑emulsion method. They sit comfortably inside the hydrophobic PLGA matrix and show little inclination to migrate into the external water phase, which means that more of the drug stays where it was placed. The consequence is an altogether more efficient encapsulation and a release profile that is smoother and more sustained over time. In short, whether a drug is hydrophilic or hydrophobic fundamentally determines which emulsion strategy is workable and how the release kinetics will unfold.
A further layer of complexity comes from the fact that the ionisation state of the drug is not fixed—it depends on the local pH. Work on the aqueous remote loading of leuprolide, a basic peptide, into pre‑formed PLGA‑COOH microspheres has shown that the peptide is taken up into the polymer matrix according to quasi‑equilibrium binding isotherms. In practical terms, how much drug can be loaded is governed by the strength and capacity of binding, the amount of water the polymer holds, and the ratio of peptide to polymer.63 When the drug carries basic amine groups—secondary or tertiary amines, for instance—raising the pH tends to make the initial burst larger and cuts the total release time shorter. The reason is that the balance of electrical forces between the drug and the polymer shifts, and at the same time the pH inside the particles changes as the polymer degrades, further altering the microenvironment within which the drug is held.
Environmental Factors
Influence of pH
As detailed in Mechanistic Pathways Governing Drug Release and Temporal Evolution of Release: The Triphasic Profile, the accumulation of acidic degradation products within PLGA microspheres leads to an autocatalytic drop in intraparticulate pH, which accelerates hydrolysis and contributes to the characteristic triphasic release profile.40 In addition to this well‑established self‑amplifying effect, the external pH also influences the chemical pathway of ester bond cleavage. Around neutrality or under mildly acidic conditions (pH 4–8), ester bonds undergo random scission along the polymer backbone, producing oligomeric fragments that further degrade to monomers. When the pH rises above approximately 10, chain‑end cleavage becomes the predominant mechanism, with terminal ester bonds hydrolyzed preferentially. Although the hydrolysis rate generally increases with pH, an anomalous acceleration also occurs at very low pH due to matrix embrittlement.64
Influence of Temperature
Temperature plays a powerful role in setting the pace of both polymer degradation and drug diffusion, which is why release studies are almost always conducted at 37°C—the temperature that best reproduces the conditions the microspheres will encounter once they are placed in the body. When the temperature is raised, two things happen simultaneously: the hydrolytic breakdown of PLGA speeds up, and the drug molecules trapped inside the matrix become more mobile. The result is a faster release and, in many cases, a slight increase in the total fraction of drug that ultimately leaves the particles. This sensitivity to temperature has a practical payoff. By applying the Arrhenius equation, researchers have been able to run release tests at elevated temperatures and use the data to estimate how the same formulation will behave in real time at 37°C. The temperature dependence that emerges from these accelerated experiments therefore offers a useful shortcut—it allows one to make quicker predictions about real-time release without waiting for weeks or months for the full profile to unfold.65 However, this accelerated approach must be interpreted with caution, as it can alter the balance between diffusion and degradation—particularly near the polymer’s glass transition temperature (Tg).
Glass Transition Temperature (Tg) has already discussed in detail why the connection between temperature and the glass transition temperature (Tg) of PLGA is mechanistically so important. To summarise, once the external temperature climbs to Tg or above it, the polymer moves out of a hard, glass-like state and enters a softer, more rubber-like phase. This conversion frees the polymer chains to move much more readily and considerably expands the free volume inside the matrix, both of which facilitate the outward diffusion of drug molecules and the inward penetration of water.51
Accelerated release testing at elevated temperature is a convenient way to speed up formulation screening, and it certainly has a practical place in early development. The method does have a well‑known shortcoming; however, the initial burst phase recorded under accelerated conditions often looks quite different from that observed under physiological conditions. The reason is straightforward—at elevated temperature, the balance between diffusion and degradation shifts, so the interplay that shapes the early part of the release curve is altered in ways that are hard to correct for. Because of this, when the specific goal is to understand, and ultimately to control, the burst release phase, there is still no substitute for running the experiment at 37°C. At that temperature, the data reflect more faithfully what will happen in the body, and the conclusions drawn from them are simply more reliable.
Influence of Enzymes
If one wants to understand why drug release from a PLGA microsphere in a living animal often looks so different from what is seen in a simple buffer, enzyme activity is one of the first places to look. The exact way in which enzymes go about accelerating PLGA degradation has not yet been completely worked out, but the experimental evidence that has accumulated makes it very hard to dismiss their importance: a variety of hydrolytic enzymes can clearly cut the ester bonds that keep the polymer backbone intact. One particularly thorough study put a series of PLGA polyesters in contact with 22 different commercially available hydrolytic enzymes—covering esterases, lipases, and proteases—and followed the release of glycolic acid with a sensitive colorimetric method. What stood out was the potency of several lipases. The enzymes that performed best—lipases isolated from Candida antarctica, Candida cylindracea, Candida rugosa, Mucor miehei, Rhizopus arrhizus, and porcine pancreas, along with the esterase from M. miehei—cleaved ester bonds up to 25 times faster than the background rate measured in enzyme‑free controls.66
What was particularly striking was the behaviour of the C. antarctica lipase: the faster the degradation, the higher the glycolic acid content of the copolymer—a clear sign that this enzyme has a strong preference for glycolide-rich sequences. The esterase from M. miehei and the lipase from R. arrhizus behaved quite differently; their activity seemed almost indifferent to the fine structural details of the PLGA chain. Looking beyond individual enzymes, a broader review of biodegradable polymer degradation has reinforced the view that, inside the body, enzymatic attack is not a minor side route—it sits alongside simple hydrolysis and oxidative breakdown as one of the principal pathways by which these polymers are eliminated.67
What these findings tell us is that the way we routinely measure drug release in the laboratory needs to be interpreted with some caution—and they have a direct bearing on how reliable an IVIVC can be. A standard in vitro release test uses a simple buffer, but the absence of enzymes from that buffer means that the degradation rate recorded in the test may well be slower than what actually occurs in the body. The discrepancy is likely to be most pronounced for formulations that are injected subcutaneously or intramuscularly, simply because those tissues are rich in lipase activity. In other words, if the in vitro method does not account for the enzymatic contribution, it will tend to give release rates that are systematically on the low side, and any IVIVC built on those data will carry that bias forward.
Strategies for Modulating Microsphere Release Behavior
Once a formulator has tuned the usual formulation parameters and accounted for the major environmental variables, there comes a point where those levers alone are no longer enough to dial in a specific release pattern. It is at this stage that a different class of approaches comes into play—deliberate, often elegant interventions that go beyond simple parameter adjustment and are designed to reshape the release profile in ways that match a particular clinical demand.
Neutralisation of autocatalytic acidity: A simple way to blunt the autocatalytic cycle is to add a mildly alkaline substance—Mg(OH)2 being a common choice—directly into the PLGA matrix. The logic is straightforward: the alkaline additive mops up acidic degradation products as they appear, preventing the local pH from falling and thereby restraining the autocatalytic surge in hydrolysis. Work published in 2025 on PLGA/Mg(OH)2 composite coatings deposited on zinc alloys confirmed that the coating managed to keep the pH steady while the polymer degraded and, at the same time, regulated the release of ions; the overall outcome was a material that remained well tolerated by cells—good cytocompatibility, in other words.68 By taking the edge off the autocatalytic feedback, the strategy effectively extends the window over which the drug is released.
Hydrogel encapsulation: Surrounding PLGA microspheres with a hydrogel jacket inserts an extra diffusional obstacle between the drug and the outside world. That additional barrier can cut down the early burst considerably and stretch out the total release time.69 The idea has been borne out, for instance, by a 2025 report on injectable alginate‑microsphere/PLGA‑PEG‑PLGA composite hydrogels, which displayed sustained release and underlined how useful hydrogel‑based hybrid systems can be.70
Surface adsorption of barrier proteins: When the surface of PLGA microspheres is coated with bovine serum albumin (BSA), the protein simply sticks to the polymer and forms a thin, temporary shield. That shield slows the rapid escape of any therapeutic protein that has been loaded into the particles, and the approach has been shown to work well for formulations that deliver growth factors.71
Modification of internal‑aqueous‑phase rheology: During the production of W/O/W microspheres, the internal aqueous phase can be replaced by a gel that flows only with difficulty. Because the gel restricts how freely water‑soluble drug molecules can move while the droplets are hardening, the early burst that often plagues this type of formulation is substantially reduced.72
Chitosan coating for pH modulation: Chitosan is a naturally occurring polycation, and when it is applied as a coating to PLGA microspheres, it actively counteracts the acidic products that are liberated as the polymer degrades. This buffering effect moderates the pH drop inside the particles and, as a direct result, lengthens the period during which the drug is released.73
Osmotic adjustment of the external aqueous phase: Adding simple salts—NaCl or CaCl2, for example—to the external aqueous phase during microsphere fabrication alters the osmotic balance across the droplet interface and also modifies electrostatic interactions. The net effect is that water‑soluble drugs are less inclined to partition out of the organic phase, so more of the drug is retained and the encapsulation efficiency rises.74
Taken together, the strategies laid out above form a flexible set of design principles that allow one to tailor the release behaviour of PLGA microspheres with a fair degree of accuracy. Being able to fine‑tune release in this way matters clinically, because it means that a single formulation platform can be adapted to meet quite different therapeutic demands. A compact overview that places these approaches side by side, comparing how they work and indicating how well each one performs, is given in Table 4.
 |
Table 4 Comparative Overview of Strategies for Mitigating Burst Release from PLGA Microspheres |
Development Trends and Research Challenges of Microspheres in the Medical Field
PLGA sustained-release microspheres have long since passed the stage of being a laboratory curiosity. They are now a clinically established delivery platform, and the past few years have seen the technology branch out in several directions at once—more diverse payloads, more precise control over release, and the first serious steps toward “smart”, stimulus-responsive systems. Clinically, these microspheres have already made a real impact, perhaps most visibly in oncology, in long-acting local anaesthesia, and in vaccine delivery. For all that progress, though, a number of stubborn problems still stand between the current state of the art and the wider clinical use that the technology deserves. Achieving the kind of spatiotemporal precision in drug release that newer therapies demand remains difficult. Ensuring that every batch of microspheres performs identically—and that the particles are stable on the shelf—continues to be a manufacturing headache. Targeting specific cells or tissues, rather than relying on passive accumulation, has not yet reached the level of reliability that would make it routine. Safety questions around immunogenicity and the long-term fate of the polymer breakdown products have not gone away. And moving from a bench-scale process that works beautifully in an academic laboratory to a robust, cost-effective production line is still a major engineering undertaking. What the field now needs is not another proof-of-concept study, but a concerted push to solve these practical, translational bottlenecks.
Development Trends of PLGA Sustained-Release Microspheres in the Medical Field
From Single-Drug to Multi-Drug Delivery Systems
For a long time, the default approach has been to load a single drug into a PLGA microsphere. Yet the clinical reality is that many of the most serious illnesses—cancer, chronic inflammatory diseases, difficult-to-treat infections—rarely respond well to a single agent; they call for combinations of drugs that hit several disease-related targets at once. It is this recognition that has pushed the field toward multi‑drug PLGA microsphere systems, where two or more therapeutic agents are housed together within the same delivery platform so that they can work in concert.
A particularly instructive example comes from glaucoma research, where a single PLGA microsphere formulation was loaded with three drugs at once—ketorolac, melatonin, and latanoprost—each chosen for a distinct therapeutic purpose. Latanoprost was included to bring down intraocular pressure, melatonin to provide antioxidant protection, and ketorolac to dampen inflammation. When the formulation was optimised, it was able to sustain the release of all three agents for well over 70 days. In a chronic glaucoma model in rats, a single intravitreal injection of these tri‑loaded microspheres produced effects that were still clearly measurable 24 weeks later: the treated animals had the lowest intraocular pressure among the groups, their bipolar and retinal ganglion cells were functioning better, and the thickness of the neuroretina was visibly greater than in the untreated glaucomatous controls.75 A different demonstration of synergy comes from the combination of metformin and Y15, which were co‑delivered from a PLGA scaffold for the treatment of platinum‑resistant ovarian cancer. When the two drugs were given together, the cytotoxic effect against the resistant cells was far stronger than when either drug was used alone, and the scaffold‑based delivery consistently outperformed the equivalent free‑drug combination.76
The same co‑delivery logic has been applied to dual‑drug porous PLGA microspheres loaded with doxorubicin and paclitaxel at an optimised 2:1 ratio, a combination that has been explored for inhalation therapy against lung cancer. Here, the two chemotherapeutics act synergistically against the tumour, while the local delivery to the lungs helps keep systemic side‑effects at a minimum.77 A quite different pairing—sorafenib, which promotes ferroptosis, and venetoclax, which drives apoptosis—has been co‑encapsulated in surface‑modified PLGA nanoparticles. Against a panel of tumour cell lines, the combination triggered a stronger response than either agent alone, with a marked increase in reactive oxygen species and a measurable loss of mitochondrial membrane potential.78 What examples such as these make clear is that multi‑drug PLGA systems are not just a way to deliver two drugs at the same time; they open the door to genuinely coordinated, mechanism‑based combination therapy, where the carrier itself contributes to the therapeutic logic rather than being a passive bystander.
Emergence of Intelligent Responsive Microspheres
The convergence of intelligent materials and nanotechnology over the past few years has given rise to a new generation of PLGA microspheres that can respond—predictably and on cue—to specific physiological signals or externally applied stimuli. A recent, wide‑ranging review of stimuli‑responsive delivery systems catalogues the triggers that have been exploited so far: endogenous cues such as local pH, temperature, enzyme activity, and redox state, as well as exogenous inputs that include light, magnetic fields, and ultrasound.79 What this intelligent design ethos brings is a step‑change in spatiotemporal precision. Instead of releasing drug at a rate that was pre‑programmed when the microspheres were fabricated, these systems can adapt their release behaviour to the conditions they encounter in the body, keeping therapeutic action tightly synchronised with the physiological or pathological state of the tissue they are meant to treat.
A good illustration of what can be achieved comes from a dual‑drug system that was built to fight infection and, at the same time, speed up wound closure. The clever part of the design is that the two drugs are released on different schedules, each triggered by a different cue. Ciprofloxacin is tucked inside a zeolite imidazolate framework that is embedded in the PLGA microspheres; because the framework is acid‑sensitive, it starts releasing the antibiotic as soon as the local pH drops—exactly the sort of acidic environment that a bacterial infection creates. Meanwhile, curcumin sits directly in the PLGA matrix and comes out only slowly, providing a sustained antibacterial assist. The real elegance of the system, however, is what happens next. The microspheres also contain Fe3O4 nanoparticles, and when near‑infrared light is shone on them, those particles generate heat. The temperature climbs to the glass‑transition point of PLGA, at which point the polymer matrix becomes more permeable and the curcumin is released in a burst. In a mouse abscess model, this two‑stage strategy worked impressively well: the microspheres not only cleared bacteria but also promoted re‑epithelialisation of the wounded tissue and kept inflammatory cell infiltration under control.80
PLGA is just one member of a larger family of programmable biopolymers whose responses to physiological cues have been studied in considerable depth. A systematic review focused on smart biopolymers has drawn attention to how effectively pH‑sensitive, temperature‑sensitive, and enzyme‑triggered release mechanisms can circumvent the toxicity problems that have long dogged conventional delivery strategies.81 As the field continues to mature, there is good reason to believe that weaving together several of these stimuli‑responsive elements within a single microsphere platform will deliver a level of spatiotemporal control over drug release that is difficult to achieve with any one trigger acting alone. The real challenge is no longer whether such multi‑stimuli systems can be built—they can—but how to integrate them in a way that is robust, reproducible, and ultimately translatable to the clinic.
Targeted Delivery Through Surface Modification
The ability to direct a drug carrier to a specific tissue or cell population—and to keep it away from healthy organs—is one of the most sought-after properties in modern drug delivery. PLGA microspheres can be endowed with this kind of biological targeting by modifying their surface with ligands that recognise particular molecular signatures on the intended target cells. A detailed review covering PEGylated PLGA nanoparticles in oncology makes clear that attaching targeting ligands to the particle surface is what enables selective delivery via ligand–receptor binding, and that this recognition step, in turn, promotes cellular uptake and intracellular drug release. The PEGylation itself creates a hydrophilic corona of polyethylene glycol chains that wraps around each nanoparticle; this coating shields the particles from opsonisation and reduces recognition by the immune system. The practical result is a longer circulation half-life and a much higher chance that an injected dose will actually reach the tissue where it is needed.82
What has become clear from work over the last few years is that the targeting concept is not confined to a single ligand or a single tumour type; it is a broadly applicable strategy. One instance is the use of hyaluronic acid-modified porous PLGA microspheres to deliver doxorubicin to hepatocellular carcinoma. The particles exploit the fact that many liver cancer cells overexpress the CD44 receptor, and the hyaluronic acid on the microsphere surface docks onto that receptor, thereby concentrating the drug in the tumour.83 Other laboratories have taken a similar line with chitosan-modified porous PLGA microspheres, also loaded with doxorubicin, but this time directed against breast cancer; the chitosan layer not only provides a handle for targeting but also improves the physical stability of the particles and their uptake by cells.84 For treating glioblastoma, researchers have designed drug-loaded PLGA microparticles that act on microglial immunosuppression. This approach tackles the major obstacle presented by the blood–brain barrier in getting therapeutic agents into the central nervous system.85
The move toward active targeting marks a fundamental departure from relying on passive accumulation through the enhanced permeability and retention effect. Instead, it embraces a strategy grounded in molecular recognition. The central idea is simple: when a ligand is conjugated to the surface of a PLGA microsphere, it seeks out its complementary receptor on the target cell and pulls the particle directly to the site where the drug is needed. As the catalogue of disease‑specific biomarkers continues to grow, so too does the collection of ligands that can be grafted onto PLGA microsphere surfaces. Antibodies, peptides, aptamers, and small molecules are all available for this purpose, and the choice among them hinges on the particular receptor being targeted, the required binding affinity, and the practical constraints of manufacturing. With every new ligand‑receptor pair that is validated, the scope for designing ever more refined targeting strategies widens, opening the door to approaches that can discriminate between different tissues and even between distinct cell populations within a single tissue.
Clinical Applications in Cancer, Pain Management, and Vaccine Delivery
PLGA sustained‑release microspheres have long since made the leap from benchtop science to something that clinicians can actually prescribe, and their impact across several therapeutic fields has been real and growing. Oncology is perhaps the most visible example. Here, the microspheres have been used to deliver a range of chemotherapeutic agents, with doxorubicin and paclitaxel standing out as two of the most studied payloads. A detailed review that surveys PLGA nanoparticles in cancer therapy draws attention to the many ways in which anti‑cancer drugs can be loaded into PLGA matrices and the therapeutic gains that follow. The review covers traditional chemotherapeutics, drugs that interfere with the cell cycle, and agents that act on microtubules. It also makes the point that PLGA nanoparticles are not confined to a single targeting strategy. On the one hand, they can take advantage of passive tumour accumulation through the enhanced permeability and retention (EPR) effect; on the other, they can be actively targeted by attaching ligands that recognise specific receptors on the tumour cell surface.86
Pain management is another area where PLGA microspheres have found a clear role, this time as carriers for long‑acting local anaesthetics. In a recent study, PLGA‑PVA nanocarriers loaded with ropivacaine formed uniform spheres around 11 nm in diameter and released the drug in a sustained fashion. The particles provided prolonged pain relief and proved to be remarkably biocompatible: even when the concentration was pushed as high as 1000 μg/mL, more than 80% of the cells remained viable.87 An earlier piece of work looked at an injectable, PLGA‑coated form of ropivacaine and found that it kept releasing the drug for at least six days in the laboratory. In animals, a single perisciatic nerve injection of that formulation eased pain caused by an incision and dampened neuropathic pain for a full week, with no signs of inflammation or tissue irritation at the injection site.88
Vaccine delivery represents yet another application where PLGA microspheres have shown considerable promise. The underlying idea is that, by controlling how quickly an antigen is released, it is possible to shape the quality and strength of the immune response that follows, ultimately making the vaccine more effective. A recent study that encapsulated nano‑engineered filamentous bacteriophage‑based vaccines inside PLGA microparticles found that the particles preserved both the structure and the immunological activity of the phage. The encapsulated vaccine triggered the activation of dendritic cells, helped antigens to be presented more efficiently, and stimulated specific T‑cell responses. What was also striking was that simply wrapping the phage in PLGA kept it stable for longer—even when the microparticles were stored at room temperature—and that the release could be tuned with the help of computer‑based predictive models.21
A comprehensive review devoted to antigen delivery systems built on PLGA microspheres for single‑step immunization draws attention to one particularly attractive feature: the ability to release antigen in a pulsatile manner. In effect, a single injection can behave more like a series of booster shots, because the microspheres are programmed to deliver the antigen in discrete bursts rather than in one continuous trickle. Achieving this, the review stresses, depends critically on three factors—choosing the right polymer, adjusting the particle size, and designing an appropriate immunization protocol.89 A further example comes from work in which recombinant SAG1 antigen was co‑delivered alongside two TLR ligands, monophosphoryl lipid A and imiquimod, using PLGA nanoparticles. The resulting immune response was not only stronger overall but also skewed toward a Th1‑biased profile, with elevations in both humoral and cellular arms. In a murine model of toxoplasmosis, this strategy led to a pronounced reduction in the burden of brain cysts.90 (Table 5 brings together a collection of representative advanced drug delivery systems that have been built on PLGA microspheres)
 |
Table 5 Representative Examples of Advanced PLGA Microsphere-Based Drug Delivery Systems |
Beyond preclinical models, PLGA microsphere-based therapies have been validated in human clinical studies across multiple therapeutic areas. The 1-month Lupron Depot® (leuprolide acetate-loaded PLGA microspheres), approved by the US Food and Drug Administration (FDA) since 1989, remains a gold standard for long-acting hormone therapy in prostate cancer, with a single intramuscular injection sustaining therapeutic drug levels for 30 days.93 A Phase III randomized, double-blind, multicenter trial (NCT00297388) demonstrated that Risperdal Consta®—risperidone-loaded PLGA microspheres administered biweekly—significantly reduced relapse rates in adult patients with schizophrenia or schizoaffective disorder.94 Furthermore, a Phase II clinical trial (NCT06199895) is currently evaluating the efficacy and safety of paclitaxel polymeric micelles for injection in patients with taxanes-resistant pancreatic adenocarcinoma, cholangiocarcinoma, lung cancer, gastric cancer, esophageal carcinoma, or breast cancer, with the primary endpoint being objective response rate.92 These registered clinical studies underscore the established and emerging clinical utility of PLGA-based sustained-release systems, providing valuable benchmarks for next-generation formulation development.
Research Challenges of PLGA Sustained-Release Microspheres
For all the progress that has been made, PLGA microsphere technology still faces a set of deeply rooted challenges that stand between laboratory demonstrations and the kind of robust, scalable products that can succeed in the clinic and in the marketplace. Perhaps the most fundamental of these is the difficulty of achieving truly precise control over drug release—being able to programme not just the average rate but the entire temporal profile, and to do so reproducibly. Closely related is the problem of consistency: ensuring that every batch of microspheres performs identically, and that the particles remain stable on the shelf for a pharmaceutically acceptable period. Targeting efficiency in the complex and dynamic environment of a living organism remains another area where expectations have often outpaced reality; the gap between elegant in vitro demonstrations and reliable in‑vivo performance is still wide. Questions around immunogenicity and the long‑term safety of microsphere‑based therapies have not gone away, particularly for chronic indications that require repeated dosing. And underpinning all of these is the formidable engineering task of scaling up a laboratory process into a cost‑effective, GMP‑compliant manufacturing line without losing the precise control over particle properties that made the formulation successful in the first place.
Precision of Drug Release Control
Being able to dictate with confidence exactly when and how fast a drug is released from a PLGA microsphere remains one of the most stubborn challenges in the field. Most-established formulations produce release curves that follow simple first‑order or zero‑order kinetics—the drug comes out at a rate that is set largely by the initial design of the particle and then simply decays, or it trickles out at a roughly constant speed. Neither pattern is particularly good at reproducing the kind of pulsatile or on‑demand delivery that would genuinely mirror the body’s own physiological rhythms, where hormone levels, for instance, rise and fall on a circadian cycle, or where a drug is needed only when a biomarker crosses a certain threshold.95 There have been steps forward. PULSED microparticle technology is one example that has shown it is possible to stretch the release window from as little as 10 days out to 5 weeks by carefully adjusting the PLGA formulation, and to do so with the help of 3D printing and non‑contact thermal sealing. But these are still early days. The leap from controlling the overall duration of release to controlling where, when, and how much drug is released at the level of a single tissue or even a single cell is a leap of a different order of magnitude, and it is this spatiotemporal dimension that remains the real frontier.
The capacity to release drugs in an on-demand fashion when triggered by external cues marks a meaningful advance toward truly individualized treatment plans. Light-responsive PLGA microparticles have been developed for on-demand vancomycin release, wherein chitosan-coated near-infrared-responsive microparticles demonstrate precise, triggered antibiotic delivery with enhanced antibacterial performance.96 Still, moving these externally controlled platforms into real-world clinical settings comes with hurdles that must be overcome, among them the limited penetration depth of the triggering signal into tissues, the difficulty of ensuring consistent triggering responses from one administration to the next, and the long-term safety profile of the stimulus-sensitive components themselves.
Stability and Batch-to-Batch Consistency
The stability and batch-to-batch consistency of microspheres are critical challenges for the large-scale production and clinical application of PLGA sustained-release microsphere technology. Fabricating PLGA microspheres usually relies on a range of process variables—polymer composition, the choice of solvent, crosslinking conditions, and emulsification settings—and even small deviations in any of these can profoundly alter the performance features of the final microspheres.
Storage stability is a practical concern that can easily be overlooked when a formulation is still at the laboratory stage. Over weeks or months on the shelf, PLGA microspheres can gradually lose their function: the polymer may begin to degrade, particles can stick together into aggregates, and drug that was meant to stay trapped can leak out prematurely. Any one of these processes can erode the therapeutic performance that was carefully built into the formulation. When production moves to a commercial scale, the bar is raised even higher. Regulators rightly demand that every batch leaving the production line behaves in the same way—the same size distribution, the same drug loading, the same release profile—because clinical reliability depends on that consistency.
It is for these reasons that Quality by Design (QbD) thinking has made its way into PLGA microsphere development. Rather than tweaking formulations by trial and error until something looks acceptable, QbD provides a structured way to map out, from the very beginning, how each formulation and process variable influences the critical quality attributes of the final product. A good illustration of what QbD can deliver comes from recent work on rapamycin‑loaded PLGA microspheres, where a QbD‑guided approach succeeded in achieving sustained drug release while keeping product characteristics consistent from batch to batch.97 Another study, this one focusing on PLGA nano‑micelles prepared with a microfluidizer, showed that the same QbD framework allowed researchers to exert precise control over particle attributes and to produce stable formulations with a level of reproducibility that would be difficult to achieve without such a systematic approach.98
Dynamic light scattering is widely used to track key parameters like particle size and the polydispersity index. Such data allow manufacturers to monitor and ensure lot-to-lot uniformity. For applications requiring specific scaffold architectures, complementary techniques to assess porosity and surface area are also essential. Nonetheless, achieving the stringent consistency requirements mandated by regulatory agencies remains a persistent challenge that necessitates continued innovation in process analytical technology and manufacturing control strategies.
Targeting Efficiency and Biological Barriers
The targeting capability of microspheres represents a critical feature of PLGA sustained-release microsphere technology, enabling increased drug concentration at the lesion site while minimizing collateral damage to normal tissues. However, achieving efficient and specific targeted delivery continues to confront numerous biological and engineering challenges. Active targeting strategies typically rely on ligand-receptor binding, yet factors such as ligand density, receptor expression levels, and receptor turnover rates can substantially influence targeting efficiency.
The body throws up a whole series of barriers that make it extraordinarily difficult to get microspheres from the injection site to the tissue where they are actually needed. Perhaps the most celebrated—and for drug delivery scientists, the most frustrating—of these barriers is the blood–brain barrier. The reason why so many promising therapies for central nervous system disorders have fallen short is because the barrier is remarkably effective at keeping particulates out of the brain. Some of the more encouraging results in this area have come from work on PEGylated PLGA nanoparticles prepared from nano‑emulsion templates. The PEG coating does two useful things: it stops the particles from clumping together, and it helps them evade recognition by the immune system, which keeps them circulating in the blood for longer. The net result is that more of the dose reaches the brain side of the barrier, and once there, the particles can interact with the target cells.85 Overcoming the BBB is only half the challenge; once inside the brain, particles must also navigate the tumor microenvironment, including immunosuppressive cells such as microglia. As earlier sections of this review have already touched upon, glioblastoma researchers have also taken a different tack. Knowing that these tumours create a profoundly suppressive immune environment that is reinforced by microglial cells, they have designed PLGA microparticles that specifically home in on those microglia. In doing so, the particles address two of the biggest obstacles to treating glioblastoma at the same time: getting across the blood–brain barrier and dialling back the local immunosuppression that allows the tumour to thrive.
Keeping a microsphere on course after it has been injected is a much harder problem than decorating its surface with a targeting ligand. Once the particles enter the bloodstream, they are immediately coated by a layer of serum proteins—the so‑called protein corona—and that layer can largely dictate where the particles end up, regardless of what ligand was originally attached. If the corona shields the ligand from view, the targeting strategy falls apart; worse, the adsorbed proteins can sometimes steer the particles toward cell types that were never meant to be reached. This problem has not gone unnoticed. A great deal of current research is focused on understanding exactly which proteins make up the corona under different physiological conditions, and on learning how to engineer particle surfaces so that the corona that forms is a predictable one—ideally, one that leaves the targeting function intact. Solving this is widely seen as one of the keys that will unlock the full clinical potential of targeted PLGA microsphere therapies.
Immunogenicity and Long-Term Safety
PLGA is generally regarded as a safe polymer, and for good reason: the lactic acid and glycolic acid monomers that it releases as it degrades are endogenous molecules that the body knows how to handle through the tricarboxylic acid cycle. Yet safety is never just about the identity of the breakdown products. When those acidic monomers are generated inside a confined microsphere core, they can accumulate locally in concentrations that would never be reached if they were free to diffuse away. The resulting pH drop can irritate the surrounding tissue and, in some circumstances, alter the behaviour of nearby immune cells. This is why the immunogenicity of PLGA microspheres needs to be looked at carefully, from the earliest stages of preclinical development all the way through to clinical evaluation. The question is not simply whether PLGA itself is safe—it is—but whether the micro‑environment that a degrading microsphere creates over its lifetime could, under certain conditions, provoke an unwanted response.
The idea of wrapping PLGA nanoparticles in a coating borrowed from the body’s own cells has attracted considerable interest, and a recent study that put this concept to the test offers a clear view of what the approach can achieve. The study looked at PLGA nanoparticles that were loaded with polysaccharides derived from Hericium erinaceus and then coated with macrophage membrane. The first thing to note is that the coating is not just a passive shell; it actively changes how the particles interact with immune cells. Macrophages exposed to the coated nanoparticles proliferated more vigorously, produced nitric oxide in a dose‑dependent manner, and took up the particles more efficiently than they did with uncoated controls. When the same nanoparticles were administered to animals, the effects were equally encouraging. Spleen and liver indices improved, splenic lymphocytes multiplied, and macrophage phagocytic activity rose—all signs of a robust and coordinated immune activation. What the coating also did was to turn the nanoparticles into a sustained‑release depot for the antigen. Because the antigen was released gradually rather than in a single pulse, IgG titres climbed higher and the humoral arm of the immune response was demonstrably strengthened.99
Whether a PLGA particle is small enough to be eaten by a macrophage or too large to be engulfed has a lot to do with how the immune system responds to it, and this relationship has been studied quite systematically. One set of experiments that looked at what happens after intra-articular injection painted a particularly clear picture. Particles that averaged 265 nm in diameter were small enough to be taken up by the macrophages that had infiltrated the synovium, and once internalised, they were carried into the deeper layers of the tissue. Particles that were a hundred times larger, with a mean diameter of 26.5 µm, were simply too big for phagocytosis; instead of being cleared, they became covered in a layer of granulation tissue that contained multinucleated giant cells. What is striking is that despite these very different fates, neither size provoked much of an inflammatory response. The local reaction was minimal in both cases, which suggests that PLGA particles, whether nano‑sized or micro‑sized, can deliver drugs into a joint space without setting off a significant immune reaction.100
The immune system does not look at a PLGA microsphere and see an inert object. Surface properties that might seem like minor formulation details—charge, hydrophobicity, roughness—are exactly the kinds of cues that immune cells have evolved to recognise, and they can set off clearance mechanisms that remove the particles from the circulation long before they have had a chance to do their job. One of the most detailed mechanistic studies to examine this problem focused on how the protein corona that forms around PLGA nanoparticles is reshaped by PEGylation. The work set out to connect three things: the surface chemistry of the particles, the proteomic fingerprint that results when those particles encounter serum, and the efficiency with which bone‑marrow‑derived macrophages then internalise them. What the data showed was that the density of PEG on the surface is not a trivial variable. By shifting the balance between opsonins, which tag particles for clearance, and dysopsonins, which help them evade it, the PEG layer effectively tunes how visible the nanoparticles are to macrophages.101 Knowing this, one can begin to think about surface design not as a trial‑and‑error exercise but as a molecular‑level conversation with the immune system, where the goal is to engineer a surface that keeps the particle below the immunological radar without compromising its therapeutic function.
Scalable Manufacturing and Industrial Translation
Laboratory-scale microsphere preparation methods are often difficult to directly translate to industrial-scale production. When the emulsion solvent evaporation method is moved to larger production scales, non-uniform stirrer shear forces, the introduction of air bubbles, and uneven temperature gradients often arise. These changes can widen the particle size distribution, alter the surface features of the microspheres, and lower drug loading efficiency.
Residual solvent is one of those practical headaches that can derail the scale‑up of a PLGA microsphere process even when the formulation itself looks perfect at the laboratory bench. A study that addressed this problem head‑on looked at twelve different formulations, systematically varying the conditions of solvent extraction, washing, and solvent evaporation to work out which process parameters really matter. What the study found was that a relatively simple change—keeping the microspheres in the ethanolic solution for longer during the second extraction step—made a surprisingly large difference. Ethyl acetate levels dropped by 93%, benzyl alcohol by 60%, and the microspheres that emerged still had the spherical shape, the geometric diameter of 75.8 μm, and the drug loading of 33.7% that the formulation was designed to deliver.5 The practical lesson is worth keeping in mind: modest adjustments to the work‑up steps can cut residual solvent levels dramatically without sacrificing the particle properties that matter, and that is precisely the kind of actionable information that helps turn a laboratory recipe into a manufacturable product.
Continuous‑flow microfluidics has been gaining ground as a serious alternative to conventional batch manufacturing, largely because it promises to deliver the kind of particle‑by‑particle consistency that is so difficult to achieve in a stirred tank. A recent paper described a process built around a split‑and‑recombine microfluidic chip, with the entire platform running under automated control. The mixing performance was impressive, reaching an efficiency of 91.87%, and the volumetric accuracy of the fluid handling came in at 99.07%. Under these conditions, the platform turned out PLGA microspheres that spanned a size range from 9.90 to 85.80 μm, and what stood out was how tight the size distribution was: the coefficient of variation was just 5.09%, which is remarkably narrow by the standards of most microsphere production methods. What these numbers point to is a platform that can combine throughput with reproducibility in a way that is genuinely attractive for pharmaceutical manufacturing, where the demand for consistent product quality is absolute.28
Another promising direction for scaling up production makes use of thermal control in a continuous stirred‑tank reactor cascade. The underlying idea is that, by rapidly solidifying nanoemulsion droplets under controlled thermal conditions, one can trap a larger fraction of the drug inside the nanosphere core. This produces a core‑loaded architecture that naturally lends itself to high drug loading and sustained release. A striking practical advantage is the speed of the process: the total synthesis time is cut from several hours down to around 40 minutes, which represents a genuine gain in efficiency for engineering nanospheres with improved release kinetics.102
Turning a laboratory‑scale PLGA microsphere formulation into a product that can be manufactured commercially is not simply a matter of making the equipment larger. It requires building what might be called an industrial knowledge framework—a comprehensive map that links the critical process parameters, the key material attributes, and the quality standards that the final product must satisfy. A systematic review that examined the feasibility of scaling up PLGA microsphere technologies made a sobering point: the gap between what academic laboratories can achieve and what pharmaceutical production demands is still wide. The review argued that bridging that gap will depend on a careful, step‑by‑step effort to recognise where academic research and industrial development diverge, and where they can be brought into closer alignment, so that translation becomes a more predictable and efficient process.103
Industrial translation also brings with it a set of practical demands that are easily overlooked when the focus is on formulation science. The production environment must remain sterile throughout the campaign, the cost of goods must be kept within a commercially viable range, and every step of the manufacturing chain must comply with Good Manufacturing Practice requirements. These are not trivial add‑ons; they are, in many respects, what determines whether a promising microsphere product ever reaches the patients for whom it was designed. There is reason to be optimistic that these challenges can be met. Continuous manufacturing platforms are beginning to bring a level of process control that is difficult to achieve in batch mode. Advanced process analytical technologies are giving operators a real‑time window into what is happening inside the reactor, rather than forcing them to rely on post‑hoc quality testing. And systematic Quality by Design frameworks are helping to ensure that quality is built into the product from the very beginning, rather than being inspected in at the end. It is the integration of these three elements that many in the field see as the most realistic path toward overcoming the manufacturing obstacles that have, for so long, kept PLGA microsphere‑based therapeutics from achieving their full clinical reach.
Future Perspectives
If one looks at the trajectory that PLGA microsphere technology has followed over the past decade, it is not difficult to see where the field is heading. The trends that are likely to drive the next wave of innovation are already visible, and they point in a direction that is intellectually exciting but also demanding. Perhaps the most transformative of these is the prospect of bringing together multi‑drug delivery and intelligent, stimuli‑responsive release within a single platform. The vision is a microsphere that does not simply release its payload at a pre‑programmed rate, but one that senses the local disease environment and adapts its release behaviour accordingly. Allied to this is the push toward more sophisticated targeting. As the catalogue of disease‑specific biomarkers grows, and as our understanding of the biological barriers that stand between an injected particle and its intended destination deepens, it becomes possible to design microspheres that home in on diseased tissue with a precision that was simply not attainable a generation ago. The third major driver will be manufacturing innovation. Continuous processing, real‑time process analytical technologies, and the maturation of Quality by Design methodologies are together beginning to close the gap between what can be made in an academic laboratory and what can be produced at scale under the rigorous conditions that regulatory authorities rightly demand. None of this, however, will happen in a vacuum. Polymer chemists, pharmaceutical engineers, immunologists, and clinicians will need to work together far more closely than has been the norm. And the regulatory framework that governs the approval of these complex, multicomponent drug‑device combinations will need to evolve in step with the science, so that appropriate quality standards are in place before the products reach the clinic. The challenges are formidable, but the direction of travel is clear, and the foundations on which the next generation of PLGA‑based therapies will be built are already being laid.
Conclusion
PLGA sustained-release microspheres have earned their place as a platform that clinicians now rely on, particularly in oncology, in the management of chronic pain, and in vaccine delivery. The aim of this review has been to step back and examine, in a systematic way, what determines how these microspheres perform—starting from the choice of materials and the method of fabrication, all the way through to the molecular events that govern how a drug is released.
Among the various ways to make PLGA microspheres, emulsion‑solvent evaporation has been the workhorse of industrial production for decades, largely because it is straightforward to operate and relatively easy to scale. The more recent arrival of membrane emulsification and microfluidic technologies has, however, opened up possibilities that go well beyond what traditional methods can offer. These newer platforms give the formulator an unprecedented degree of command over particle size, monodispersity, and internal architecture—qualities that translate directly into more predictable and reproducible drug release. The obstacles that still stand in the way of their wider adoption are well known: throughput remains lower, and the capital cost of equipment is higher. But as these barriers are gradually lowered, it is not difficult to foresee a future in which precision fabrication becomes the default route for manufacturing the next generation of PLGA microspheres, especially for high‑value biologics where the cost of the active pharmaceutical ingredient justifies a more sophisticated delivery system.
What has also become clear from the body of work reviewed here is that the molecular character of PLGA itself is not a fixed given; it is a set of variables that can be deliberately tuned. The distribution of molecular weights, the balance between lactide and glycolide, and the chemistry of the terminal groups all leave a clear and largely predictable signature on the rate at which the polymer degrades and on the shape of the release curve that results. Once these structure–property relationships are understood, they become powerful tools. They allow the formulation scientist to move beyond empirical optimisation and toward the rational design of polymer matrices that can sustain drug release for weeks or months, depending on what the clinical situation demands. The triphasic release mechanism that is so characteristic of PLGA microspheres—diffusion, then a lag, then a surge driven by erosion and autocatalysis—is not merely an academic observation. It provides a mechanistic framework within which one can diagnose why a particular formulation is performing the way it is, and then intervene to correct problems such as an unacceptably large initial burst.
For all that has been achieved, the field is not standing still, nor should it. Challenges that have been recognised for years—building robust in vitro–in vivo correlations, delivering consistent product quality at production scale, and demonstrating long‑term safety and low immunogenicity in chronic indications—continue to require sustained attention. At the same time, the direction of travel is unmistakable. The convergence of multi‑drug delivery, stimulus‑triggered release, and genuinely active targeting is pushing PLGA microsphere technology toward a future in which therapeutic interventions can be tailored to the needs of individual patients with a precision that was difficult to imagine when the first PLGA products were approved. Whether that future materialises will depend, in no small part, on how effectively polymer chemistry, intelligent formulation design, and continuous manufacturing can be brought together. The foundations for that integration are already being laid, and the progress reviewed here suggests that the next generation of PLGA‑based long‑acting injectables will be substantially more capable than the one that came before.
Funding
This study was supported by the Opening Foundation of Hubei Key Laboratory of Tumor Microenvironment and Immunotherapy (2024ZLKF2-36).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siepmann J, Siepmann F. Release mechanisms of PLGA-based drug delivery systems: a review. Int J Pharm X. 2025;10:100440. doi:10.1016/j.ijpx.2025.100440
2. Kakavandi S, Zare I, Esmaeilnejad N, et al. PLGA-integrated micro/nanoparticles for cargo delivery and nanomedicine of lung cancer. Int J Pharm. 2026;687:126339. doi:10.1016/j.ijpharm.2025.126339
3. Lee JW, Park JH, Yu GW, et al. Sustained-release intra-articular drug delivery: PLGA systems in clinical context and evolving strategies. Pharmaceutics. 2025;17(10):1350. doi:10.3390/pharmaceutics17101350
4. Qi F, Wu J, Li H, et al. Recent research and development of PLGA/PLA microspheres/nanoparticles: a review in scientific and industrial aspects. Front Chem Sci Eng. 2019;13(1):14–32. doi:10.1007/s11705-018-1729-4
5. Khanmohammadi S, Sadeghi S, Ansari S, et al. Reducing residual solvent levels in poly (D, L-lactic-co-glycolic acid) microspheres: a roadmap for scalable industrial production. Drug Dev Ind Pharm;2025. 1–10. doi:10.1080/03639045.2025.2514214
6. Wang Y, Otte A, Park H, et al. In vitro-in vivo correlation (IVIVC) development for long-acting injectable drug products based on poly(lactide-co-glycolide). J Controlled Release. 2025;377:186–196. doi:10.1016/j.jconrel.2024.11.021
7. Stevanović MM, Qian K, Huang L, et al. PLGA-based co-delivery nanoformulations: overview, strategies, and recent advances. Pharmaceutics. 2025;17(12):1613. doi:10.3390/pharmaceutics17121613
8. Danhier F, Ansorena E, Silva JM, et al. PLGA-based nanoparticles: an overview of biomedical applications. J Controlled Release. 2012;161(2):505–522. doi:10.1016/j.jconrel.2012.01.043
9. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377
10. Wang J, Xiao E, Zhang L, et al. Research progress of poly(lactic-co-glycolic acid). Synthetic Fiber China. 2023;52(5):1–5.
11. Grillo A, Rusconi Y, D’alterio MC, et al. Ring opening polymerization of six- and eight-membered racemic cyclic esters for biodegradable materials. Int J Mol Sci. 2024;25(3):1647. doi:10.3390/ijms25031647
12. Gentile P, Chiono V, Carmagnola I, et al. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int J Mol Sci. 2014;15(3):3640–3659. doi:10.3390/ijms15033640
13. Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Poly Ed. 2006;17(3):247–289. doi:10.1163/156856206775997322
14. Huang YT, Huang HY, Cheng JL, et al. A regio- and stereoselective ring-opening polymerization approach to isotactic alternating Poly(lactic-co-glycolic acid) with stereocomplexation. Angewandte Chemie. 2025;64(9):e202422147. doi:10.1002/anie.202422147
15. Li M, Rouaud O, Poncelet D. Microencapsulation by solvent evaporation: state of the art for process engineering approaches. Int J Pharm. 2008;363(1–2):26–39. doi:10.1016/j.ijpharm.2008.07.018
16. Alvarez I, Gutiérrez C, Gracia I, et al. Synthesis of gemcitabine-loaded PLGA microparticles with green solvents. ACS omega. 2025;10(30):33946–33958. doi:10.1021/acsomega.5c06385
17. Chereddy KK, Vandermeulen G, Préat V. PLGA based drug delivery systems: promising carriers for wound healing activity. Wound Repair Regeneration. 2016;24(2):223–236. doi:10.1111/wrr.12404
18. Schiller S, Hanefeld A, Schneider M, et al. Towards a continuous manufacturing process of protein-loaded polymeric nanoparticle powders. AAPS Pharm Sci Tech. 2020;21(7):269. doi:10.1208/s12249-020-01814-w
19. Su Y, Zhang B, Sun R, et al. PLGA-based biodegradable microspheres in drug delivery: recent advances in research and application. Drug Delivery. 2021;28(1):1397–1418. doi:10.1080/10717544.2021.1938756
20. Kakkar V, Wani S, Gautam SP, et al. Role of microspheres in novel drug delivery systems: preparation methods and applications. Int J Curr Pharmaceutical Res. 2020;12(3):10–15. doi:10.22159/ijcpr.2020v12i3.38326
21. Jamaledin R, Sartorius R, Di Natale C, et al. PLGA microparticle formulations for tunable delivery of a nano-engineered filamentous bacteriophage-based vaccine: in vitro and in silico-supported approach. J Nanostruct Chem;2023. 1–16. doi:10.1007/s40097-022-00519-9
22. Guo Y, Liu L, Zheng M, et al. Formulation optimization and evaluation of leuprolide acetate-loaded PLGA microspheres. Cent South Pharm. 2023;21(2):421–426.
23. Li J, Wei G, Yuan Y, et al. New direction in antimicrobial delivery system: preparation and applications of hydrogel microspheres. Pharmaceutics. 2025;17(4):529. doi:10.3390/pharmaceutics17040529
24. Garms BC, Poli H, Baggley D, et al. Evaluating the effect of synthesis, isolation, and characterisation variables on reported particle size and dispersity of drug loaded PLGA nanoparticles. Mater Adv. 2021;2(17):5657–5671. doi:10.1039/D1MA00410G
25. Haque S, Boyd BJ, Mcintosh MP, et al. Suggested procedures for the reproducible synthesis of Poly(d,l-lactide-co-glycolide) nanoparticles using the emulsification solvent diffusion platform. Curr Nanosci. 2018;14(5):448–453. doi:10.2174/1573413714666180313130235
26. Zhang C, Zhang R, Liu L, et al. Designing long-acting injectable formulations using PLGA via spray-drying. Int J Pharm. 2025;683:126083. doi:10.1016/j.ijpharm.2025.126083
27. Michaelides K, Al Tahan MA, Zhou Y, et al. New insights on the burst release kinetics of spray-dried PLGA microspheres. Mol Pharmaceut. 2024;21(12):6245–6256. doi:10.1021/acs.molpharmaceut.4c00686
28. Renganathan K, Jain R, Chheda D, et al. Development of a continuous-flow microfluidic process for microsphere production. Mater Lett. 2026;404:139599. doi:10.1016/j.matlet.2025.139599
29. Sivakumar M, Muthu Y, Elumalai K. Advancements in drug delivery systems: a focus on microsphere-based targeted delivery. Biomed Mat Devices. 2025;3(2):1030–1057. doi:10.1007/s44174-024-00245-6
30. Ito F, Honnami H, Kawakami H, et al. Preparation and properties of PLGA microspheres containing hydrophilic drugs by the SPG (shirasu porous glass) membrane emulsification technique. Colloids Surf B. 2008;67(1):20–25. doi:10.1016/j.colsurfb.2008.07.008
31. Pöttgen S, Wischke C. Alternative techniques for porous microparticle production: electrospraying, microfluidics, and supercritical CO2. Pharm Res. 2025;42(9):1461–1480. doi:10.1007/s11095-025-03923-2
32. Wang B, Ma C, Wang K, et al. Preparation of poly(lactide-co-glycolide) microspheres via microfluidic technology and analysis of their degradation and drug release characteristics. Chin J Anal Chem. 2025;53(4):621–630.
33. Zhang B, Liu H, Li Y, et al. Innovative centrifugal microfluidic approach for risperidone-loaded PLGA microsphere production. Int J Pharm. 2025;674:125484. doi:10.1016/j.ijpharm.2025.125484
34. Liu Q, Liu H, Wu H, et al. Continuous preparation of long-acting hydromorphone PLGA microspheres using an automatic and scalable microfluidic process system. Int J Pharm. 2025;674:125459. doi:10.1016/j.ijpharm.2025.125459
35. Wei R, Dou J, Wu Y, et al. Microfluidic regulation of core-shell PLGA microspheres for sustained release of leuprolide acetate. Langmuir. 2025;41(27):17893–17901. doi:10.1021/acs.langmuir.5c01667
36. Liu X-A, Hussain Y, Wang J, et al. Engineering long-acting apixaban loaded PLGA microspheres for the treatment of stroke caused by atrial fibrillation. Chem Eng J. 2025;518:164321. doi:10.1016/j.cej.2025.164321
37. Zhang C, Yan D, Chang G, et al. Research progress on the effect of PLGA molecular properties on its microsphere properties. Chin J Pharm. 2023;54(9):1294–1301.
38. Liang D, Walker J, Schwendeman PS, et al. Effect of PLGA raw materials on in vitro and in vivo performance of drug-loaded microspheres. Drug Delivery Transl Res. 2025;15(1):185–202. doi:10.1007/s13346-024-01577-y
39. Bakhrushina EO, Sakharova PS, Konogorova PD, et al. Burst release from in situ forming PLGA-based implants: 12 effectors and ways of correction. Pharmaceutics. 2024;16(1):115. doi:10.3390/pharmaceutics16010115
40. Mayer JB, Patil SM, Shin SH, et al. Internal water-induced acceleration, chemical pathways, and contributing factors in the degradation of Poly(lactic-co-glycolic acid) (PLGA) microparticles and devices. ACS Biomater Sci Eng. 2025;11(7):3932–3948. doi:10.1021/acsbiomaterials.5c00419
41. Frota Reis AV, De Sousa ACC, De Freitas JVB, et al. Effect of PLGA composition on nanoencapsulation, fluorescence stability and cellular internalization of R-phycoerythrin in colorectal cancer cells. Int J Pharm. 2025;682:125966. doi:10.1016/j.ijpharm.2025.125966
42. Alshammari F, Alshammari B, Alshamari AK, et al. Dual-phase ocular insert with bromfenac-loaded PLGA MPs in a PVA matrix for sustained postoperative anti-inflammatory delivery. Pharmaceutics. 2025;17(8):1066. doi:10.3390/pharmaceutics17081066
43. Borrelli MA, Warunek JJP, Ravikumar T, et al. End group chemistry modulates physical properties and biomolecule release from biodegradable polyesters. J Mat Chem B. 2025;13(34):10621–10634. doi:10.1039/D5TB00816F
44. Zhao Q, Yang J, Liu J, et al. Impact of terminal groups on PLGA degradation rate and their role in sustained release of cyproterone acetate. European J Pharmaceutics Biopharmaceutics. 2026;218:114914. doi:10.1016/j.ejpb.2025.114914
45. Wang L, Wang P, Liu Y, et al. The effect of different factors on Poly(lactic-co-glycolic acid) nanoparticle properties and drug release behaviors when co-loaded with hydrophilic and hydrophobic drugs. Polymers. 2024;16(7):865. doi:10.3390/polym16070865
46. Muddineti OS, Omri A. Current trends in PLGA based long-acting injectable products: the industry perspective. Expert Opin Drug Delivery. 2022;19(5):559–576. doi:10.1080/17425247.2022.2075845
47. Chhetri P. Polymeric nanocarriers: advances, challenges, and future perspectives in targeted drug delivery. Nanomed Nanotechnol Biol Med. 2026;72:102896. doi:10.1016/j.nano.2025.102896
48. Singh A, Gupta C, Godse S, et al. Design, synthesis, and evaluation of lipoyl ester conjugated Star PLGA for sustained drug delivery systems. Curr Applied Polymer Sci. 2024;7(1):33–45. doi:10.2174/0124522716306935240614081407
49. Sunazuka Y, Ueda K, Higashi K, et al. Mechanistic analysis of temperature-dependent curcumin release from Poly(lactic-co-glycolic acid)/Poly(lactic acid) polymer nanoparticles. Mol Pharmaceut. 2024;21(3):1424–1435. doi:10.1021/acs.molpharmaceut.3c01066
50. Liu G, Martinez R, Bhatnagar A, et al. Influence of surfactant on glass transition temperature of poly(lactic-co -glycolic acid) nanoparticles. Soft Matter. 2023;19(28):5371–5378. doi:10.1039/D3SM00082F
51. Park K, Otte A, Sharifi F, et al. Potential roles of the glass transition temperature of PLGA microparticles in drug release kinetics. Mol Pharmaceut. 2021;18(1):18–32. doi:10.1021/acs.molpharmaceut.0c01089
52. Thakkar A, Bhattacharya S. Advancing structure-property insights of poly (lactic-co-glycolic acids): a mini review on the biodegradable polymers applications in drug delivery. J Macromol SciPart B. 2026;65(5):774–788. doi:10.1080/00222348.2025.2468118
53. Serizawa M, Snijders D, Van Berkel S, et al. Quantifying Poly (lactide-co-glycolide) residual dimer-alternating structure and block-length distribution using controlled chemical degradation and reverse-phase liquid chromatography–mass spectrometry analysis. Macromolecules. 2026;59(7):4473–4484. doi:10.1021/acs.macromol.5c03061
54. Walker J, Albert J, Liang D, et al. In vitro degradation and erosion behavior of commercial PLGAs used for controlled drug delivery. Drug Delivery Transl Res. 2023;13(1):237–251. doi:10.1007/s13346-022-01177-8
55. Ngo GT, Nghiem THL, Tran THY. Extended drug release characteristics of biodegradable Poly (lactic-co-glycolic acid) microspheres: overview. VNU J Sci. 2025;41(2):4768.
56. Park K. PLGA-based long-acting injectable (LAI) formulations. J Control Release. 2025;382:113758. doi:10.1016/j.jconrel.2025.113758
57. Fredenberg S, Wahlgren M, Reslow M, et al. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems--a review. Int J Pharm. 2011;415(1–2):34–52. doi:10.1016/j.ijpharm.2011.05.049
58. Kamaly N, Yameen B, Wu J, et al. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. doi:10.1021/acs.chemrev.5b00346
59. Rahmani F, Naderpour S, Nejad BG, et al. The recent insight in the release of anticancer drug loaded into PLGA microspheres. Med Oncol. 2023;40(8):229. doi:10.1007/s12032-023-02103-9
60. Costa MS, Ramos AM, Cardoso MM. Drug release kinetics of PLGA-PEG microspheres encapsulating aclacinomycin A: the influence of PEG content. Processes. 2025;13(1):112. doi:10.3390/pr13010112
61. Somayaji MR, Das D, Przekwas A. A new level a Type IVIVC for the rational design of clinical trials toward regulatory approval of generic polymeric long-acting injectables. Clin Pharmacokinet. 2016;55(10):1179–1190. doi:10.1007/s40262-016-0388-1
62. Giao Thong N, Ha BTN, Thuong BT, et al. Effect of processing parameters on characteristics of biodegradable extended-release microspheres containing leuprolide acetate. Drug Dev Ind Pharm. 2024;50(12):981–994. doi:10.1080/03639045.2024.2433621
63. Giles MB, Walker J, Schwendeman SP. Optimization of aqueous remote loading of leuprolide in poly(lactic-co-glycolic acid) microspheres. Int J Pharm. 2025;685:126206. doi:10.1016/j.ijpharm.2025.126206
64. López-Muñoz R, Chevallier P, Copes F, et al. pH-sensitive release of functionalized chiral carbon dots from PLGA coatings on titanium alloys for biomedical applications. Polymers. 2025;17(19):2667. doi:10.3390/polym17192667
65. Shafiee K, Bazraei S, Mashak A, et al. The impact of temperature on the formation, release mechanism, and degradation of PLGA-based in-situ forming implants. J Polym Environ. 2024;32(8):3591–3608. doi:10.1007/s10924-023-03173-6
66. Kemme M, Prokesch I, Heinzel-Wieland R. Comparative study on the enzymatic degradation of poly(lactic-co-glycolic acid) by hydrolytic enzymes based on the colorimetric quantification of glycolic acid. Polym Test. 2011;30(7):743–748. doi:10.1016/j.polymertesting.2011.06.009
67. Pustak A, Maršavelski A. Enzymatic degradation of biopolymers in amorphous and molten states: mechanisms and applications. FEBS Open Bio. 2026;16(4):670–685. doi:10.1002/2211-5463.70177
68. Jiao Q, Xu H, Yang L, et al. PLGA and Mg(OH)2 composite coatings on zinc alloy for improving degradation resistance and cell compatibility. Metals. 2025;15(11):1187. doi:10.3390/met15111187
69. Ji YB, Lee S, Ju HJ, et al. Preparation and evaluation of injectable microsphere formulation for longer sustained release of donepezil. J Controlled Release. 2023;356:43–58. doi:10.1016/j.jconrel.2023.02.024
70. Zhao J, Guo B, Ma PX. Injectable alginate microsphere/PLGA-PEG-PLGA composite hydrogels for sustained drug release. RSC Adv. 2014;4(34):17736. doi:10.1039/c4ra00788c
71. Lee M, Chen TT, Iruela-Arispe ML, et al. Modulation of protein delivery from modular polymer scaffolds. Biomaterials. 2007;28(10):1862–1870. doi:10.1016/j.biomaterials.2006.12.006
72. Jiang W, Gao X, Wang Q, et al. The modified exenatide microspheres: PLGA-PEG-PLGA gel and zinc-exenatide complex synergistically reduce burst release and shorten platform stage. AAPS Pharm Sci Tech. 2023;24(8):251. doi:10.1208/s12249-023-02705-6
73. Shatri AN, Lemmer Y, Kalombo L, et al. Synthesis of chitosan-modified poly (lactic-co-glycolic acid) microparticles with pH-dependent controlled-release kinetics to enhance the delivery of potential antidiarrheal medicinal plant extract to the lower gastrointestinal tract. Sci Afr. 2025;e03153.
74. Sekhar Nanda H, Kawazoe N, Chen G. Ionic salt induced morphology and drug release control of insulin incorporated biodegradable PLGA microspheres. Adv Mater Lett. 2016;7(11):866–871. doi:10.5185/amlett.2016.6907
75. Ana González-Cela-Casamayor M, Rodrigo MJ, Brugnera M, et al. Ketorolac, melatonin and latanoprost tri-loaded PLGA microspheres for neuroprotection in glaucoma. Drug Delivery. 2025;32(1):2484277. doi:10.1080/10717544.2025.2484277
76. Jordan E, Arriaga MA, Obregon H, et al. Dual delivery of metformin and Y15 from a PLGA scaffold for the treatment of platinum-resistant ovarian cancer. Future Med Chemistry. 2025;17(3):301–312. doi:10.1080/17568919.2025.2458457
77. Kim Y, Kwak J, Lim M, et al. Advances in PCL, PLA, and PLGA-based technologies for anticancer drug delivery. Pharmaceutics. 2025;17(10):1354. doi:10.3390/pharmaceutics17101354
78. Patel D, Bhardwaj S, Yadav V, et al. Exploring the co-delivery of sorafenib and venetoclax to induce ferroptosis and apoptosis mediated cancer cell death using surface modified PLGA nanoparticles. AAPS Pharm Sci Tech. 2025;27(1):58. doi:10.1208/s12249-025-03304-3
79. Tiwari S, Bhattacharya S. Understanding advanced poly (ethylene glycol)-disulphide-poly (lactic-co-glycolic acid) (PEG-SS-PLGA) nanoparticles for cutting-edge innovations, applications in smart drug delivery systems and targeted cancer therapy. Biomed Mater. 2026;21(1):012006. doi:10.1088/1748-605X/ae3933
80. Wu Y, Wei G, Cao X, et al. Stimuli-responsive dual-drug loaded microspheres with differential drug release for antibacterial and wound repair promotion. Colloids Surf B. 2025;248:114455. doi:10.1016/j.colsurfb.2024.114455
81. Mazidi Z, Garshasbi HR, Naghib SM, et al. Multifunctional smart nano biopolymers for programmed controlled release of biomolecules and therapeutic agents: an overview on modern emerging systems. Mini-Rev Organic Chemistry. 2025;22(6):565–589. doi:10.2174/0118756298304506240628062045
82. Kesharwani P, Kumar V, Goh KW, et al. PEGylated PLGA nanoparticles: unlocking advanced strategies for cancer therapy. Mol Cancer. 2025;24(1):205. doi:10.1186/s12943-025-02410-x
83. Lee SY, Choi JW, Lee JY, et al. Hyaluronic acid/doxorubicin nanoassembly-releasing microspheres for the transarterial chemoembolization of a liver tumor. Drug Delivery. 2018;25(1):1472–1483. doi:10.1080/10717544.2018.1480673
84. Zhang Q, Liu T, Qiao F, et al. Hyaluronic acid–chitosan conjugated PLGA nanoparticles for dual chemo-photothermal therapy of triple-negative breast cancer. Sci Rep. 2025;16(1):1910. doi:10.1038/s41598-025-31485-1
85. Lopez-Mitjavila JJ, Palma-Florez S, Lagunas A, et al. PEGylated PLGA nanoparticles prepared from nano-emulsion templates as versatile platforms to cross blood-brain barrier models. J Drug Delivery Sci Technol. 2025;110:107057. doi:10.1016/j.jddst.2025.107057
86. Lakshmikanthan M, Muthu S, Krishnan K, et al. Poly(d,l-lactide-co-glycolide) (PLGA) nanoparticles in oncological therapeutics: mechanisms of targeted delivery and encapsulation strategies for chemotherapeutics, gene modulators, and phytochemicals. Nanomed Nanotechnol Biol Med. 2026;74:102935. doi:10.1016/j.nano.2026.102935
87. Kong JR, Mo WY, Yao H, et al. Enhancing local anesthetic efficacy: controlled release of ropivacaine using Poly(lactic-co-glycolic) acid-polyvinyl alcohol nanocarriers. Curr Pharm Biotechnol. 2026;27. doi:10.2174/0113892010394956251027114744
88. Tian X, Zhu H, Du S, et al. Injectable PLGA-coated ropivacaine produces a long-lasting analgesic effect on incisional pain and neuropathic pain. J Pain. 2021;22(2):180–195. doi:10.1016/j.jpain.2020.03.009
89. Kyeong YM, Wook CY. Improved antigen delivery systems with PLGA microsphere for a single-step immunization. J Pharm Invest. 2004;34(1):1–14.
90. Allahyari M, Golkar M, Fard-Esfahani P, et al. Co-delivery of PLGA nanoparticles loaded with rSAG1 antigen and TLR ligands: an efficient vaccine against chronic toxoplasmosis. Microb Pathogenesis. 2022;162:105312. doi:10.1016/j.micpath.2021.105312
91. Adediran E, Arte T, Pasupuleti D, et al. Delivery of PLGA-loaded influenza vaccine microparticles using dissolving microneedles induces a robust immune response. Pharmaceutics. 2025;17(4):510. doi:10.3390/pharmaceutics17040510
92. Clinical efficacy and safety of paclitaxel polymeric micelles for injection in the treatment of patients with taxanes-resistant pancreatic adenocarcinoma, cholangiocarcinoma, lung cancer, gastric cancer, esophageal carcinoma, or breast cancer. NCT06199895. Bethesda (MD): National Library of Medicine (US); 2023.
93. AbbVie Inc. Lupron Depot (leuprolide acetate for depot suspension) [prescribing information]. Bethesda (MD): National Library of Medicine (US); 2023.
94. Alphs L, Nasrallah HA, Bossie CA, et al. Factors associated with relapse in schizophrenia despite adherence to long-acting injectable antipsychotic therapy. Int Clin Psychopharmacol. 2016;31(4):202–209. doi:10.1097/YIC.0000000000000125
95. Omidian H, Wilson RL. PLGA implants for controlled drug delivery and regenerative medicine: advances, challenges, and clinical potential. Pharmaceuticals. 2025;18(5):631. doi:10.3390/ph18050631
96. Pokharel M, Neron A, Dey AK, et al. Light-responsive PLGA microparticles for on-demand vancomycin release and enhanced antibacterial efficiency. Pharmaceutics. 2025;17(8):1007. doi:10.3390/pharmaceutics17081007
97. Zhang Q, Jin G, Li L, et al. Rapamycin-loaded Poly(lactic-co-glycolic) acid microspheres optimized by a quality by design approach for atherosclerosis therapy: preparation, characterization, and in vitro evaluation. J Drug Delivery Sci Technol. 2025;107:106759. doi:10.1016/j.jddst.2025.106759
98. Develi Arslanhan EN, Bahadori F, Eskandari Z, et al. Optimization of microfluidizer-produced PLGA nano-micelles for enhanced stability and antioxidant efficacy: a quality by design approach. Pharmaceutics. 2025;18(1):631. doi:10.3390/pharmaceutics18010025
99. Qin T, Hong A, Lin Y, et al. Physicochemical properties and immunological effects of macrophage membrane-coated PLGA nanoparticles loaded with Hericium erinaceus polysaccharides. Colloids Surf B. 2026;259:115366. doi:10.1016/j.colsurfb.2025.115366
100. Horisawa E, Kubota K, Tuboi I, et al. Size-dependency of DL-lactide/glycolide copolymer particulates for intra-articular delivery system on phagocytosis in rat synovium. Pharm Res. 2002;19(2):132–139. doi:10.1023/A:1014260513728
101. Spinelli L, D’anna P, Morretta E, et al. PEGylation-driven remodeling of the protein corona on PLGA nanoparticles: implications for macrophage recognition. Biomacromolecules. 2025;26(12):8522–8534. doi:10.1021/acs.biomac.5c01369
102. Chen H, Udepurkar AP, Clasen C, et al. Synthesizing porous nanospheres with highly efficient drug loading and sustained release through a thermal-controlled continuous stirred-tank reactor cascade. Nanoscale Adv. 2026;8(4):1281–1290. doi:10.1039/D5NA00897B
103. Li X, Zhang Z, Harris A, et al. Bridging the gap between fundamental research and product development of long acting injectable PLGA microspheres. Expert Opin Drug Delivery. 2022;19(10):1247–1264. doi:10.1080/17425247.2022.2105317
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.