Back to Journals » OncoTargets and Therapy » Volume 13
HMGB1 in Radiotherapy: A Two Headed Signal Regulating Tumor Radiosensitivity and Immunity
Authors Liao Y ![]() , Liu S, Fu S
, Liu S, Fu S ![]() , Wu J
, Wu J
Received 13 March 2020
Accepted for publication 19 June 2020
Published 14 July 2020 Volume 2020:13 Pages 6859—6871
DOI https://doi.org/10.2147/OTT.S253772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Yin Liao,* Shuya Liu,* Shaozhi Fu, Jingbo Wu
Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingbo Wu; Shaozhi Fu
Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, People’s Republic of China
Email [email protected]; [email protected]
Abstract: Radiotherapy (RT) is a mainstay of cancer treatment. Recent studies have shown that RT not only directly induces cell death but also has late and sustained immune effects. High mobility group box 1 (HMGB1) is a nuclear protein released during RT, with location-dependent functions. It is essential for normal cellular function but also regulates the proliferation and migration of tumor cells by binding to high-affinity receptors. In this review, we summarize recent evidence on the functions of HMGB1 in RT according to the position, intracellular HMGB1 and extracellular HMGB1. Intracellular HMGB1 induces radiation tolerance in tumor cells by promoting DNA damage repair and autophagy. Extracellular HMGB1 plays a more intricate role in radiation-related immune responses, wherein it not only stimulates the anti-tumor immune response by facilitating the recognition of dying tumor cells but is also involved in maintaining immunosuppression. Factors that potentially affect the role of HMGB1 in RT-induced cytotoxicity have also been discussed in the context of possible therapeutic applications, which helps to develop effective and targeted radio-sensitization therapies.
Keywords: autophagy, DNA damage repair, high mobility group box 1, immune modulation, tumor radiosensitivity
Introduction
Radiotherapy (RT) remains the mainstay of cancer treatment because of the ability to induce DNA double strand breaks (DSBs), which can result in direct cancer cell death.1 Recently, research into improving outcomes of RT focused on changes of the tumor cell phenotype, and the complex biological interactions between tumor cells and tumor-associated stroma in the tumor microenvironment (TME).2,3 RT-induced anti-tumor immunity is generated by the interaction of both immune-activating signals and immune suppressive factors.4,5 After tumor cells are damaged by RT, the release of tumor antigens and damage-associated molecular patterns (DAMPs) can change the TME into an immune-stimulatory profile, thereby inducing an effective anti-tumor immune response.6–8 The major contributor to this “all-sided” response is RT-induced immunogenicity cell death (ICD). Typically, ICD facilitates the uptake of tumor antigens by dendritic cells (DCs) and promotes T cell activation and infiltration, which transforms a tumor into in-situ vaccines.9,10 However, in some cases, the intricacy of tumor resistance to radiation may cause ICD to be unsuccessful. Therefore, a second stimulus,such as hyperthermia and necroptosis inducers, is necessary to better induce ICD in irradiated tumors, which results in better tumor control. For example, Podolska and colleagues demonstrated that graphene-induced hyperthermia in combination with RT resulted in higher levels of ICD in B16F10 melanoma cells.11 Moreover, a specific immune response was implemented in immunocompetent animals that were inoculated with tumor cells undergoing ICD, which was associated with immunological memory.12 Thus, it can be concluded that successful induction of ICD may directly influence the efficacy of RT.
One of the key hallmarks of ICD is the release of high mobility group box 1 (HMGB1), a histone-chromatin binding protein that belongs to DAMPs. However, HMGB1 plays a contradictory role in RT, and the function of HMGB1 changes with its location.13,14 Inside the cell, nuclear HMGB1 binds loosely with histones (H1 and H5) to stabilize chromosomal structure and facilitate nucleosome sliding, which involves DNA transcription, recombination, and repair.15 Cytoplasmic HMGB1 drives autophagy by promoting lysosomal degradation and maintains cell homeostasis.16 Outside the cell, HMGB1 can activate and mobilize antigen-presenting cells by binding to Toll-like receptors (TLRs), and can drive inflammatory responses by activating downstream inflammatory cytokines.17 Notably, the presence of extracellular HMGB1 is a two-edged sword: while a transient increase in secreted levels of HMGB1 can induce immune responses against tumor cells,18 chronic accumulation in the extracellular space can result in abnormal pathophysiological conditions, such as cancer.19,20 In previous studies, it was shown that HMGB1 can combine with specific ligands to induce chronic inflammation, thereby driving malignant transformation by inducing immunosuppression, activation of oncogenes, and inhibition of tumor suppressors.21,22 Moreover, HMGB1 can directly promote the production of cytokines, such as vascular endothelial growth factor (VEGF), transforming growth factor β (TGF-β), and metalloproteinase (MMP) to favor tumor angiogenesis, invasion, and metastasis.23–25
HMGB1 has been implicated in tumor radio-resistance based on its DNA damage repair and autophagy functions, and radio-sensitization through immune-mediated tumor destruction.26–28 Thus, it is essential to elucidate the mechanisms underlying the action of HMGB1 in RT, to use it as a therapeutic target to increase radio-sensitivity. In the following sections, the pivotal role of both intracellular and extracellular HGMB1 in RT is discussed, along with the potential underlying mechanisms influencing the effect of RT to provide novel ideas for improving radiation effects.
Extracellular: HMGB1 and RT-Related Immune Response
Immune Recognition
The immune system can distinguish between “self” and “non-self” antigens based on pathogen associated molecular patterns (PAMPs) and between “danger” and “non-danger” based on DAMPs.8,29 PAMPs often refer to the recognition of common components of bacteria and viruses, such as peptidoglycans and lipoproteins. In general, aberrant clones from normal cells, such as tumor cells, carry mostly autoantigens, and thus are not recognized by PAMPs. Accordingly, recognition of tumor cells by the immune system is more dependent on DAMPs, which refer to endogenously derived danger signals or alarmins released by dying cells.30 RT is known to cause fatal damage to tumor cells, which improves recognition of the immune system through the release of DAMPs. In general, RT-induced DAMPs contain the following cellular hallmarks: calreticulin, heat shock proteins (HSPs), adenosine 5-triphosphate, and HMGB1.31 Several DAMPs, including HSPs, are increased at both exposed surface expression and are released after RT treatment.32,33 Stimulated by these danger signals, RT-induced systemic antitumor immunity not only shrinks local tumors but also controls distant tumors, a phenomenon known as the “abscopal effect”.34,35 However, in several studies, it has been reported that a relative increase in the number of immune suppressive properties in the TME after RT, including TGF- β and immune checkpoint molecules, can potentially impair RT-induced anti-tumor immune responses.36 Therefore, RT-induced immune responses against tumors can be considered a newly alternative radio-sensitizing modality.
The immune system can recognize and respond to dying tumor cells, and HMGB1 serves as a “necrotic marker” for recognition by immune cells. Importantly, in this role, immune stimulation characteristics of HMGB1 presuppose that they should be released by dead cells. The lethal damage caused by RT is sufficient to result in loss of cell membrane integrity, and HMGB1 must be released from inside to outside the cell. Due to lack of a guiding peptide, active secretion of HMBG1 is mediated via vesicles rather than via the (ER)/Golgi secretion pathway, whereas loss of membrane integrity and necrosis passively releases cytosolic HMGB1.37 Of note, the release of HMGB1 from necrotic tumor cells has been reported to also recruit macrophages and neutrophils, resulting in tumor growth and metastasis.38,39 Figure 1 presents the myriad immune-regulatory roles of HMGB1.
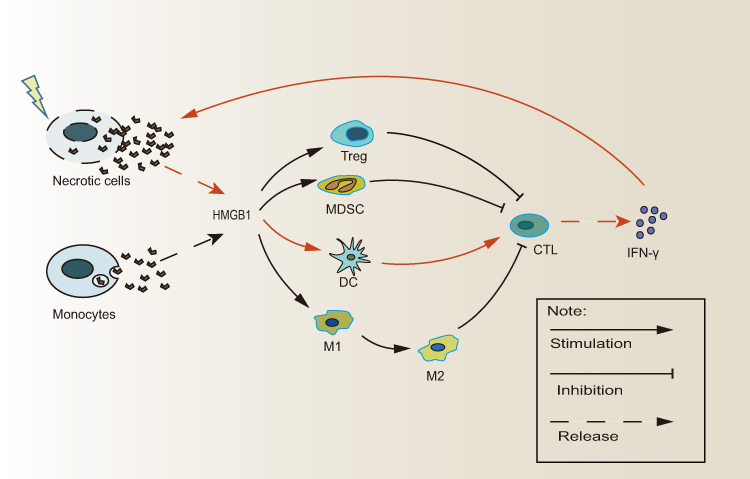 |
Figure 1 The paradoxical role of extracellular HMGB1 in RT-related immune effect. HMBG1 can be released in two ways: Monocytes actively secrete HMBG1 through vesicles, while necrotic cells passively leak cytoplasmic HMGB1. Extracellular HMGB1 stimulates DC-mediated activation of CD8+T cells and subsequent IFN-γ secretion, which further increases the number of necrotic tumor cells and promote more HMGB1 release. However, HMGB1 facilitates the growth of immunosuppressive cell such as Tregs, MDSCs, and promotes the polarization of M1-type macrophages to M2-type macrophages. (DCs, Dendritic cells; HMGB1, High Mobility Group Box 1; IFN-γ, Interferon-γ; MDSCs, Marrow-derived inhibitory cells; Treg,T regulatory cell). |
Inflammation
Solid tumors tend to be immunosuppressive and resist infiltration of inflammatory cells.40 However, RT can induce an acute inflammatory reaction in tumors by upregulating multiple pro-inflammatory cytokines, including TNF, IL-1α, and IL-1β.41,42 In these inflammatory conditions, binding of HMGB1 with receptors for advanced glycation end-products (RAGEs) triggers the RAGE/JNK/NF-κB inflammatory signaling pathway and plays a role in maintaining inflammation.43 Once out of the cell, HMGB1 induces the release of TNFα, IL-1α and other pro-inflammatory cytokines from monocytes in an autocrine manner, thereby creating an inflammatory environment.44 Furthermore, HMGB1 can form complexes with DNA, lipopolysaccharide (LPS), il-1β, and nucleosomes that interact with different receptors to promote inflammation.45 Especially, HMGB1/DNA complexes released from necrotic tissue are thought to be highly pro-inflammatory, and act as a death signal to promote cytokine production through binding with TLR9.46 As a cytokine, HMGB1 can directly activate endothelial cells to up-regulate the adhesion molecule Mac-1, which leads to rapid recruitment, adhesion, and migration of neutrophils.47,48 Moreover, the binding of HMGB1 to TLRs is conducive for the infiltration and chemotaxis of immune effectors, including Th1 cells, cytotoxic T-lymphocytes (CTLs), and natural killer (NK) cells. The activation of TLRs increases secretion of CCL2, CCL5, and CXCL10, and the resulting chemokine gradient attracts cytolytic immune cells into tumor tissues.49 In addition to this, the interaction of HGMB1 with TLR2 can activate NK cells via NF-κB, STAT3, and Smad3 signaling pathways to induce anti-tumor immune responses.50 Taken together, HMGB1 accelerates RT-mediated transformation of tumors into “acute inflammatory” tissues by activating cytokines, which is critical for initiation of the adaptive immune response.
Adaptive Immune Response
Maturation of Dendritic Cells
DCs are potent antigen presenting cells (APCs) that mediate RT-induced adaptive immune responses by engulfing, processing, and presenting tumor antigens to specific T-cells.51 Immature DCs differentiate into mature cells in response to antigenic stimuli, which is accompanied by upregulation of CD40, CD80, CD86, and MHC that function as T-cell activators and co-stimulatory factors.52,53 However, immunosuppressive factors in the TME inhibit DC maturation. In a previous report, it was identified that HMGB1 mediates activation of DCs and enhances the expression of CD80 and CD86 on DCs in vitro in a dose-dependent manner.54 Furthermore, upon maturation DCs secrete high levels of HMGB1, which up-regulates CXCR4 and CCR7 chemokine receptors, and therefore can respond to chemokine receptor ligands.55 In this regard, the autocrine/paracrine release of HMGB1 and the HMGB1/RAGE pathway are essential for the migratory function of DCs.55 All events, including the surface expression of chemokine receptors as well as DC migration, abate in the presence of antagonists of HMGB1 or its receptor, RAGE.56 Moreover, HMGB1 can enhance the production of various cytokines by DCs, including IL-6, IL-12p70, and TNF-α.56 Importantly,Kanegasaki’s group showed that radiation-induced HMGB1 inhibited tumor growth in non-irradiated sites by targeting ECI301 (eMIP), a single amino-acid substituted CCL3, to form a DC-activating complex.57 These findings suggested that HMGB1 contributed to a strong tumor control mediated by DCs. In a subsequent study, it was hypothesized that the HMGB1-IFN-γ loop was the regulatory mechanism of DC-mediated anti-tumor immune responses against lung cancer.58 HMGB1 stimulates the DC-mediated activation of CD8+T cells and therefore IFN-γ secretion, which further increases the release of HMGB1 from tumor cells and promotes DC maturation.59 Taken together, HMGB1 may be one of the significant mediators augmenting RT-mediated cancer immune responses by stimulating DC maturation.
Recruitment of T Lymphocytes
As discussed previously, the clinical outcome of local RT is dependent on the tumor infiltration of effector immune cells.60 HMGB1 directly mediates adaptive immunity as a proliferation signal of activated T lymphocytes.61 In addition, release of HMGB1 by DCs controls clonal expansion, survival, and the functional polarization of naive T cells via RAGE.62 The interaction between HMGB1 and RAGE activates MAPKs (p38 and ERK1/2) and NF-κB pathways to promote maturation of DCs, and subsequent Th1 polarization of pre-activated T cells.62 Moreover, HMGB1 was essential to the surface expression of cytokine lymphotoxinα1β2 on T-cells, which contributes to their activation and intratumoral accumulation.63 To test whether HMGB1 is required for the activation of T-cells, He’s group first blocked HMGB1 in mice and observed a weakened priming of adoptively transferred T-cells.64 Also, HMGB1 modulates the expression of CXCL11 to enhance the infiltration of T-cells.65 Suzuki and colleagues reported that the HMGB1 that is released following chemo-radiotherapy is necessary for immune reactions in patients with esophageal squamous cell carcinoma (ESCC), and may affect clinical outcomes.66 Tumor antigen–specific T-cell responses were confirmed to coexist with elevated serum levels of HMGB1.66 Importantly, HMGB1 released by dying tumor cells promotes the activation of tumor-antigen specific T-cells and increases the expression of IFN-γ, a key player in driving anti-tumor immunity.67 Taken together, these findings indicate that HMGB1 may be one of the significant mediators linking antigen-specific T-cell responses to RT-induced cell death.
Opposite: Immunosuppressive Cell Accumulation
RT is known to transform the TME by mobilizing T-cell responses.68 Paradoxically, HMGB1 is involved in maintaining the TME under certain conditions that might contribute to the suppression of RT-mediated antitumor immunity.69 HMGB1 assists the proliferation, survival, and function of several pro-tumor immunosuppressive cells that are described below to facilitate immune tolerance and tumor escape.49
Macrophages play an important role in immunomodulation. It has previously been shown that HMGB1 expression positively correlated with peritumoral macrophage infiltration, which represented a poor prognosis in hepatocellular carcinoma patients.70 Tumor infiltrating macrophages (TAMs) can be subdivided into two distinct functional phenotypes: M1-type macrophages that are activated through classical pathways and mediate T-cells responses by secreting various cytokines and expressing major histocompatibility complex (MHC) class I and MHC-II. M2-type macrophages are activated alternatively and account for most TAMs that contribute to T-cell suppression via IL-10 and transforming and growth factor β (TGFβ).71,72 Interestingly, in a bladder cancer model, RT in combination with the HMGB1 inhibitor glycyrrhizin significantly increased the number of anti-tumor M1-type macrophages when compared with RT alone.73 Thus, this suggests that HMGB1 promotes HMGB1-mediated inhibition of M1-type macrophages. Moreover, tumor-derived HMGB1 can efficiently trigger the differentiation of monocytes into PD1+ TAMs with a substantial enhancement in CD206 and IL-10 expression, thereby inhibiting CD8+ T-cell proliferation and promoting the development of ESCC.19 Moreover, He and coworkers reported that although HMGB1 induced adaptive immunity in a prostate cancer model, and the ensuing T-cell response resulted in further recruitment of macrophages and promoted tumor progression, and the blockade of HMGB1 could hinder macrophage infiltration.63 These findings confirmed that HMGB1 could dampen RT-mediated immune responses by infiltration of macrophages and skewing the polarization of macrophages.
In the TME formation process, regulatory T cells (Tregs) produce significant IL-10 to mediate immune suppression. In addition, Tregs showed more resistance to irradiation compared to other lymphocytes, which may be responsible for the immune evasion of tumor cells after RT.74 In activated CD4+CD25+T cells, HMGB1 can upregulate the transcription factor Foxp3 to enhance the differentiation of Tregs and dominantly control the suppressive capacity of Tregs in the neuroblastoma microenvironment in vitro.69 Moreover, HMGB1 was found to trigger the production of thymic stromal lymphopoietin by tumor cells, which is necessary for the activation of Tregs.75 Tregs significantly express a high level of RAGE on the cell surface, and HMGB1 directly enhances the suppressive capacity of Tregs in a RAGE-dependent manner.76 HMGB1 may combine with RAGE on Tregs and activate transcriptional factors (AP-1 and NF-kB) for IL-10 production in Tregs.77 Tumor cell-derived HMGB1 facilitates Tregs to produce IL-10, which promotes Tregs-mediated suppression of CD8+ T cell anti-tumor responses in vitro and in vivo.78 In addition, HMGB1 acts as a chemoattractant for Tregs and prolongs their survival by mediation of TLR4 and RAGE.79 Therefore, it is possible that the interaction of HMGB1 with Treg receptors increases infiltration of the latter into tumor tissues, and shifts RT-induced antitumor responses in favor of the tumor.
Myeloid-derived suppressor cells (MDSCs) represent a heterogeneous population of immature myeloid cells, including precursors of granulocytes, DCs, and macrophages that accumulate during tumor progression and chronic inflammation.80 MDSC expansion may be a possible factor driving tumor metastasis and RT-induced secondary growth. In particular, HMGB1 promotes the survival of MDSC in an anoxic and nutritionally deficient tumor microenvironment.81 In a previous study on renal cell carcinoma, it was found that HMGB1 did not directly inhibit the proliferation of T cells and B cells, but instead, induced the proliferation of MDSCs to mediate a pro-tumor effect. By down-regulating HMGB1, the differentiation and proliferation of MDSCs were significantly inhibited, both in vitro and in vivo.82 Parker and coworkers reported that HMGB1 regulates the quantity and quality of MDSCs in murine tumor systems through activation of the NF-κB signal transduction pathway. HMGB1 stimulates bone marrow progenitor cells to differentiate into MDSC, thereby increasing the MDSC-mediated production of IL-10 as well as suppression on expression of the naive T cell homing receptor L-selectin.83 Therefore, it can be speculated that HMGB1 secretion can dampen the effects of RT through MDSC-mediated immunosuppression.
Intracellular: HMGB1 and RT-Related DNA Damage
According to traditional radiobiology, RT-induced DNA damage includes base and sugar damage, single-strand breaks (SSBs), and DSBs.84 X-rays emit high-energy photons that transfer energy to cellular molecules, resulting in the formation of unstable and highly reactive oxygen species (ROS) that oxidize chemical bonds and induce DNA damage.85 The cell-cycle checkpoints are then activated to allow cells to repair the damaged DNA before entering the next cycle. However, if the level of radiation-induced DNA damage exceeds its repair ability, the apoptotic pathways are triggered, leading to cell death.86 Thus, the DNA repair ability of tumor cells is the key factor to determine survival and radio-resistance under nonlethal radiation with cell membranes maintaining integrity.
HMGB1 is involved in four major DNA repair pathways: nucleotide excision repair (NER), mismatch repair (MMR), base excision repair (BER) and double strand break repair (DSBR).87,88 DNA repair proteins cannot fully access DSBs due to heterochromatin factors that limit the flexibility of nucleosomes.89 HMGB1 has a high affinity for non-standard DNA structures,90,91 and can bind to and bend the helix at the site of radiation-induced damage, resulting in greater distortion of DNA.91–93 This allows NER proteins, such as XPA, RPA, and RAD23B to be removed from the nucleosomes and facilitate accessibility of the damaged DNA.91 The proposed model of HGMB1-induced NER is named “Access-Repair-Restore”.87 HMGB1 recognizes the injury in the access phase through proteins such as ACF, TFTC, STAGA and CBP, which leads to damage repair and restoration. HMGB1 also plays a role in non-homologous end joining of DSBs repair and V(D)J recombination by enhancing intra- and inter-DNA ligation, and recruiting DNA-dependent protein kinase catalytic subunit (PKcs) to the DNA break ends. Even in the absence of complementarity, HMGB1 can increase the proximity of the DNA duplexes and their termini to facilitate break end ligation.94,95 Furthermore, HMGB1 also increases the stability of DNA by bending and looping the linear DNA strands into a more compact structure.96
The crucial role of HMGB1 in promoting error-free DNA repair is underscored by the significant increase in DNA damage and mutations in HMGB1-knockout mice after exposure to DNA damaging agents.91 The suppression of HMGB1 also sensitized cancer cells to radiation by inducing cell cycle arrest at the G0/G1 phase, thereby enhancing apoptosis.97 In HMGB1 knockdown urothelial carcinoma cell lines, the increase of post-radiation DNA damage was induced, and showed a three-fold increase in γH2AX foci after irradiation.26 Likewise, HMGB1 knockdown inhibited the repair of DNA damage in MCF-7 cells by decreasing the levels of telomerase reverse transcriptase (hTERT) and cyclin D1, leading to accumulation of p-ATM, p-ATR, and γH2AX foci.98 In another study, it was shown that the interaction between HMGB1 and retinoblastoma (RB) protein increased genomic stability post-radiation, and promoted radio-resistance.99 This radiation-protective behavior is achieved inside cells with intact membranes. Taken together, HMGB1 meaningfully mediates DNA damage repair and is critical to cancer cell radio-resistance under certain circumstances.
Intracellular: HMGB1 and RT-Related Autophagy
Autophagy is a catabolic pathway that involves lysosomal degradation of damaged organelles and misfolded proteins that recycles cellular components, and sustains energy levels.100 In other words, autophagy is a self-protective response of cells to RT-induced various stresses, including hypoxia, nutrient deprivation, and therapeutic side effects. Autophagy is regulated by a group of evolutionarily conserved genes that are collectively known as autophagy-related genes (ATG).101–103 Target molecules are first surrounded by cytoplasmic vesicles, and the resulting autophagosomes fuse with lysosomes to form autophagolysosomes in which these molecules are digested and released into the cytoplasm.104 Cancer cells exposed to irradiation show accumulation of autophagosomes, and a marked increase in the levels of Beclin-1 and the microtubule-associated protein LC3-II.105,106 In addition, induction of autophagy can affect RT-mediated radiobiological effects in solid tumors.107 For example, the autophagic pathway is stimulated in radioresistant MDA-231 breast cancer cells following irradiation, while autophagy levels are only slightly altered in radiosensitive HBL-100 cells.108 Thus, autophagy greatly influences the therapeutic effect of irradiation and can contribute to radio-tolerance.
HMGB1 is a pivotal regulator of autophagy and presumably a promoter of tumor radio-resistance.109 Cytosolic HMGB1 binds to Beclin1 through an intra-molecular disulfide bridge (C23/45), which frees Beclin1 from its complex with Bcl2 and subsequently induces autophagy.110 Disassociation of the Bcl2–Beclin1 complex, the molecular switch of autophagy, is negatively regulated by p53.111 In bladder cancer UM-UC3 cells, the association between HMGB1 and Beclin1 increased four-fold and that between HMGB1 and p53 decreased three-fold after radiation exposure.26 In addition, nuclear HMGB1 can also induce autophagy via transcriptional activation of heat shock protein B1 (HSPB1) through the Pink1/Parkin pathway.112 Phosphorylated HSPB1 (Ser15 and Ser86) plays a key role in actin polymerization and recombination, which is necessary for autophagy-related intracellular transport. In this regard, the absence of HSPB1 limits the fusion of autophagy vacuoles and lysosomes.113,114 The nuclear HMGB1/HSP-induced autophagy pathway likely contributes to cellular survival and radio-resistance. In a breast cancer study, downregulated HMGB1 mRNA and protein significantly suppressed RT-induced autophagy and sensitized cells to irradiation.115 Ma and coworkers also reported increased LC3 II puncta and accumulation of autophagosomes in siHMGB1-transfected ESCC cells.116 Taken together, high levels of HGMB1 induced by radiation exposure enhance autophagy and most likely protects the cells from radiotoxicity (Figure 2).
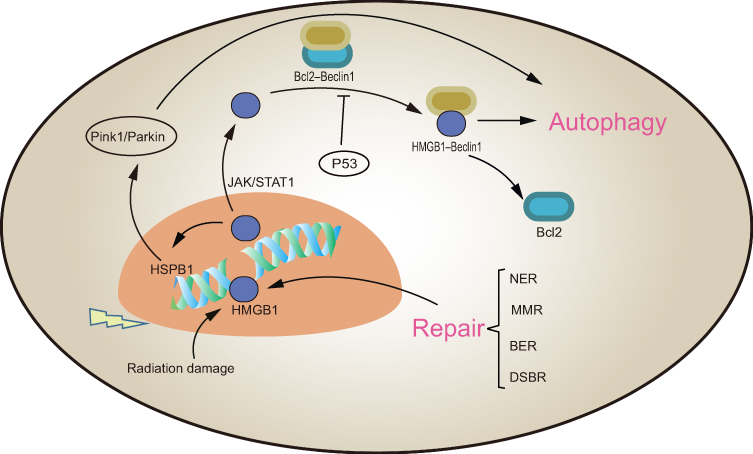 |
Figure 2 The role of intracellular HMGB1 in RT. HMGB1 is involved in the identification of DNA damage and four major DNA repair processes: nucleotide excision repair (NER), mismatch repair (MMR), base excision repair (BER) and double strand break repair (DSBR). Nuclear HMGB1 promotes the transcription of HSPB1 to induce autophagy. Cytoplasmic HMGB1 binds with Bclin1 separated from the Bcl2-Bclin1 complex to promote autophagy. Both DNA damage repair and autophagy promote tumor radio-resistance. |
Discussion: Potential Factors Altering the Role of HMGB1 in RT
The role of HMGB1 in RT is highly paradoxical. A non-lethal level of radiation leaves the cell membrane intact, and HMGB1 that is retained inside the cell promotes tumor survival and radio-resistance by repairing damaged DNA and inducing autophagy. This might explain that increased level of nuclear and cytoplasmic HMGB1 is a marker of poor prognosis in the early stages of cancer.117 Because lethal radiation causes cell necrosis and cell membrane fragmentation, HMGB1 is released to the outside of the cell and subsequently induces complex immune responses. As discussed above, HMGB1 can facilitate both tumor elimination and immune evasion by respectively mediating anti- and pro-tumor responses. The final outcome might depend on which side the balance tilts.
The dichotomy between physiological and RT-induced tumor cell death that respectively leads to tumor progression and elimination hinges on the release of HMGB1 that links chronic inflammatory responses to acute adaptive immunity. Campana’s group concluded that post-radiation immune responses are dependent on the nature of the HGMB1 release.118 Since the rapid growth of tumors exceeds their nutrient supply, the core of solid tumors is hypoxic and has an extreme low pH.119,120 Therefore, most cells in the core of the tumor are necrotic and continuously secrete HMGB1. Steady accumulation of HMGB1 in advanced tumors induces chronic inflammation, and aggravates tumor growth and metastasis.121 This can explain why HMGB1 expression was significantly higher in tumor tissues compared to normal tissues and associated with larger tumor volumes, higher rates of lymphatic invasion, and more frequent lymph node metastases.122 While active secretion of HMGB1 in physiological conditions or chronic passive secretion from dying cells facilitates neo-angiogenesis and tumor invasion/metastasis,19,20 pulsed acute release in response to lethal radiation promotes an anti-tumor immune response.18,118,123 The proportion of various immune cells in the TME is the major difference between chronic and acute inflammatory responses, and is also one of the reasons for HMGB1-mediated contradictory effects on the tumor.124 Immunosuppressive cells, such as Tregs are dominant during chronic inflammation, and TLR-activated CD8+T cells and NK cells prevailed in the acute stage and eliminated tumor cells (Figure 3).125,126
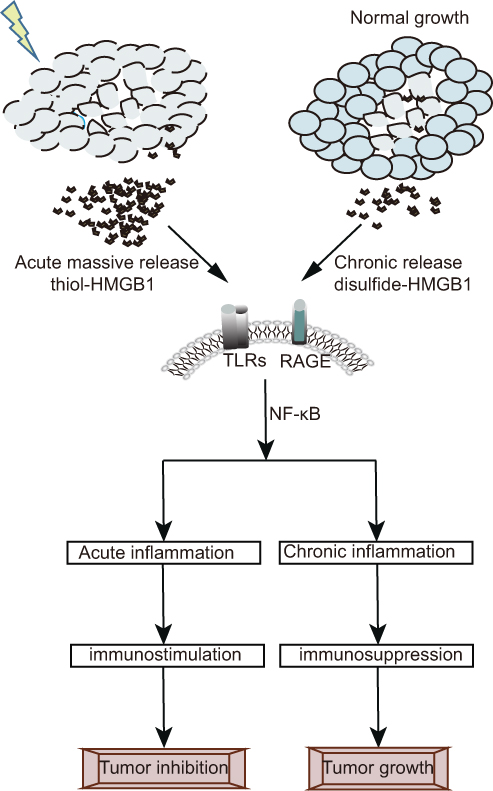 |
Figure 3 Post-radiation immune response is dependent on the nature of HGMB1 release. Necrotic cells release bursts of pre-formed cytosolic non-acetylated thiol-HMGB1, while dying cells with transient biological activity slowly release newly synthesized or modified disulfide-HMGB1.The pulsed acute release of thiol-HMGB1 is more likely to form heterocomplexes which act as strong immune stimulators to enhance leukocyte recruitment and promote acute inflammation dominated by immunologic effector cell. On the contrary, the chronic release of disulfide-HMGB1 promotes the tumor growth with the formation of immunosuppressive chronic inflammation. |
Chronic and acute HMGB1 release is characterized by distinct post-translational modifications and distinct binding receptors, and determines immune outcome. For instance, necrotic cells release bursts of pre-formed cytosolic non-acetylated thiol-HMGB1, while dying cells with transient biological activity slowly release newly synthesized or modified disulfide-HMGB1.127,128 It is worth noting that reductive proteins are more likely to bind CXCL12 to form heterocomplexes, which act as strong immune stimulators to enhance leukocyte recruitment via the chemokine receptor CXCR4.129 The interaction between HMGB1 and different receptors may determine key biological effects in HMGB1-mediated paradoxical immune responses. TLRs are a large group of HMGB1 receptors, which are involved in host HMGB1-mediated inflammation and immune responses. TLRs/HMGB1 recognition predominantly transmits signals in RT through two pathways, including myeloid differentiation primary response protein-88 (MyD88) and Toll/IL-1R domain–containing adaptor inducing IFNα (TRIF), which plays an important anti-tumor role.18 RAGE is another trans-membrane receptor that is possibly involved in promoting HMGB1-mediated maintenance of chronic inflammatory states after RT.130 It has previously been demonstrated that mice lacking RAGE resist the induction of skin cancer.131 Moreover, the interaction of HMGB1 and RAGE promotes tumor angiogenesis to favor tumor growth.132
In addition to the HMGB1 isoform and receptor type, the amount of extracellular HMGB1 also influences its immune effects. Ma and coworkers showed that extracellular HMGB1 downregulated the tumor suppressor SAM and SH3 domain-containing 1 gene (SASH1), but further supplementation with recombinant HMGB1 upregulated SASH1 and inhibited tumor growth in a concentration-dependent manner.133 Thus, the type and amount of HMGB1 affects the direction of RT-induced immune responses, however this needs to be validated further.
The amount and nature of HMGB1 release also depend on the type and potency of external stress. For instance, low levels of H2O2 induced cytoplasmic translocation of HMGB1, which was actively released as the oxidative stress increased. Finally, cytotoxic doses of the oxidant resulted in both active and passive release of HMGB1.134 This may explain the dose-dependent variation in radiation in anti-tumor immune responses.135 While low doses of radiation result in inflammation due to the inability to induce immunogenic tumor cell death, high doses of radiation have shown to induce in situ vaccination and stimulate the immune system. In several studies, it has been shown that high doses of RT achieve better tumor inhibition and clinical effects when compared to low doses by increasing T-cell activity.136–138 RT induced HMGB1 in a dose-dependent manner and once the immunogen signal from dying tumor cells reached a certain threshold, the anti-tumor immune response was activated.123 However, continuous up-regulation of HMGB1 can aggravate tissue damage and release more danger signals, thereby resulting in a vicious cycle of chronic inflammation and tumor progression.139 This is a potential factor in RT failure and may even lead to tumor relapse and metastasis. Therefore, RT-related immune responses mediated by HMGB1 need to be further elucidated.
Conclusions and Outlook
In recent years, the pathological role of HMGB1 in various diseases has gained significant attention. In many experimental studies that focused on infectious and inflammatory diseases, HMGB1 as a successful target was confirmed.140,141 However, the ambiguous role of HMGB1 in cancer complicates any potential therapeutic use. Unrepaired DSBs of DNA are lethal to the cell, therefore, combining RT with HMGB1 inhibitors can enhance DNA damage repair and improve radio-sensitivity. Given the role of HMGB1 in the maintenance of cell homeostasis, HGMB1-induced autophagy can be targeted to reverse radiation tolerance. HMGB1 inhibitors can improve the efficacy of RT, and systemic inhibition of HMGB1 can impair immune responses against infection and other physiological functions. Therefore, targeted delivery of HMGB1 in RT to tumors is worth exploring in the future. The introduction of nanomedicine may offer novel opportunities for targeting HMGB1 treatment of cancer. Nanoparticles can encapsulate various drugs and multivalent surface modifications with targeting ligands and efficiently improve current drug delivery standards for biological distribution and intra-tumor uptake in vivo in a complex environment.142
Although much research has been done on the anti-tumor role of HMGB1 that acts as an alarm molecule, cancer cells have the ability to secrete HMGB1 to maintain the inflammatory TME and favor their own growth.124,126 The biological characteristics of HMGB1-maintained chronic inflammation are dominated by the recruitment and persistence of MDSCs and Tregs, the promotion of fibrosis and angiogenesis, and the production of other individual inflammatory factors, including IL-1β and LPS.126 This immunosuppressive network, composed of an array of cytokines, immune inhibitory cells, and the extracellular matrix, mutually blocks antigen presentation by DCs and inhibits T-cell-mediated antitumor immune effects. Alternately, HMGB1 is a signature DAMP that is released from dying tumor cells after RT, and is likely associated with radiation-induced anti-tumor immune responses that are dominated by immune effector cells.57,143 Lethal doses skew the immune landscape from suppressive cells to effector cells, altering the inflammatory status from chronic to acute, and optimizing immune responses. Mature DCs and activated CD8+T cells prevail in this acute inflammation and arrive at the tumor site under the guidance of homing chemokines to remove tumors in an immune-activated environment.59 Thus, extracellular HMGB1 can be harnessed to inhibit tumor growth by triggering cell damage, which releases high amounts of HMGB1 in acute pulses. However, this approach carries the risk of chronic inflammation, tumor progression, and organ failure. Future studies will focus on selecting the correct RT dose and fractionation frequency to optimize the stimulation of HMGB1 in RT.
Abbreviations
APCs, antigen-presenting cells; ATG, autophagy-related genes; BER, base excision repair; CRT, calreticulin; CTLs, cytotoxic T lymphocytes; DAMPs, damage-associated molecular patterns; DCs, dendritic cells; DDR, DNA damage repair; DSBs,double strand breaks; ERK, extracellular- regulated kinase; HMGB1, high mobility group box 1; HSP70, heat shock protein; ICD, immunogenicity cell death; IFN-γ, interferon-γ; IL-1α, interleukin-1α; IL-2, interleukin-2; IR, ionizing radiation; JAK, Janus Kinase; MAPK, mitogen-activated protein kinase; MDSCs, marrow-derived inhibitory cells; MHC, major histocompatibility complex; MMR, mismatch repair; MyD88, myeloid differentiation primary response protein-88; NER, nucleotide excision repair; NF- κB, nuclear factor-κB; NK cells, natural killer cells; PAMPs, pathogen associated molecular Patterns; RAGE, receptor for advanced glycation end-products; RT, radiotherapy; STAT3, signaltransducers and activators of transcription 3; TAMs, tumor-associated macrophages; TLRs, toll-like receptors; TRIF, Toll/IL-1R domain-containing adaptor inducing IFNα; Treg, T regulatory cell; TME, tumor microenvironment.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Lomax ME, Folkes LK, O’neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol. 2013;25(10):578–585. doi:10.1016/j.clon.2013.06.007
2. Jarosz-Biej M, Smolarczyk R, Cichoń T, Kułach N. Tumor microenvironment as a “game changer” in cancer radiotherapy. Int J Mol Sci. 2019;20(13). doi:10.3390/ijms20133212
3. Shahabi V, P MA, T D, W JD. Immune-priming of the tumor microenvironment by radiotherapy: rationale for combination with immunotherapy to improve anticancer efficacy. Am J Clin Oncol. 2015;38(1):90–97. doi:10.1097/COC.0b013e3182868ec8
4. Muroyama Y, Nirschl TR, Kochel CM, et al. Stereotactic radiotherapy increases functionally suppressive regulatory t cells in the tumor microenvironment. Cancer Immunol Res. 2017;5(11):992–1004. doi:10.1158/2326-6066.Cir-17-0040
5. Ozpiskin O, Zhang L, L JJ. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics. 2019;9(5):1215–1231. doi:10.7150/thno.32648
6. Deloch L, Derer A, Hartmann J,et al. Modern radiotherapy concepts and the impact of radiation on immune activation. Front Oncol. 2016;6:141. doi:10.3389/fonc.2016.00141
7. Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. 2015;9(10):1966–1981. doi:10.1016/j.molonc.2015.10.016
8. Garg AD, A P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280(1):126–148. doi:10.1111/imr.12574
9. Gameiro SR, Ardiani A, Kwilas A, Hodge JW. Radiation-induced survival responses promote immunogenic modulation to enhance immunotherapy in combinatorial regimens.. Oncoimmunology. 2014;3(5):e28643. doi:10.4161/onci.28643
10. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11–17. doi:10.1016/j.semradonc.2014.07.005
11. Pinho AR, Martins F, Elisabete VA, et al. Graphene oxide nanosheets for localized hyperthermia-physicochemical characterization, biocompatibility, and induction of tumor cell death. Cells. 2020;9(3). doi:10.3390/cells9030776
12. Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Letters. 2018;438:17–23. doi:10.1016/j.canlet.2018.08.028
13. Xin Y, Jiang F, Yang C, et al. Role of autophagy in regulating the radiosensitivity of tumor cells. J Cancer Res Clin Oncol. 2017;143(11):2147–2157. doi:10.1007/s00432-017-2487-2
14. Di X, H G, H H, et al. High-mobility group box 1 protein modulated proliferation and radioresistance in esophageal squamous cell carcinoma. J Gastroenterol Hepatol. 2019;34(4):728–735. doi:10.1111/jgh.14371
15. Bonaldi T. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 2002;21(24):6865–6873. doi:10.1093/emboj/cdf692
16. Tang D, Kang R, Livesey KM, Zeh HJ
17. Kono K, Mimura K, Kiessling R. Immunogenic tumor cell death induced by chemoradiotherapy: molecular mechanisms and a clinical translation. Cell Death & Disease. 2013;4(6):e688. doi:10.1038/cddis.2013.207
18. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059. doi:10.1038/nm1622
19. Li B, Song T-N, Wang F-R, et al. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1+ TAM expansion. Oncogenesis. 2019;8(3):17. doi:10.1038/s41389-019-0126-2
20. A E, P C, A C, Papavassiliou AG. Pivotal role of high-mobility group box 1 (HMGB1) signaling pathways in glioma development and progression. J Mol Med. 2016;94(8):867–874. doi:10.1007/s00109-016-1435-y
21. Sato Y, Goto Y, Narita N, Hoon DS. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron. 2009;205–214. doi:10.1007/s12307-009-0022-y
22. Pikarsky E, Porat RM, S I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi:10.1038/nature02924
23. Wu C-Z, Zheng -J-J, Bai Y-H, et al. HMGB1/RAGE axis mediates the apoptosis, invasion, autophagy, and angiogenesis of the renal cell carcinoma. OncoTargets and Therapy. 2018;11(1):4501–4510. doi:10.2147/OTT.S167197
24. Zhu L, Li X, Chen Y, Fang J, Ge Z. High-mobility group box 1: a novel inducer of the epithelial-mesenchymal transition in colorectal carcinoma. Cancer Letters. 2015;357(2):527–534. doi:10.1016/j.canlet.2014.12.012
25. P X, Zhang Y, Zhang S. High-mobility group box 1 is overexpressed in cervical carcinoma and promotes cell invasion and migration in vitro. Oncology Reports. 2017;37(2):831–840. doi:10.3892/or.2016.5317
26. Shrivastava S, Mansure JJ, Almajed W, et al. The role of HMGB1 in radioresistance of bladder cancer. Mol Cancer Ther. 2016;15(3):471–479. doi:10.1158/1535-7163.MCT-15-0581
27. He S, Cheng J, Sun L, et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018;9(6):648. doi:10.1038/s41419-018-0626-6
28. Zhou H, Jin C, Cui L, et al. HMGB1 contributes to the irradiation-induced endothelial barrier injury through receptor for advanced glycation endproducts (RAGE). J Cell Physiol. 2018;233(9):6714–6721. doi:10.1002/jcp.26341
29. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805(1):53–71. doi:10.1016/j.bbcan.2009.08.003
30. Pitt JM, Kroemer G, Zitvogel L. Immunogenic and non-immunogenic cell death in the tumor microenvironment. 2017;1036:65.
31. Rapoport BL, Anderson R. Realizing the clinical potential of immunogenic cell death in cancer chemotherapy and radiotherapy. Int J Mol Sci. 2019;20(4). doi:10.3390/ijms20040959
32. Schildkopf P, Frey B, Ott OJ, et al. Radiation combined with hyperthermia induces HSP70-dependent maturation of dendritic cells and release of pro-inflammatory cytokines by dendritic cells and macrophages. Radiother Oncol. 2011;101(1):109–115. doi:10.1016/j.radonc.2011.05.056
33. E Schmid T, Multhoff G. Radiation-induced stress proteins - the role of heat shock proteins (HSP) in anti- tumor responses. Curr Med Chem. 2012;19(12):1765–1770. doi:10.2174/092986712800099767
34. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin. 2017;67(1):65–85. doi:10.3322/caac.21358
35. Chicas-Sett R, Morales-Orue I, Rodriguez-Abreu D, Lara-Jimenez P. Combining radiotherapy and ipilimumab induces clinically relevant radiation-induced abscopal effects in metastatic melanoma patients: a systematic review. Clin Transl Rad Oncol. 2018;9(1):5–11. doi:10.1016/j.ctro.2017.12.004
36. Frey B, Rückert M, Deloch L, et al. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunological Reviews. 2017;280(1):231–248. doi:10.1111/imr.12572
37. Raucci A, Palumbo R, Bianchi ME. HMGB1: a signal of necrosis. Autoimmunity. 2007;40(4):285–289. doi:10.1080/08916930701356978
38. Lv W, Na N, Lin Y, et al. Macrophage migration inhibitory factor promotes breast cancer metastasis via activation of HMGB1/TLR4/NF kappa B axis. Cancer Letters. 2016;375(2):245–255. doi:10.1016/j.canlet.2016.02.005
39. Chen X, Z L, J Y, et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J Exp Clin Cancer Res. 2018;37(1):77. doi:10.1186/s13046-018-0726-2
40. Cassim S, Pouyssegur J. Tumor microenvironment: a metabolic player that shapes the immune response. Int J Mol Sci. 2019;21(1). doi:10.3390/ijms21010157
41. Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol. 2013;23(4):273–280. doi:10.1016/j.semradonc.2013.05.009
42. Mukherjee D, Coates PJ, Lorimore SA, Wright EG. Responses to ionizing radiation mediated by inflammatory mechanisms. J Pathol. 2014;232(3):289–299. doi:10.1002/path.4299
43. Zhao J, Sun T, W S, L Y. High Mobility Group Box 1: an immune-regulatory protein. Curr Gene Ther. 2019;19(2):100–109. doi:10.2174/1566523219666190621111604
44. Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192(4):565–570. doi:10.1084/jem.192.4.565
45. Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86(3):573–576. doi:10.1189/jlb.1008585
46. Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi:10.1038/ni1457
47. Wu X, Mi Y, Yang H, Hu A, Zhang Q, Shang C. The activation of HMGB1 as a progression factor on inflammation response in normal human bronchial epithelial cells through RAGE/JNK/NF-kappaB pathway. Mol Cell Biochem. 2013;380(1–2):249–257. doi:10.1007/s11010-013-1680-0
48. Orlova VV, Choi EY, Xie C, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26(4):1129–1139. doi:10.1038/sj.emboj.7601552
49. Patidar A, Selvaraj S, Sarode A, Chauhan P, Chattopadhyay D, Saha B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine. 2018;104:114–123. doi:10.1016/j.cyto.2017.10.004
50. Conti L, Lanzardo S, Arigoni M, et al. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013;27(12):4731–4744. doi:10.1096/fj.13-230201
51. Persa E, Szatmári T, Sáfrány G, Lumniczky K. In vivo irradiation of mice induces activation of dendritic cells. Int J Mol Sci. 2018;19(8). doi:10.3390/ijms19082391
52. Choi C, Jeong MH, Park Y-S, Son C-H, Lee H-R, Koh K. Combination treatment of stereotactic body radiation therapy and immature dendritic cell vaccination for augmentation of local and systemic effects. Cancer Res Treat. 2019;51(2):464–473. doi:10.4143/crt.2018.186
53. Messmer D, Yang H, Telusma G, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173(1):307–313. doi:10.4049/jimmunol.173.1.307
54. Manfredi AA, Capobianco A, Esposito A, et al. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol. 2008;180(4):2270–2275. doi:10.4049/jimmunol.180.4.2270
55. Dumitriu IE, Bianchi ME, B M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81(1):84–91. doi:10.1189/jlb.0306171
56. Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81(1):59–66. doi:10.1189/jlb.0306180
57. Kanegasaki S, Tsuchiya T. Alarmins released during local antitumor treatments play an essential role in enhancing tumor growth inhibition at treated and non-treated sites via a derivative of CCL3. Oncoimmunology. 2014;3(10):e958956. doi:10.4161/21624011.2014.958956
58. Gao Q, Li F, Wang S, et al. A cycle involving HMGB1, IFN-γ and dendritic cells plays a putative role in anti-tumor immunity. Cellular Immunology. 2019;343:103850. doi:10.1016/j.cellimm.2018.08.011
59. Gao Q, Li F, Wang S, et al. A cycle involving HMGB1, IFN-gamma and dendritic cells plays a putative role in anti-tumor immunity. Cell Immunol. 2019;343:103850. doi:10.1016/j.cellimm.2018.08.011
60. Li XY, Xiao L, Zhang JH, Chen L. The irradiated-fibroblasts elicit immune response against tumor in a murine colon cancer model. Eur Rev Med Pharmacol Sci. 2015;19(15):2812–2817.
61. S E, Fasth AER, P K, Harris H, Andersson U. High mobility group box chromosomal protein 1 acts as a proliferation signal for activated T lymphocytes. Immunobiology. 2009;214(4):303–309. doi:10.1016/j.imbio.2008.09.006
62. Dumitriu IE, B P, V B, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174(12):7506–7515. doi:10.4049/jimmunol.174.12.7506
63. He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013;73(2):629–639. doi:10.1158/0008-5472.Can-12-2704
64. He Y, Zha J, Wang Y, et al. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013;73(2):629–639. doi:10.1158/0008-5472.Can-12-2704
65. Gao Q, Wang S, Chen X, et al. Cancer-cell-secreted CXCL11 promoted CD8+ T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer. 2019;7(1):42. doi:10.1186/s40425-019-0511-6
66. Suzuki Y, Mimura K, Yoshimoto Y, et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72(16):3967–3976. doi:10.1158/0008-5472.Can-12-0851
67. Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220(1):47–59. doi:10.1111/j.1600-065X.2007.00573.x
68. Demaria S, Formenti SC. Role of T lymphocytes in tumor response to radiotherapy. Front Oncol. 2012;(2):95. doi:10.3389/fonc.2012.00095
69. V T, W C, J P, U A, S C, S H. In vitrosecretory High-Mobility Group Box 1 protein affects regulatory T cell differentiation in neuroblastoma microenvironment. J Oncol. 2018;2018:7946021. doi:10.1155/2018/7946021.
70. Zhang Q-B, Jia Q-A, Wang H, et al. High-mobility group protein box1 expression correlates with peritumoral macrophage infiltration and unfavorable prognosis in patients with hepatocellular carcinoma and cirrhosis. BMC Cancer. 2016;16(1):880. doi:10.1186/s12885-016-2883-z
71. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi:10.3389/fimmu.2014.00614.
72. Verreck FAW, de Boer T, Langenberg L, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101(13):4560–4565. doi:10.1073/pnas.0400983101
73. Ayoub M, Shinde-Jadhav S, Mansure JJ, et al. The immune mediated role of extracellular HMGB1 in a heterotopic model of bladder cancer radioresistance. Sci Rep. 2019;9(1):6348. doi:10.1038/s41598-019-42864-w
74. Kachikwu EL, Iwamoto KS, Liao Y-P, et al. Radiation enhances regulatory T cell representation. Int J Rad Oncol Biol Phys. 2011;81(4):1128–1135. doi:10.1016/j.ijrobp.2010.09.034
75. Zhang Y, Liu, Liu Z, et al. Tumor-derived high-mobility group box 1 and thymic stromal lymphopoietin are involved in modulating dendritic cells to activate T regulatory cells in a mouse model. Cancer Immunol Immunother. 2017;67(3):353–366. doi:10.1007/s00262-017-2087-7
76. Wild CA, Bergmann C, Fritz G, et al. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24(8):485–494. doi:10.1093/intimm/dxs051
77. R A, F H, M E. Fueling inflammation at tumor microenvironment: the role of multiligand/RAGE axis. Carcinogenesis. 2010;31(3):334–341. doi:10.1093/carcin/bgp322
78. Liu Z, Falo LD
79. Wild CA, Brandau S, Lotfi R, et al. HMGB1 is overexpressed in tumor cells and promotes activity of regulatory T cells in patients with head and neck cancer. Oral Oncol. 2012;48(5):409–416. doi:10.1016/j.oraloncology.2011.12.009
80. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid-derived suppressor cells: important contributors to tumor progression and metastasis. J Cell Physiol. 2018;233(4):3024–3036. doi:10.1002/jcp.26075
81. Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA. Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother. 2019. doi:10.1007/s00262-019-02388-8
82. Li J, Sun J, Li L, et al. HMGB1 promotes myeloid-derived suppressor cells and renal cell carcinoma immune escape. Oncotarget. 2017;8(38):63290–63298. doi:10.18632/oncotarget.18796
83. Parker KH, Sinha P, Horn LA, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74(20):5723–5733. doi:10.1158/0008-5472.Can-13-2347
84. Morgan MA, Lawrence TS. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin Cancer Res. 2015;21(13):2898–2904. doi:10.1158/1078-0432.CCR-13-3229
85. Wang L, He L, Bao G, He X, Fan S, Wang H. Ionizing radiation induces HMGB1 cytoplasmic translocation and extracellular release. Guo Ji Fang She Yi Xue he Yi Xue Za Zhi. 2016;40(2):91–99. doi:10.3760/cma.j.issn.1673-4114.2016.02.002
86. Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers. 2019;11(6):862. doi:10.3390/cancers11060862
87. Lange SS, Vasquez KM. HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinogen 2009;48(7):571–580. doi:10.1002/mc.20544
88. M P, Vasquez KM. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: implications in DNA repair and immune responses. DNA Repair (Amst). 2019;83(1):102701. doi:10.1016/j.dnarep.2019.102701
89. Goodarzi AA, Noon AT, Deckbar D, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31(2):167–177. doi:10.1016/j.molcel.2008.05.017
90. Bidney DL, Reeck GR. Purification from cultured hepatoma cells of two nonhistone chromatin proteins with preferential affinity for single-stranded DNA: apparent analogy with calf thymus HMG proteins. Biochem Biophys Res Commun. 1978;85(3):1211–1218. doi:10.1016/0006-291x(78)90671-x
91. Lange SS, Mitchell DL, Vasquez KM. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci U S A. 2008;105(30):10320–10325. doi:10.1073/pnas.0803181105
92. Reddy MC, Christensen J, Vasquez KM. Interplay between human high mobility group protein 1 and replication protein A on psoralen-cross-linked DNA. Biochemistry. 2005;44(11):4188–4195. doi:10.1021/bi047902n
93. Lange SS, Reddy MC, Vasquez KM. Human HMGB1 directly facilitates interactions between nucleotide excision repair proteins on triplex-directed psoralen interstrand crosslinks. DNA Repair (Amst). 2009;8(7):865–872. doi:10.1016/j.dnarep.2009.04.001
94. Stros M, Cherny D, Jovin TM. HMG1 protein stimulates DNA end joining by promoting association of DNA molecules via their ends. Eur J biochem. 2000;267(13):4088–4097. doi:10.1046/j.1432-1327.2000.01450.x
95. Yuan F, Gu L, Guo S, Wang C, Li GM. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem. 2004;279(20):20935–20940. doi:10.1074/jbc.M401931200
96. Yamanaka S, Katayama E, Yoshioka K, Nagaki S, Yoshida M, Teraoka H. Nucleosome linker proteins HMGB1 and histone H1 differentially enhance DNA ligation reactions. Biochem Biophys Res Commun. 2002;292(1):268–273. doi:10.1006/bbrc.2002.6647
97. Zhang X, Yang X, Zhu S, Li Q, Zou N. Radiosensitization of esophageal carcinoma cells by knockdown of HMGB1 expression. Oncol Rep. 2019;41(3):1960–1970. doi:10.3892/or.2018.6923
98. Ke S, Zhou F, Yang H, et al. Downregulation of high mobility group box 1 modulates telomere homeostasis and increases the radiosensitivity of human breast cancer cells. Int J Oncol. 2015;46(3):1051–1058. doi:10.3892/ijo.2014.2793
99. Jiao X, Zhang S, Jiao J, et al. Promoter methylation of SEPT9 as a potential biomarker for early detection of cervical cancer and its overexpression predicts radioresistance. Clinical Epigenetics. 2019;11(1):120. doi:10.1186/s13148-019-0719-9
100. Xin Y, Jiang F, Yang C, et al. Role of autophagy in regulating the radiosensitivity of tumor cells. Journal of Cancer Research and Clinical Oncology. 2017;143(11):2147–2157. doi:10.1007/s00432-017-2487-2
101. Zarzynska JM. The importance of autophagy regulation in breast cancer development and treatment. Biomed Res Int. 2014;2014:1–9. doi:10.1155/2014/710345
102. Zhu X, Messer JS, Wang Y, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125(3):1098–1110. doi:10.1172/jci76344
103. Chai W, Ye F, Zeng L, Li Y, Yang L. HMGB1-mediated autophagy regulates sodium/iodide symporter protein degradation in thyroid cancer cells. J Exp Clin Cancer Res. 2019;38(1):325. doi:10.1186/s13046-019-1328-3
104. Kelekar A. Introduction to the review series Autophagy in Higher Eukaryotes–a matter of survival or death. Autophagy. 2008;4(5):555–556. doi:10.4161/auto.6396
105. Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi:10.4161/auto.19496
106. Chen YS, Song HX, Lu Y, et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus. 2011;24(6):437–443. doi:10.1111/j.1442-2050.2010.01156.x
107. Lomonaco SL, Finniss S, Xiang C, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125(3):717–722. doi:10.1002/ijc.24402
108. Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99(3):287–292. doi:10.1016/j.radonc.2011.06.002
109. Min HJ, Suh KD, Lee YH, Kim K, Mun S-K, Lee SY. Cytoplasmic HMGB1 and HMGB1-Beclin1 complex are increased in radioresistant oral squamous cell carcinoma. Br J Oral Maxillofacial Surg. 2019;57(3):219–225. doi:10.1016/j.bjoms.2019.01.011
110. Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. doi:10.1083/jcb.200911078
111. Livesey KM, Kang R, Vernon P, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72(8):1996–2005. doi:10.1158/0008-5472.Can-11-2291
112. Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37(6):315–324. doi:10.1016/j.tins.2014.03.004
113. Tang D, Kang R, Livesey KM, et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13(6):701–711. doi:10.1016/j.cmet.2011.04.008
114. Xu T, Jiang L, Wang Z. The progression of HMGB1-induced autophagy in cancer biology. Onco Targets Ther. 2019;12:365–377. doi:10.2147/OTT.S185876
115. Luo J, Chen J, He L. mir-129-5p attenuates irradiation-induced autophagy and decreases radioresistance of breast cancer cells by targeting HMGB1. Med Sci Monit. 2015;21:4122–4129. doi:10.12659/msm.896661
116. Ma H, Zheng S, Zhang X, et al. High mobility group box 1 promotes radioresistance in esophageal squamous cell carcinoma cell lines by modulating autophagy. Cell Death Dis. 2019;10(2):136. doi:10.1038/s41419-019-1355-1
117. Xu Y, Chen Z, Zhang G, et al. HMGB1 overexpression correlates with poor prognosis in early-stage squamous cervical cancer. Tumour Biol. 2015;36(11):9039–9047. doi:10.1007/s13277-015-3624-7
118. Campana L, Bosurgi L, Rovere-Querini P. HMGB1: a two-headed signal regulating tumor progression and immunity. Curr Opin Immunol. 2008;20(5):518–523. doi:10.1016/j.coi.2008.04.012
119. Lotfi R, Kaltenmeier C, Lotze MT, Bergmann C. Until death do us part: necrosis and oxidation promote the tumor microenvironment. Transfus Med Hemother. 2016;43(2):120–132. doi:10.1159/000444941
120. Zhang Y, Pan N, Sheng Y, et al. Hypoxia enhances IL-10-producing B cell generation through upregulating high-mobility group B1 on tumor cell-released autophagosomes. Immunol Lett. 2019;216:36–42. doi:10.1016/j.imlet.2019.09.005
121. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34(5):637–650. doi:10.1016/j.immuni.2011.05.006
122. Ueda M, T Y, S Y, et al. Prognostic significance of high mobility group box 1 (HMGB1) expression in patients with colorectal cancer. Anticancer Res. 2014;34(10):5357–5362.
123. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3(4):e28518. doi:10.4161/onci.28518
124. Rojas A, Delgado-Lopez F, Perez-Castro R, et al. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016;37(3):3321–3329. doi:10.1007/s13277-015-3940-y
125. Ameri AH, Moradi Tuchayi S, Zaalberg A, et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc Natl Acad Sci U S A. 2019;116(7):2646–2651. doi:10.1073/pnas.1815016116
126. Gorgulho CM, Romagnoli GG, Bharthi R, Lotze MT. Johnny on the spot-chronic inflammation is driven by HMGB1. Front Immunol. 2019;10:10. doi:10.3389/fimmu.2019.01561.
127. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol. 2017;17(4):262–275. doi:10.1038/nri.2017.9
128. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534–544. doi:10.1016/j.phrs.2016.06.031
129. Fassi EMA, Sgrignani J, D’Agostino G, et al. Oxidation state dependent conformational changes of HMGB1 regulate the formation of the CXCL12/HMGB1 heterocomplex. Comput Struct Biotechnol J. 2019;17:886–894. doi:10.1016/j.csbj.2019.06.020
130. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. An Rev Med. 2018;69(1):349–364. doi:10.1146/annurev-med-041316-085215
131. Gebhardt C, Riehl A, Durchdewald M, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205(2):275–285. doi:10.1084/jem.20070679
132. Schlueter C, Weber H, Meyer B, et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166(4):1259–1263. doi:10.1016/s0002-9440(10)62344-9
133. Ma C, Chen H, Zhang S, et al. Exosomal and extracellular HMGB1 have opposite effects on SASH1 expression in rat astrocytes and glioma C6 cells. Biochem Biophys Res Commun. 2019;518(2):325–330. doi:10.1016/j.bbrc.2019.08.057
134. Tang D, Shi Y, Kang R, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81(3):741–747. doi:10.1189/jlb.0806540
135. de la Cruz-merino L, Illescas-Vacas A, Grueso-Lopez A, Barco-Sanchez A, Miguez-Sanchez C. Cancer Immunotherapies Spanish G. radiation for awakening the dormant immune system, a promising challenge to be explored. Front Immunol. 2014;5. doi:10.3389/fimmu.2014.00102.
136. Sologuren I, Rodríguez-Gallego C, Lara PC. Immune effects of high dose radiation treatment: implications of ionizing radiation on the development of bystander and abscopal effects. Transl Cancer Res. 2014;3(1):18–31. doi:10.3978/j.issn.2218-676X.2014.02.05
137. Lippitz BE, Harris RA. A translational concept of immuno-radiobiology. Radiother Oncol. 2019;140(2019):116–124. doi:10.1016/j.radonc.2019.06.001
138. Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res. 2010;173(4):406–417. doi:10.1667/RR1931.1
139. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:1–21. doi:10.1155/2010/672395
140. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Ann Rev Immunol. 2011;29(1):139–162. doi:10.1146/annurev-immunol-030409-101323
141. Schaper F, Heeringa P, Bijl M. Inhibition of high-mobility group box 1 as therapeutic option in autoimmune disease: lessons from animal models. Curr Opin Rheumatol. 2013;25(2):254–259. doi:10.1097/BOR.0b013e32835cee2d
142. Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21(4):223–232. doi:10.1016/j.molmed.2015.01.001
143. Yoshimoto Y, Oike T, Okonogi N, et al. Carbon-ion beams induce production of an immune mediator protein, high mobility group box 1, at levels comparable with X-ray irradiation. J Radiat Res. 2015;56(3):509–514. doi:10.1093/jrr/rrv007
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.