Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
How Do Innate Immune Cells Contribute to Airway Remodeling in COPD Progression?
Authors Bu T ![]() , Wang LF, Yin YQ
, Wang LF, Yin YQ
Received 17 October 2019
Accepted for publication 19 December 2019
Published 10 January 2020 Volume 2020:15 Pages 107—116
DOI https://doi.org/10.2147/COPD.S235054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Tegeleqi Bu, Li Fang Wang, Yi Qing Yin
Department of Anesthesiology, China-Japan Friendship Hospital, Beijing, People’s Republic of China
Correspondence: Yi Qing Yin
Department of Anesthesiology, China-Japan Friendship Hospital, Ying Hua East Road No. 2, Chaoyang District, Beijing, People’s Republic of China
Tel +86 135 2126 2509
Email [email protected]
Abstract: Recently, the therapeutic potential of immune-modulation during the progression of chronic obstructive pulmonary disease (COPD) has been attracting increasing interest. However, chronic inflammatory response has been over-simplified in descriptions of the mechanism of COPD progression. As a form of first-line airway defense, epithelial cells exhibit phenotypic alteration, and participate in epithelial layer disorganization, mucus hypersecretion, and extracellular matrix deposition. Dendritic cells (DCs) exhibit attenuated antigen-presenting capacity in patients with advanced COPD. Immature DCs migrate into small airways, where they promote a pro-inflammatory microenvironment and bacterial colonization. In response to damage-associated molecular patterns (DAMPs) in lung tissue affected by COPD, neutrophils are excessively recruited and activated, where they promote a proteolytic microenvironment and fibrotic repair in small airways. Macrophages exhibit decreased phagocytosis in the large airways, while they demonstrate high pro-inflammatory potential in the small airways, and mediate alveolar destruction and chronic airway inflammation. Natural killer T (NKT) cells, eosinophils, and mast cells also play supplementary roles in COPD progression; however, their cellular activities are not yet entirely clear. Overall, during COPD progression, “exhausted” innate immune responses can be observed in the large airways. On the other hand, the innate immune response is enhanced in the small airways. Approaches that inhibit the inflammatory cascade, chemotaxis, or the activation of inflammatory cells could possibly delay the progression of airway remodeling in COPD, and may thus have potential clinical significance.
Keywords: innate immune cell, airway remodeling, lung immunity, immune tolerance, chronic obstructive pulmonary disease
Introduction
Chronic obstructive pulmonary disease (COPD) has, for many years, been a leading cause of morbidity and mortality worldwide. Even in patients who have not deliberately inhaled noxious substances, airway remodeling and lung function decline continue. The 2017 Global Initiative for Chronic Obstructive Lung Disease (GOLD) Guidelines has defined COPD as an inflammatory disorder of the respiratory system.1 The innate immune system in the lung functions acts as a front-line defense against noxious particles and infections.2 The manner in which innate immune cells contribute to airway remodeling is similar to “covering up arsonists” in the lung. Immune tolerance and an exhausted defense mechanism “pamper” pathogen colonization, while “boosting” the flare-up of inflammation in the small airways. Studies in the therapeutic modulation of innate immune cells hope to reduce both structural and functional airway remodeling. In this article, we review how innate immune cells contribute to airway remodeling during COPD progression.
Airway Remodeling in COPD
Airway remodeling is a determinant of COPD progression. During COPD progression, airway remodeling involves both the small and large airways.3 In the large airways, structural changes include: (1) reduction in type I collagen and elastic fibers; and (2) deposition of the extracellular matrix (ECM) (e.g. α-smooth muscle actin and proteoglycans) in the subepithelium of the lumen of non-cartilaginous airways.4 Regarding the small airways, the bronchial wall thickness measured by high-resolution computed tomography (HRCT) is correlated both with the severity of emphysema, as well as the decline in the forced expiratory volume in 1 s (FEV1).5 In comparison with lung function in asthma, COPD creates a significantly larger airway luminal area in the third to sixth generation of each airway.6 In addition, an association exists between structural alterations of the bronchi and pulmonary hypertension in COPD.7
Structural changes that cause airflow obstruction in COPD are induced by two-major processes: (1) thickening and narrowing of respiratory airways and small bronchioles;8 and (2) destruction of the lung parenchyma and alveolar walls. Thickening of the airway walls is due to hypertrophy of the airway smooth muscle and excessive ECM deposition.9 According to Michaeloudes et al,10 airway smooth muscle cell (ASMC) hyperplasia could be stimulated by transforming growth factor beta (TGF-β), which is unregulated during the course of COPD. Proliferation of ASMCs is negatively correlated with FEV1/forced vital capacity (FVC).
Jones et al9 demonstrated that an increase in the ECM within the smooth muscle layer is responsible for fixed airflow obstruction. A change in the composition of the ECM is also evident in COPD, and ECM-related airway stiffness is resistant to existing treatments. The study of Limjunyawong et al,11 among other similar studies, demonstrates that emphysema could be induced in an animal model by the overexpression of interferon-gamma (IFN-γ) and activation of M2 macrophages. Such studies have indicated that an aberrant immune response could promote airway remodeling, which is characteristic of COPD.
“Battle and Exhaustion” – Overview of the Change in Cell Components During COPD Progression
Predisposing factors related to an aberrant immune response (such as low body weight, childhood respiratory infections, underlying antibody deficiency syndrome, and depression) could increase susceptibility to COPD and lead to more frequent exacerbations.1,12,13 Hoonhorst et al14 showed that the response to corticosteroids in the skin was comparable among non-smokers, healthy smokers, and patients with GOLD stages I and II COPD. However, patients with GOLD stages III and IV COPD exhibit significant decline in corticosteroid response of the skin, suggesting the possibility of genetic factors that affect airway immunity and promote COPD development.
Innate immune cells directly resist exogenous noxious stimuli in the large airways, and set a “fire” against a potentially hazardous microenvironment that gradually “burns out” during progression of the disease. The major structural and cellular changes in the large airways include: (1) the reduction of FEV1 and increase in the levels of pro-inflammatory cytokines in the lung, which are both positively correlated with squamous cell and goblet cell hyperplasia, mucous gland hypertrophy, and increased thickness of the smooth muscle layer in non-cartilaginous airways and pulmonary arteries;15,16 and (2) the accumulation of T cells, macrophages, and neutrophils within the wall of the airway, which amplifies the immune response – tissue injury cycle.17,18 Overall, hypocellularity in the large airways has been confirmed in non-smokers, as well as in patients with mild to moderate COPD. Furthermore, a reduction in the number of neutrophils, CD8+ and CD4+ T cells, macrophages,19 as well as mature DCs20 can be observed in large airway specimens and bronchoalveolar lavage fluid. Yet, there is limited supporting evidence indicating that the large airways may exhibit an exhausted innate immune response in COPD. Similarly, studies on the differences in cellular activities between the large and small airways in lungs affected by COPD are lacking.
In the small airways, innate immune cells are more exposed to the regional microenvironment. The innate immune cells surround the respiratory membranes and interact with the pulmonary microcirculation. The major differences in cell components of the small airways include: (1) senescence of epithelial cells,21 destruction and fibrosis of peribronchiolar and alveolar walls;22 (2) a marked increase in the number of neutrophils, which is correlated with the severity of airflow obstruction;23 (3) increased number of macrophages with impaired phagocytosis and efferocytosis;24,25 (4) increased number of CD8+ T and CD4+ T lymphocytes;26 and (5) increased number and activation of epithelial DCs.27
In the small airways, lymphoid aggregates (LAs) can be found in the outer layer of the small vessels, as well as in the adventitia of bronchioles, and the alveolar lumen. The LAs share extensive interfaces with the alveolar parenchyma in lungs affected by COPD. Activated DCs migrate to the lymph nodes, present antigens, and activate T cells to form lymphoid aggregates.28 In COPD, the DC-rich alveolar-lymphoid epithelium exhibits enhanced antigen uptake in the small airways. Although an exhausted immune attack seems to occur in the large airways, a persistent agitated immune response is evident in the small airways during COPD progression.29
Innate Immune Cells Contribute to Airway Remodeling in COPD
Epithelial Barrier
The airway epithelial barrier is the first-line defense of the innate immune system in the airway that protects against noxious particles and infection. The bronchiolar epithelial tissue and epithelial cells in patients with COPD exhibit a retarded wound repair process and inhibited epithelial cell proliferation, which are correlated with the severity of airflow obstruction and emphysema.30 Both in vitro and in vivo experiments in epithelial cells from patients with COPD have shown that cigarette smoking and viral infections could induce the downregulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and promote the release of interleukin 8 (IL-8), indicating non-specific airway inflammation in the airway epithelium.31
Epidermal growth factor receptor (EGFR) is known as the primary regulator of epithelial cell function. Extensive upregulation of EGFR in epithelial cells is evident in the bronchial walls of lungs affected by COPD in the stable state, and could be exaggerated by infection and proinflammatory cytokines.32,33 Overexpression of EGFR leads to epithelial phenotypic alterations, ie, goblet cell production, mucus hypersecretion, epithelial cell depolarization, and epithelial-mesenchymal transition (EMT), which exacerbate pathological changes in COPD.34 Mucus hypersecretion from epithelial cells and the impairment of mucociliary clearance are associated with progressive airflow obstruction, exacerbation of dyspnea, and a poor outcome.35 In brief, the microenvironment of lungs affected by COPD makes epithelium more susceptible to mucus hypersecretion, depolarization, aberrant repair, and the loss of epithelial integrity.
Innate Immune Cells
Innate immune cells in patients with COPD are responsible for the progression of airway remodeling. A noxious microenvironment and impaired immune activities, pathogen dissolution, and structural injury to the airways could all induce the release of damage-associated molecular patterns (DAMPs). Pattern recognition receptors (PRRs), like toll-like receptors (TLRs) or the Nod-like receptor family, are receptors of DAMPs.
DAMPs can recruit and activate innate immune cells both systemically and locally.36 The PRRs in immune cells are overexpressed in patients with COPD.37 However, it is the activated immune cells, and not the level of PRR expression, that determine COPD progression and severity.38 Halper-Stromberg et al39 have shown that systemic immune cell counts could be predictive of the 3-year change in lung function, indicating a strong correlation between immune cell components in the lung and airway remodeling.
Dendritic Cells
The DCs in the lung are distributed along the epithelium and parenchyma. The loss of epithelial integrity directly exposes intraepithelial DCs to antigens. The DCs take up antigens, and then migrate toward draining lymph nodes. In addition to the local DCs in lymphoid tissue, DCs within lung lymphoid follicles activate naive T and B cells.40,41 In COPD, an increased number of DCs are recruited to the lungs. However, long-term exposure to noxious particles impairs DC maturation, which inhibits their antigen-presenting capacity (also called “non-functional DCs”).42
As the number of mature CD83+ DCs is decreased in the tissue of lungs affected by COPD lung tissue, the number of immature DCs is increased in the small airways. Immature DCs in the lung release more CCL3 and CXCL2, both of which could promote neutrophil recruitment.41 They also induce T helper 1 and T helper 17 responses, and participate in elastin degradation in the lung parenchyma.43 In COPD, the increase in the number of immature DCs in the small airways is proportionate to the FEV1.21 During COPD progression, DCs participate in bacterial colonization44 and immune tolerance,40 and facilitate lung remodeling.20
Macrophages
Alveolar macrophages are directly exposed to antigens and noxious particles, and accumulate in areas of persistent inflammation (eg emphysema).45 The number of macrophages in the lung is increased in COPD, and this increase is more prominent in larger airways than in small airways.46 Most studies have shown that the major change observed in macrophages in COPD is a shift toward the M2 phenotype. The M2 macrophages release lower levels of pro-inflammatory mediators, such as tumor necrosis factor alpha (TNF-α), IL-1β, and IL-6, when stimulated by lipopolysaccharide.47
According to Dewhurst et al,48 in patients with COPD, the total number of macrophages is increased; a marked increase is evident in the number of large macrophages, which produce fewer pro-inflammatory cytokines and have reduced phagocytic ability; and fewer small macrophages can be observed, which present higher levels of pro-inflammatory gene expression than found in normal individuals.
Berenson et al49 studied alveolar macrophages from patients with COPD, and these cells exhibited impaired phagocytosis of respiratory pathogens. Impairment of phagocytosis is strongly correlated with FEV1% predicted in patients with COPD. Although small in number, M1 macrophages exhibit enhanced pro-inflammatory effects, produce more TNF-α, and enhance ECM deposition in the lung in COPD; and M1 macrophages are responsible for matrix metalloproteinase (MMP) production in COPD, which is related to elastin breakdown and excessive accumulation of collagen or fibronectin in the parenchyma.50,51 Thus, macrophages in COPD exhibit phenotypic alteration, attenuate phagocytosis, and are involved in airway injury and remodeling.
Neutrophils
In some studies, COPD has been described as “chronic neutrophilic airway inflammation.”52 Under the stimulus of antigen presentation and inflammatory chemotaxis, neutrophils act as the first responders to inflammation. The neutrophil count and level of neutrophil chemoattractant in the sputum of patients are regarded as diagnostic markers of COPD progression. Furthermore, IL-8, released by injured epithelial cells, and leukotrienes (LTB4), released by agitated macrophages, are major neutrophil chemoattractants in the lungs of patients with COPD.53 Patients with high levels of IL-8 are more susceptible to emphysema.54
According to Tulah et al,55 polymorphisms within genes that control LTB4 production could determine baseline FEV1 and FEV1/FVC, as well as susceptibility to airflow obstruction in smokers. Microbial overload in the airways could also induce the release of myeloperoxidase from neutrophils, generate cell debris and reactive oxygen species (ROS), and aggravate damage to pulmonary structures.56,57 In response to DAMPs, PRRs such as TLRs on neutrophils in peripheral blood and lung tissue are also over-expressed, thereby amplify neutrophil recruitment and activation.36,58 Excessive infiltration of neutrophils in the lungs leads to the release of MMPs and neutrophil elastase (stored in azurophilic granules), which mediate a protease-antiprotease imbalance in the lung.54 The activation of neutrophils provokes dissolution of the alveolar wall, injury to the ciliated epithelium and connective tissue matrix, mucus hypersecretion, and squamous metaplasia in epithelial cells, thereby leading to emphysema and fibrotic repair in the small airways.54,59
Natural Killer T Cells
Rijavec et al60 demonstrated that in induced sputum, bronchoalveolar lavage fluid, and bronchial-biopsy specimens, the proportion of natural killer T (NKT) cells in the airway is low, but slightly increased in COPD. The NKT cells play a supplementary role in the pathogenesis of COPD.61 Urbanowicz et al62 found that NKT cells in patients with COPD are activated, marked by increased expression of granzyme B and perforin, and are involved in epithelial and parenchymal destruction.63 Activated NKT cells are also the main sources of IFN-γ and TNF-α64 they also enhance the function of DCs, T cells, and B cells,65 and affect COPD progression.
However, unlike NKT cells in the lung, a functional defect is evident in circulating natural killer (NK) and NKT cells in patients with COPD. Tang et al66 found that the numbers of CD3−CD56+ NK cells and CD3+CD56+ NKT-like cells in peripheral blood are greater in patients with COPD; CD158a+ and CD158b+ NK cells and CD158b+ NKT-like cells are significantly increased. The number of CD158b+ NK cells is negatively correlated with FEV1% prediction and FEV1/FVC, representing attenuated cytotoxicity potential in long-term disease progression.
Eosinophils
Eosinophilic inflammation in COPD is less extensive than it is in asthma, but eosinophilia could become more prominent in severe COPD. It is still debatable whether subgroups of COPD exist as it relates to eosinophils and the response of corticosteroids. Higher blood eosinophil counts might be related to frequent exacerbations of COPD.67 Some patients would present with eosinophilia during COPD exacerbation, and tend to respond better to treatment and have better outcomes.68 Eosinophilic COPD is related to relatively mild alveolar damage.69 The influx and activation of eosinophils in the lungs in COPD is seen in some patients with chronic bronchitis, rather than emphysema. However, whether this eosinophilia is independent of asthmatic etiology during COPD progression is still controversial. Most patients with eosinophilic airway inflammation respond well to corticosteroid treatment, although they may not show other clinical features resembling asthma.70 Little is known about this unique subset of COPD.
Mast Cells
Few studies have been focused on the manner in which mast cells contribute to the pathogenesis of COPD. Mast cells facilitate the release of pro-inflammatory cytokines, mainly via the immunoglobulin E (IgE)-mediated pathway or degranulation.71 Mast cells in COPD could express TLRs, protease activated receptor 2, and calcium-independent phospholipase A2, and the activation of mast cells promotes the secretion of IL-8 and TNF-α, etc. Mast cells could interact with neutrophils, T cells, and smooth muscle cells, and thereby contribute to inflammatory cascades, angiogenesis,72 and airway remodeling.71
To summarize, throughout COPD progression, excessive airway inflammation gradually weakens the lungs. Innate immune cells are involved in persistent infections, airway destruction, and aberrant repair, and contribute to airway remodeling during COPD progression (Table 1).
 |
Table 1 Major Cell Types in the Innate Immune System That Contribute to Airway Remodeling in COPD |
Adaptive Immune Cells in COPD
The adaptive immune system is activated during COPD progression, and marked by the activation of CD8+ T cells, Th17 CD4+ T cells, Th4+ T cells, and B cells, as well as the inhibition of regulatory T cells (Tregs). In short, activation of the adaptive immune system is gradually exhausted as the disease progresses. The manifestation and course of COPD exacerbations reflect the residue capacity of the pulmonary adaptive immune system in the lungs.
The FEV1% predicted in COPD is correlated with the tissue and circulating CD4+ and CD8+ T cell levels, as well as the ratio of CD4+/CD8+ T cells.73 As the prime effector cells, the percentage of apoptotic CD8+ T cells is strongly correlated with FEV1% predicted, as well as the FEV1/FVC.74 The CD4+ T cells are especially associated with the severity of emphysema,75 regardless of smoking habits.76,77 However, Geerdink et al78 reported that patients with frequent exacerbations and rapid disease progression have fewer CD4+ and CD8+ memory T cells in the peripheral blood than more stable patients with COPD. In the lungs of patients with COPD, when CD8+ T cells are excessively activated, an increase in the blood CD8+/CD4+ ratio indicates the progression of airflow limitation.79 The CD4+ T cells in COPD mainly facilitate the functions of B cells and CD8+ T cells in the lungs, thereby both amplifying and prolonging the inflammatory response in the lung microenvironment. The Tregs in the lung maintain immune tolerance. In COPD models, the numbers of Tregs are typically decreased both in patients with COPD80 and in animal models.81
Immune-Modulatory Approaches to Delay COPD Airway Remodeling
Current treatment for COPD mainly consists of bronchodilatation, anti-inflammation, and anti-infection. These therapeutic strategies have remained unchanged for a relatively long time. The latest 2017 GOLD guidelines proposed immune-modulatory treatment, such as corticosteroids, for overlap syndrome. We have demonstrated that long-term aberrations in the microenvironment of the lung also results in long term over-active, but sloppy, defense mechanisms, which lead to chronic inflammatory stress and bacterial colonization. As none of the current approaches could reverse airway remodeling and the subsequent irreversible obstructive airflow, modulation of the immune-imbalance in the lungs of patients with COPD may have potential clinical significance.
A few studies have been focused on therapeutics that modulate immune function and attempt to control airway remodeling in COPD (Table 2). The therapeutic dosage of an inhaled corticosteroid could reduce reticular basement membrane thickening and inflammatory cell infiltration in the lung parenchyma of patients with COPD;82 and could also inhibit angiogenesis,83 and the EMT process.84 Corticosteroids have been considered as standard treatment for airway inflammation, and protection against airway remodeling in COPD.
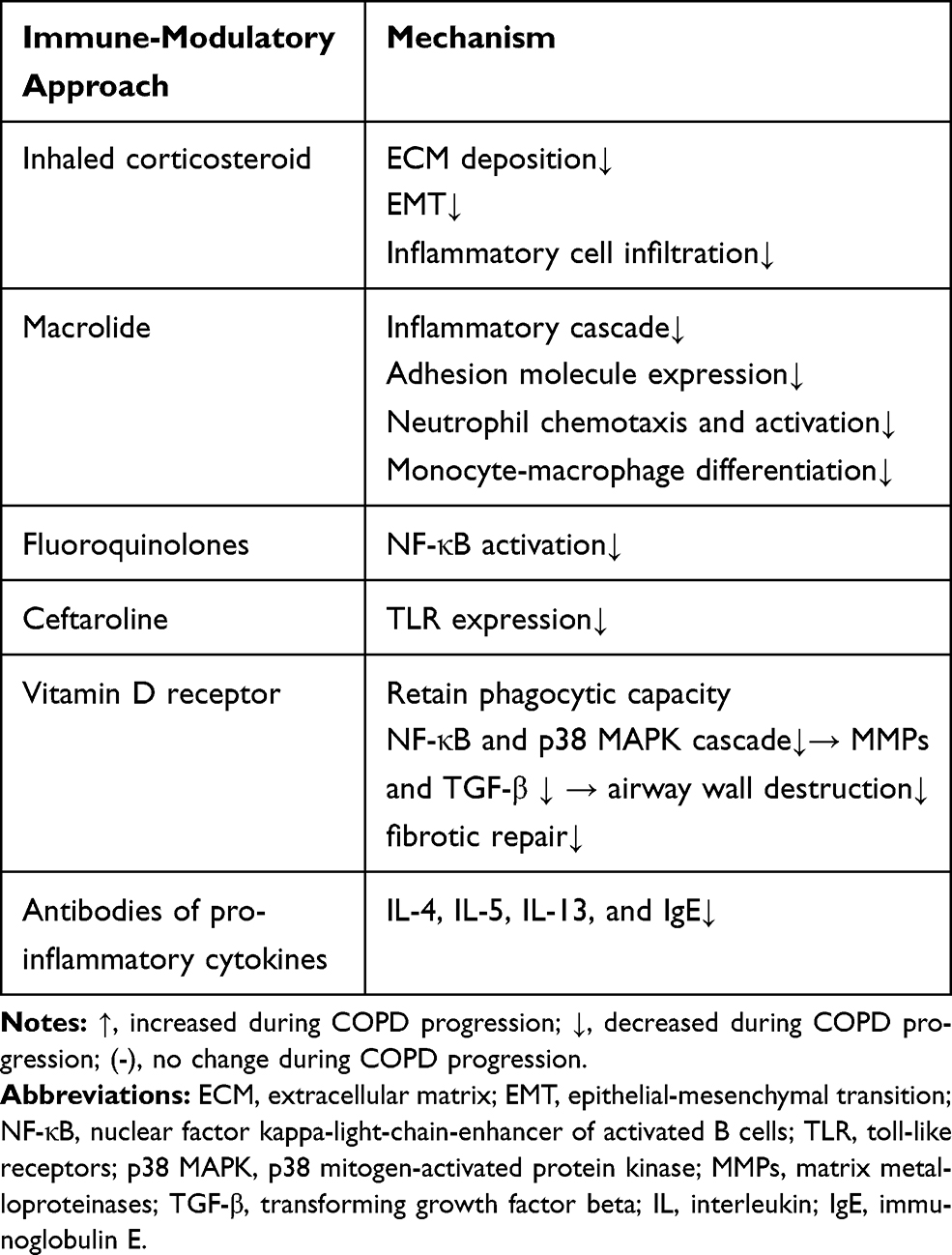 |
Table 2 Immune-Modulatory Approaches to Delay COPD Airway Remodeling |
Bacterial load is correlated with the extent of airway inflammation in stable COPD.85 Aberrant immune host responses and the bacterial-viral load are reciprocal causes during COPD progression, and are typically accompanied by the presence of C-reactive protein (CRP), IL-1β, IL-8, TNF-α, etc, in lung tissue. The long-term use of antibiotics could reduce bacterial load in the airway.86 Besides their antimicrobial effects, certain antibiotics are now known to have immune-modulatory effects on airway remodeling and have been attracting increasing research interest.
Macrolides are widely known to have immune-modulatory effects in the lung. New macrolide derivatives have been recently developed to reduce COPD exacerbation rate and severity. Macrolides suppress activator protein 1 (AP1) and the NF-κB-mediated inflammatory cascade, which is marked by reduced levels of IL-1, IL-6, IL-8, and TNF-α in patients with COPD.87 Macrolides also reduce the expression of adhesion molecules in the pulmonary epithelium and endothelium. Macrolides are known to reduce neutrophil chemotaxis and monocyte-macrophage differentiation, as well as MMP production and protease activity.87
Fluoroquinolones have been found to inhibit NF-κB activation, and thereby reduce the levels of IL-1, IL-6, and TNF-α in an animal model of COPD.87 Giusti et al88 conducted a randomized controlled trial to determine whether fluoroquinolones could exert additional benefits in patients with acute exacerbations of COPD, apart from its anti-infective effects.89 Other antibiotics, such as ceftaroline, which could counteract the effects of cigarette smoking, induces TLR expression in bronchial epithelial cells and monocytes, and may increase the effectiveness of the innate immune response to pathogen activity in the lungs of patients with COPD.90
According to Heulens et al,91 the vitamin D receptor is widely expressed in alveolar epithelial cells, neutrophils, and macrophages in the lung. Vitamin D inactivates transcription of NF-κB and p38 MAPK in epithelial cells and macrophages, has anti-inflammatory effects, and protects the phagocytic capacity of alveolar macrophages and neutrophils in the lungs of patients with COPD.91 Vitamin D has also been found to reduce the expression of MMPs and TGF-β in the lungs, and consequently to inhibit airway remodeling and the decline in lung function, both in an animal model and in clinical trials.91
Other innovative studies, such as those focused on IL-4, IL-5, IL-13, and the IgE antibody, as well as the prostaglandin (PG) D2 blocker, have shown that in some clinical trials, deteriorating lung function can be ameliorated in patients with COPD, who present with eosinophilic airway inflammation. However, heterogeneity in the treatment responses and a lack of reproducible biomarkers highlight the need for further research.92
Conclusion
Long-term exposure to noxious particles leads to a heightened immune response and continuous structural and functional changes in the airways. Innate immune cells regulate airway inflammation, bacterial colonization, and airway remodeling, which lead to progressive airflow obstruction during COPD progression. Immune tolerance and exhaustion of innate immune cells promote inflammatory flare-up in the airways. Immune modulation against the remodeling process could be a promising therapeutic strategy to retard, and possibly even reverse, the progression of the disease.
Abbreviations
ASMC, airway smooth muscle cell; COPD, chronic obstructive pulmonary disease; DAMP, damage-associated molecular patterns; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; HRCT, high-resolution computed tomography; ICS, inhaled corticosteroids; LA, lymphoid aggregates; NK, natural killer; PRR, pattern recognition receptors; ROS, reactive oxygen species; TLR, toll-like receptors.
Acknowledgment
This review was funded by National Natural Science Foundation of China (No. 30500482).
Author Contributions
Both Tegeleqi Bu and Li Fang Wang contributed equally in drafting and revising the article. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors declare that there are no conflicts of interest regarding the publication of this paper.
References
1. Montes OM, Pérez-Padilla R. Global initiative for Chronic Obstructive Lung Disease (GOLD)-2017: the alat perspective. Arch Bronconeumol. 2017;53(3):87.
2. Derek EB, Kangyun W, Geoffrey DV, et al. Triggering receptor expressed on myeloid cells-2 expression tracks with M2-like macrophage activity and disease severity in COPD. Chest. 2015;153(1):77–86.
3. Burgel PR, Bourdin A, Pilette C, et al. Structural abnormalities and inflammation in COPD: a focus on small airways. Rev Mal Respir. 2011;28(6):749–760. doi:10.1016/j.rmr.2011.01.009
4. Pini L, Pinelli V, Modina D, et al. Central airways remodeling in COPD patients. Int J Chron Obstruct Pulmon Dis. 2014;927–933. doi:10.2147/COPD.S52478
5. Tho NV, Ryujin Y, Ogawa E, et al. Relative contributions of emphysema and airway remodelling to airflow limitation in COPD: consistent results from two cohorts. Respirology. 2015;20(4):594–601. doi:10.1111/resp.12505
6. Shimizu K, Hasegawa M, Makita H, et al. Comparison of airway remodeling assessed by computed tomography in asthma and COPD. Respir Med. 2011;105(9):1275–1283. doi:10.1016/j.rmed.2011.04.007
7. Dournes G, Laurent F, Coste F, et al. Computed tomographic measurement of airway remodeling and emphysema in advanced chronic obstructive pulmonary disease. Correlation with pulmonary hypertension. Am J Respir Crit Care Med. 2015;191(1):63. doi:10.1164/rccm.201408-1423OC
8. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4(1):435–459. doi:10.1146/annurev.pathol.4.110807.092145
9. Jones RL, Noble PB, Elliot JG, et al. Airway remodelling in COPD: it’s not asthma. Respirology. 2016;21(8):1347–1356. doi:10.1111/resp.12841
10. Michaeloudes C, Kuo CH, Haji G, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. 2017;50(5):1700202. doi:10.1183/13993003.00202-2017
11. Limjunyawong N, Craig JM, Lagassé HA, et al. Experimental progressive emphysema in BALB/cJ mice as a model for chronic alveolar destruction in humans. Am J Physiol Lung Cell Mol Physiol. 2015;309(7):L662–L676. doi:10.1152/ajplung.00214.2015
12. Mccullagh BN, Comellas AP, Ballas ZK, et al. Antibody deficiency in patients with frequent exacerbations of Chronic Obstructive Pulmonary Disease (COPD). PLoS One. 2017;12(2):e0172437. doi:10.1371/journal.pone.0172437
13. Moisieieva NV, Burya LV, Kapustianskaya AA, et al. Comprehensive patterns of comorbidity: copd and depression. Aspects of treatment. Wiad Lek. 2018;71:588–591.
14. Hoonhorst SJM, Hacken NHTT, Adèle TLTL, et al. Lower Corticosteroid skin blanching response is associated with severe COPD. PLoS One. 2014;9(3):e91788. doi:10.1371/journal.pone.0091788
15. Löfdahl M, Kaarteenaho R, Lappi-Blanco E, et al. Tenascin-C and alpha-smooth muscle actin positive cells are increased in the large airways in patients with COPD. Respir Res. 2011;12(15):48. doi:10.1186/1465-9921-12-48
16. Victor K, Michelle O, Heba D, et al. Chronic bronchitis and current smoking are associated with more goblet cells in moderate to severe COPD and smokers without airflow obstruction. PLoS One. 2015;10(2):e0116108. doi:10.1371/journal.pone.0116108
17. Aoshiba K, Nagai A. Differences in airway remodeling between asthma and chronic obstructive pulmonary disease. Clin Rev Allergy Immunol. 2004;27(1):35–43. doi:10.1385/CRIAI:27:1
18. Rutgers SR, Postma DS, Ten HNHT, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax. 2000;55:12–18. doi:10.1136/thorax.55.1.12
19. Eapen MS, Mcalinden KD, Tan D, et al. Inflammation paradox–a detailed analysis of inflammatory profile in the large and small airway wall in mild to moderate chronic obstructive pulmonary disease (COPD) patients. Eur Respir J. 2016;48:120.
20. Zanini A, Spanevello A, Baraldo S, et al. Decreased maturation of dendritic cells in the central airways of COPD patients is associated with VEGF, TGF-β and vascularity. Respiration. 2014;87(3):234–242. doi:10.1159/000356749
21. Birch J, Johnson G, Jiwa K, et al. S65 Large and small airway epithelial cell senescence present in COPD and bronchiectasis. Thorax. 2013;68(Suppl 3):35. doi:10.1136/thoraxjnl-2013-204457.72
22. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi:10.1056/NEJMoa032158
23. Abo-Zeid A, Sabry I, Hany H. Effect of smoking cessation on airway inflammation in COPD. Egypt J Bronchol. 2013;7(1):13–20.
24. Eapen MS, Hansbro PM, Mcalinden K, et al. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci Rep. 2017;7(1):13392. doi:10.1038/s41598-017-13888-x
25. Eltboli O, Bafadhel M, Hollins F, et al. COPD exacerbation severity and frequency is associated with impaired macrophage efferocytosis of eosinophils. BMC Pulm Med. 2014;14(1):1–10. doi:10.1186/1471-2466-14-112
26. Stefano AD, Caramori G, Ricciardolo FLM, et al. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. 2004;34(8):1156–1167. doi:10.1111/j.1365-2222.2004.02030.x
27. Stoll P, Ulrich M, Bratke K, et al. Imbalance of dendritic cell co-stimulation in COPD. Respir Res. 2015;16(1):19. doi:10.1186/s12931-015-0174-x
28. Mori M, Andersson CK, Svedberg KA, et al. Appearance of remodelled and dendritic cell-rich alveolar-lymphoid interfaces provides a structural basis for increased alveolar antigen uptake in chronic obstructive pulmonary disease. Thorax. 2013;68(6):521. doi:10.1136/thoraxjnl-2012-202879
29. Pace E, Ferraro M, Minervini MI, et al. Innate immunity responses are differently altered in central and distal airways of COPD patients.
30. Perotin JM, Adam D, Vella-Boucaud J, et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. 2014;15(1):1–9. doi:10.1186/s12931-014-0151-9
31. Manzel LJ, Shi L, O’Shaughnessy PT, Thorne PS, Look DC. Inhibition by cigarette smoke of nuclear factor-κB-dependent response to bacteria in the airway. Am J Respir Cell Mol Biol. 2011;44:155–165. doi:10.1165/rcmb.2009-0454OC
32. Sohal SS, Eapen MS, Ward C, et al. Epithelial mesenchymal transition (EMT): a necessary new therapeutic target in COPD? Am J Respir Crit Care Med. 2017;196(3):393. doi:10.1164/rccm.201704-0771LE
33. Ganesan S, Sajjan US. Repair and remodeling of airway epithelium after injury in Chronic Obstructive Pulmonary Disease. Respir Care. 2013;2(3):145–154.
34. Saied EM, Bediwy AS. Expression of epidermal growth factor receptor (EGFR) in the bronchial epithelium of patients with chronic obstructive pulmonary disease (COPD). Eur Respir J. 2011;125(55):3823.
35. Martin C, Frija-Masson J, Burgel PR, et al. Targeting mucus hypersecretion: new therapeutic opportunities for COPD? Drugs. 2014;74(10):1073–1089. doi:10.1007/s40265-014-0235-3
36. Pouwels SD, Heijink IH, Hacken NHT, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215–226. doi:10.1038/mi.2013.77
37. Pouwels SD, Van Geffen WH, Jonker MR, et al. Increased neutrophil expression of pattern recognition receptors during COPD exacerbations. Respirology. 2017;22(2):401–404. doi:10.1111/resp.12912
38. Kinose D, Ogawa E, Kudo M, et al. Association of COPD exacerbation frequency with gene expression of pattern recognition receptors in inflammatory cells in induced sputum. Clin Respir J. 2016;10(1):11–21. doi:10.1111/crj.2016.10.issue-1
39. Halper-Stromberg E, Yun JH, Parker MM, et al. Systemic markers of adaptive and innate immunity are associated with COPD severity and spirometric disease progression. Am J Respir Cell Mol Biol. 2018;58(4):500–509. doi:10.1165/rcmb.2017-0373OC
40. Freeman CM, Curtis JL. Lung dendritic cells: shaping immune responses throughout chronic obstructive pulmonary disease progression. Am J Respir Cell Mol Biol. 2017;56(2):152.
41. Givi ME, Redegeld FA, Folkerts G, et al. Dendritic cells in pathogenesis of COPD. Curr Pharm Des. 2012;18(16):2329–2335. doi:10.2174/138161212800166068
42. Givi ME, Folkerts G, Wagenaar GTM, et al. Cigarette smoke differentially modulates dendritic cell maturation and function in time. Respir Res. 2015;16(1):1–10. doi:10.1186/s12931-015-0291-6
43. Shan M, Cheng HF, Song LZ, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1(4):4ra10. doi:10.1126/scitranlsmed.3000154
44. Ma YJ. Correlation between small intestinal bacterial overgrowth and dendritic cell phenotype and function in cirrhotic patients with hepatitis B. World J Gastroenterol. 2016;24(3):443.
45. Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1:59–70. doi:10.1081/COPD-120028701
46. Stewart JI, Criner GJ. The small airways in chronic obstructive pulmonary disease: pathology and effects on disease progression and survival. Curr Opin Pulm Med. 2013;19(2):109–115. doi:10.1097/MCP.0b013e32835ceefc
47. Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76(3):509–513. doi:10.1189/jlb.0504272
48. Dewhurst JA, Lea S, Hardaker E, et al. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci Rep. 2017;7(1):7143. doi:10.1038/s41598-017-07101-2
49. Berenson CS, Kruzel RL, Eberhardt E, et al. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. 2013;208(12):2036–2045. doi:10.1093/infdis/jit400
50. Eurlings IM, Dentener MA, Mercken EM, et al. A comparative study of matrix remodeling in chronic models for COPD; mechanistic insights into the role of TNF-α. Am J Physiol Lung Cell Mol Physiol. 2014;307(7):557–565. doi:10.1152/ajplung.00116.2014
51. Eurlings IM, Reynaert NL, van de Wetering C, et al. Involvement of c-Jun N-terminal kinase in TNF-α-driven remodeling. Am J Respir Cell Mol Biol. 2017;56(3):393. doi:10.1165/rcmb.2015-0195OC
52. Pouwels SD, Van Geffen WH, Jonker MR, et al. Increased neutrophil expression of pattern recognition receptors during COPD exacerbations. Respirology. 2016;22(2):401. doi:10.1111/resp.12912
53. Ellingsen J, Hedenström H, Högman M, et al. The relationship between blood neutrophils, blood eosinophils and exacerbations of COPD–results from the TIE-study. Eur Respir J. 2016;48(suppl 60):PA1013.
54. Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121(5):151S–155S. doi:10.1378/chest.121.5_suppl.151S
55. Tulah AS, Parker SG, Moffatt MF, et al. The role of ALOX5AP, LTA4H and LTB4R polymorphisms in determining baseline lung function and COPD susceptibility in UK smokers. BMC Med Genet. 2011;12(1):173. doi:10.1186/1471-2350-12-173
56. Sng JJ, Prazakova S, Thomas PS, et al. MMP-8, MMP-9 and neutrophil elastase in peripheral blood and exhaled breath condensate in COPD. COPD. 2017;14(2):238–244. doi:10.1080/15412555.2016.1249790
57. Echevarría LU, Leimgruber C, González JG, et al. Evidence of eosinophil extracellular trap cell death in COPD: does it represent the trigger that switches on the disease? Int J Chron Obstruct Pulmon Dis. 2017;12:885–896. doi:10.2147/COPD
58. Kaur M, Beardsall M, Singh D TLR stimulation induces glucocorticoid insensitive production of functional neutrophil chemoattractant from COPD alveolar macrophages.
59. Sin D, van Eeden SF. Neutrophil-mediated lung damage: a new COPD phenotype? Respiration. 2012;83(2):103–105. doi:10.1159/000334178
60. Rijavec M, Volarevic S, Osolnik K, et al. Natural killer T cells in pulmonary disorders. Respir Med. 2011;105(supplement 1):S20. doi:10.1016/S0954-6111(11)70006-3
61. Vijayanand P, Seumois G, Pickard C, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. 2007;356(14):1410–1422. doi:10.1056/NEJMoa064691
62. Urbanowicz RA, Lamb JR, Todd I, et al. Altered effector function of peripheral cytotoxic cells in COPD. Respir Res. 2009;22:53. doi:10.1186/1465-9921-10-53
63. Hodge G, Mukaro V, Holmes M, et al. Enhanced cytotoxic function of natural killer and natural killer T-like cells associated with decreased CD94 (Kp43) in the chronic obstructive pulmonary disease airway. Respirology. 2013;18(2):369–376. doi:10.1111/j.1440-1843.2012.02287.x
64. Tsao CC, Tsao PN, Chen YG, et al. Repeated activation of lung invariant NKT cells results in Chronic Obstructive Pulmonary Disease-like symptoms. PLoS One. 2016;11(1):e0147710. doi:10.1371/journal.pone.0147710
65. Emoto M, Kaufmann SH. Liver NKT cells: an account of heterogeneity. Trends Immunol. 2003;24:364–369. doi:10.1016/S1471-4906(03)00162-5
66. Tang Y, Li X, Wang M, et al. Increased numbers of NK cells, NKT-like cells, and NK inhibitory receptors in Peripheral blood of patients with chronic obstructive disease. Clin Dev Immunol. 2013;2013:721–782.
67. Zeiger RS, Tran TN, Butler RK, et al. Relationship of blood eosinophil count to exacerbations in Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol Pract. 2018;6(3):944–954. doi:10.1016/j.jaip.2017.10.004
68. Villalobos RE, Magallanes J, Davidwang A. Blood eosinophilia as predictor for patient outcomes in COPD exacerbations: a systematic review and meta-analysis. Thorax. 2017;71(Suppl 3):A160–A160. doi:10.1136/thoraxjnl-2016-209333.286
69. Baloira VA, Pallaries SA. Chronic obstructive pulmonary disease with eosinophilia, an emerging phenotype? Arch Bronconeumol. 2016;52(4):177–178. doi:10.1016/j.arbr.2016.02.012
70. Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697–1700. doi:10.1183/09031936.00162414
71. Mortaz E, Folkerts G, Redegeld F. Mast cells and COPD. Pulm Pharmacol Ther. 2011;24(4):372. doi:10.1016/j.pupt.2011.03.007
72. Soltani A, Ewe YP, Lim ZS, et al. Mast cells in COPD airways: relationship to bronchodilator responsiveness and angiogenesis. Eur Respir J. 2012;39(6):1361–1367. doi:10.1183/09031936.00084411
73. Glader P, Wachenfeldt KV, Löfdahl CG. Systemic CD4+ T-cell activation is correlated with FEV 1, in smokers. Respir Med. 2006;100(6):1088–1093. doi:10.1016/j.rmed.2005.09.025
74. Siena L, Gjomarkaj M, Elliot J, et al. Reduced apoptosis of CD8+ T-lymphocytes in the airways of smokers with mild/moderate COPD. Respir Med. 2011;105(10):1491–1500. doi:10.1016/j.rmed.2011.04.014
75. Sullivan AK, Simonian PL, Falta MT, et al. Oligoclonal CD4+ T cells in the lungs of patients with severe emphysema. Am J Respir Crit Care Med. 2005;172:590–596. doi:10.1164/rccm.200410-1332OC
76. Wen Y, Reid DW, Zhang D, et al. Assessment of airway inflammation using sputum, BAL, and endobronchial biopsies in current and ex-smokers with established COPD. Int J Chron Obstruct Pulmon Dis. 2010;5:327–334.
77. Lapperre TS, Postma DS, Gosman MME, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115. doi:10.1136/thx.2005.040519
78. Geerdink JX, Simons SO, Pike R, et al. Differences in systemic adaptive immunity contribute to the ‘frequent exacerbator’ COPD phenotype. Respir Res. 2016;17(1):140. doi:10.1186/s12931-016-0456-y
79. Caramori G, Ruggeri P, Di SA, et al. Autoimmunity and COPD: clinical implications. Chest. 2018;153(6):1424–1431. doi:10.1016/j.chest.2017.10.033
80. Chiappori A, Folli C, Balbi F, et al. CD4+CD25 high DC 127-regulatory T-cells in COPD: smoke and drugs effect. World Allergy Organ J. 2016;9(1):5. doi:10.1186/s40413-016-0095-2
81. Duan MC, Zhang JQ, Liang Y, et al. Infiltration of IL-17-producing T cells and Treg cells in a mouse model of smoke-induced emphysema. Inflammation. 2016;39(4):1334–1344. doi:10.1007/s10753-016-0365-8
82. Toczyska I, Zwoliå„Ska E, Chcialowski A. Influence of inhaled corticosteroids on bronchial inflammation and pulmonary function in Chronic Obstructive Pulmonary Disease with moderate obstruction. Adv Exp Med Biol. 2018;1047:41–52.
83. Soltani A, Walters EH, Reid DW, et al. Inhaled corticosteroid normalizes some but not all airway vascular remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2016;22(11):2359–2367. doi:10.2147/COPD.S113176
84. Sohal SS, Soltani A, Reid D, et al. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study. Int J Chron Obstruct Pulmon Dis. 2014;27(9):533–542. doi:10.2147/COPD.S63911
85. Singh R, Mackay AJ, Patel AR, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res. 2014;15(1):1–10. doi:10.1186/s12931-014-0114-1
86. D’Anna SE, Balbi B, Cappello F, et al. Bacterial–viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. 2016;11(1):445. doi:10.2147/COPD.S93398
87. Blasi F, Mantero M, Aliberti S. Antibiotics as immunomodulant agents in COPD. Curr Opin Pharmacol. 2012;12(3):293–299. doi:10.1016/j.coph.2012.01.006
88. Giusti M, Blasi F, Iori I, et al. Prulifloxacin vs levofloxacin for exacerbation of COPD after failure of other antibiotics. COPD. 2016;13(5):555–560. doi:10.3109/15412555.2016.1152236
89. Patel A, Wilson R. Newer fluoroquinolones in the treatment of acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2012;1(3):243–250.
90. Bruno A, Cipollina C, Di VS, et al. Ceftaroline modulates the innate immune and host defense responses of immunocompetent cells exposed to cigarette smoke. Toxicol Lett. 2017;279:9–15. doi:10.1016/j.toxlet.2017.07.878
91. Heulens N, Korf H, Janssens W. Innate immune modulation in COPD: moving closer towards vitamin D therapy. J Pharmacol Exp Ther. 2015;353(2):360–368. doi:10.1124/jpet.115.223032
92. Bel EH, Ten BA. New Anti-eosinophil drugs for asthma and COPD: targeting the trait! Chest. 2017;152(6):1276–1282. doi:10.1016/j.chest.2017.05.019
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.