Back to Archived Journals » Neurobehavioral HIV Medicine » Volume 7
Neurodevelopmental delay in pediatric HIV/AIDS: current perspectives
Authors Blokhuis C, Kootstra N, Caan M, Pajkrt D
Received 1 July 2015
Accepted for publication 24 October 2015
Published 13 January 2016 Volume 2016:7 Pages 1—13
DOI https://doi.org/10.2147/NBHIV.S68954
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Stephen Ferrando
Video abstract presented by Dr Dasja Pajkrt.
Views: 575
Charlotte Blokhuis,1 Neeltje A Kootstra,2 Matthan WA Caan,3 Dasja Pajkrt1
1Department of Pediatric Hematology, Immunology and Infectious Diseases, Emma Children's Hospital, 2Department of Experimental Immunology, 3Department of Radiology, Academic Medical Center, Amsterdam, the Netherlands
Abstract: The effects of HIV on the developing nervous system of perinatally HIV (PHIV)-infected children are substantial, yet poorly understood. While the introduction of combination antiretroviral therapy (cART) reduced the prevalence of HIV encephalopathy, many cART-treated PHIV-infected children still present with neurodevelopmental delays and cognitive impairment. The underlying pathogenesis may partially differ from that in adults, as HIV not only causes direct injury to the central nervous system (CNS) but may also impede the development of the pediatric brain. Increasing evidence implies significant roles for ongoing neuroinflammation, vascular dysfunction and hypercoagulability induced by HIV despite adequate viral suppression. Subsequent manifestations of HIV in the pediatric brain include gray-matter volume reduction, white-matter lesions, and basal ganglia calcifications, but even in the absence of such macrostructural changes, the detrimental effects of HIV are clearly revealed in widespread microstructural changes, such as poorer white-matter integrity and alterations in cerebral metabolites and blood flow. Viral suppression on cART evidently does not fully protect against cerebral injury, but knowledge concerning the pharmacokinetics and toxicity of cART in the pediatric CNS is lacking. Investigations combining neuropsychological assessment, multimodal neuroimaging, and laboratory evaluation of inflammatory and neurodegenerative markers could greatly increase our understanding of the pathogenesis of HIV in the developing nervous system and improve treatment and prevention strategies. Longitudinal research will be crucial to observe the long-term consequences of CNS exposure to HIV and cART in PHIV-infected children as they survive into adulthood.
Keywords: perinatally HIV-infected children, cognitive functioning, neuroinflammation, cerebral injury, cART
Introduction
HIV affects an estimated 3.2 million children worldwide, and the majority of the 240,000 newly infected children are perinatally HIV (PHIV)-infected.1 The virus can enter the central nervous system (CNS) within days after primary infection, posing a major threat to neurodevelopment.2 Indeed, without combination antiretroviral therapy (cART), 20%–50% of PHIV-infected children developed HIV-related encephalopathy.3 These children showed severe progressive neurological impairment or arrested neurodevelopment, including failure to attain or loss of developmental milestones, impaired brain growth, and motor deficits.4 Initiation of cART, which combines multiple antiretroviral agents from at least two drug classes, has been shown to decrease the incidence of HIV encephalopathy to under 2%.3 Nonetheless, many PHIV-infected children still show neurodevelopmental delay or neurocognitive impairment, which are thought to represent milder forms of HIV encephalopathy.5
The pathogenesis of neurodevelopmental delay in PHIV-infected children is not yet fully understood. As the nervous system and immune system, which should control viral replication, are still developing in children,6 and the clinical features of pediatric HIV encephalopathy differ from adult HIV-associated dementia (HAD), the underlying mechanisms of neurological pathogenesis may in part differ from that of adults.4,7 To identify further the devastating effects of HIV infection in the developing CNS in children, recent investigations have focused on inflammation, coagulation, and vascular dysfunction, as well as microstructural deviations assessed by multimodal neuroimaging. This review seeks to summarize current insights in the pathogenesis and evaluation of neurodevelopmental delay in PHIV-infected children and adolescents in the era of cART.
Clinical features
Neurodevelopmental delay spans motor, language, and cognitive domains, and affects HIV-infected children in both high-resource and low-resource settings. However, reported rates and severity of developmental delay are highly variable, due to inconsistency among tests and outcome measures, as well as population differences in age, immune status, and cART regimens.8,9 Furthermore, PHIV-infected children are often exposed to multiple other risk factors for neurodevelopmental delay, both physical (eg, prematurity, low birth weight or head circumference, stunting, wasting) and psychosocial (eg, low socioeconomic status, parental or caregiver unemployment, lower education level, or drug abuse), emphasizing the necessity of a well-matched control group for comparisons on HIV-related disease.8,10
Impaired motor development is found in up to 67% of PHIV-infected infants stable on cART when compared to either HIV-exposed but uninfected children, HIV-unexposed children, or national norms.11–14 The severity of these motor inadequacies varies from within normal range to more than two standard deviations below the population mean.12–14 Loss of motor function is less evident in older children, but subtle strength and fine-motor impairments have been reported in clinically stable school-age PHIV-infected children, both cART-naïve and -experienced.15,16
General cognition is significantly affected from an early age onward. PHIV-infected infants, school-age children, and adolescents show poorer cognitive performance when compared to controls or national norms, with rates of severe impairment (IQ <70 or scores more than two standard deviations below population mean) ranging from 16% to 46%.5,15,17 Additionally, PHIV-infected children display variable deficits in all subdomains of cognitive functioning, ie, processing speed, attention, and working memory, perceptual and visuospatial performance, and executive functioning,5,16,17 even when general cognition is within the normal range.18–20 Overall, processing speed, memory, and attention appear to be most severely affected.21
Language abatement is also seen in cART-treated PHIV-infected children of all ages. Both expressive and receptive language skills are poorer when compared to controls.15,22–25 Of note, this specific language disorder is independent of the presence of hearing deficits and lower general cognitive functioning as risk factors for language impairment.26
Impact
Few studies describe the consequences of impaired neurodevelopment on daily functioning of PHIV-infected children. Generally, a negative impact of HIV infection on school performance is implied by poor school results, failing or repeating grades, and high rates of special education.5,24,27 However, school performance is strongly influenced by many other factors, such as increased absence from school due to illness, hearing loss, family circumstances, socioeconomic factors, and caregiver education or intelligence, making the direct effect of HIV difficult to determine.21,27 There is also a strong association between poorer neurocognitive functioning and depression, conduct disorders, and increased risk-taking behavior in PHIV-infected adolescents, which may further negatively affect their health and academic performances.21,28
HIV-related risk factors
In utero exposure
Pediatric HIV studies often use HIV-exposed but uninfected children as a control group. However, several studies show that these children also have poorer cognitive performance when compared to HIV-unexposed children, suggesting that in utero exposure to HIV during pregnancy may already negatively affect later neurodevelopment.23,24,29 It is known that HIV can invade the fetal CNS,30 although it is unclear how this relates to future HIV serostatus or cerebral injury. PHIV-infected children that developed encephalopathy later in life were found to have a smaller head circumference at birth compared to nonencephalopathic PHIV-infected children, also indicative of a prenatal disease process.4 Studies evaluating a potential protective effect of in utero exposure to maternal cART use report contrasting outcomes regarding neurodevelopment.12,31 Research on the consequences of in utero exposure to HIV and cART is further complicated by the frequent presence of confounders, such as maternal substance abuse, prematurity, and low birth weight.32,33
HIV disease severity
The poorest neurocognitive outcomes are seen in HIV-infected children with a history of higher disease severity, indicating a significant contribution of residual HIV-induced cerebral injury to neurodevelopmental delay at a later age. Indeed, having experienced a Centers for Disease Control and Prevention class C event has been associated with lower scores in general cognition, processing speed, and verbal performance.5,24,34 This is in line with the finding of poorer outcomes in HIV-infected children with lower nadir CD4+ T-cell measurements and higher zenith viral load, although this association is not consistently found, possibly due to small study groups.5,34,35 Younger age at HIV diagnosis is also associated with higher rates of neurodevelopmental delay,36,37 and encephalopathy was found to occur more frequently in PHIV-infected children when compared to adults after similar duration of HIV infection.4 Overall, these findings strongly suggest that perinatally infected children are more susceptible to CNS damage caused by HIV infection than older children or adults.
In contrast to historical markers of HIV disease severity, HIV load and CD4+ T-cell count show less consistent correlations with neurocognitive performance,15,26,38 and some recent studies in children stable on cART did not detect this correlation at all.5,24 While HIV load and CD4+ T-cell count are commonly used as indicators of systemic HIV disease severity, to which the CNS may be exposed, they may not be a reliable indicator of HIV impact on the CNS. Systemic HIV load does not always represent cerebrospinal fluid (CSF) HIV load, which may be a better marker for cerebral HIV disease. However, studies have failed to show an association between CSF viral load and neurocognitive functioning in the setting of adequate viral suppression with cART.39,40 This could be due to insensitivity of commonly used detection tests or CSF viral load being an imperfect proxy for brain-tissue HIV replication, but may also indicate more prominent roles for other pathogenic mechanisms in chronic HIV-induced cerebral injury.40
Neuropathogenesis
Neurodevelopmental delay in pediatric HIV disease could be considered the mild end of the spectrum of HIV encephalopathy, with similar pathogenic mechanisms, but limited in severity by the improved treatment strategies that have become available over the last few decades.32 Several hypotheses concerning the neuropathogenesis of HIV exist, albeit largely based on histopathological studies in pre-cART adult populations.
Early HIV infection may lead to a degree of irreversible damage before the initiation of effective cART, which could account for some of the neurological deficits persisting over time.41 The contribution of this legacy effect is supported by the predictive value of historical HIV disease markers. In a chronic state, HIV infection may continue to injure the CNS even after HIV load suppression is achieved with cART. Firstly, the CNS is an anatomically distinct compartment in which HIV can escape therapy, allowing independent replication, genetic evolution, and continuing injury within the CNS. Detectable intrathecal HIV load despite systemic viral suppression has indeed been reported in up to 10% of both asymptomatic and neurologically symptomatic adults on cART, and discordance between plasma and CSF HIV genetic strains has been detected in adults and children on cART.39,42,43 Secondly, HIV infection – even with undetectable HIV load – may induce a chronic low-grade inflammatory state, consisting of persistent monocyte and macrophage activation, vascular endothelial activation, and a hypercoagulable state. Markers of inflammation and vascular dysfunction are indeed elevated in cohorts of cART-experienced children, correlating with poorer neurocognitive functioning.44,45 Furthermore, inflammation appears to be a better predictor for neurocognitive impairment than HIV load or CD4+ T-cell measurements in adults.46 Finally, chronic exposure to cART may itself induce neurotoxicity, contrasting with the evidenced protective effects.47
HIV invasion of the CNS
To invade the CNS, HIV must penetrate the blood–brain barrier (BBB) (Figure 1A), which is composed of a layer of human brain microvascular endothelial cells (HBMECs) linked by tight junctions. These cells work in close communication with pericytes and astrocytes in regulating bidirectional traffic of cells and substances between the systemic circulation and the CNS.48 The most feasible mechanism by which HIV crosses the BBB is by transmigration of infected monocytes and to a lesser degree lymphocytes.49 HIV proteins, such as gp120 and Tat, activate HBMECs, which then release inflammatory mediators, including TNFα and MCP-1, and express specific adhesion molecules, such as ICAMs and VCAMs, facilitating invasion of infected monocytes into the brain.50,51 The permeability of the BBB is further increased by HIV-induced oxidative injury, functional impairment of astrocytes and pericytes, and damage to tight junctions via upregulation of proteolytic MMPs, consequently allowing free HIV virions and viral proteins to pass through the BBB via transcellular and paracellular pathways.52–54 Productive infection of HBMECs is minimally evidenced, and thus unlikely to contribute significantly to HIV entry into the CNS.55
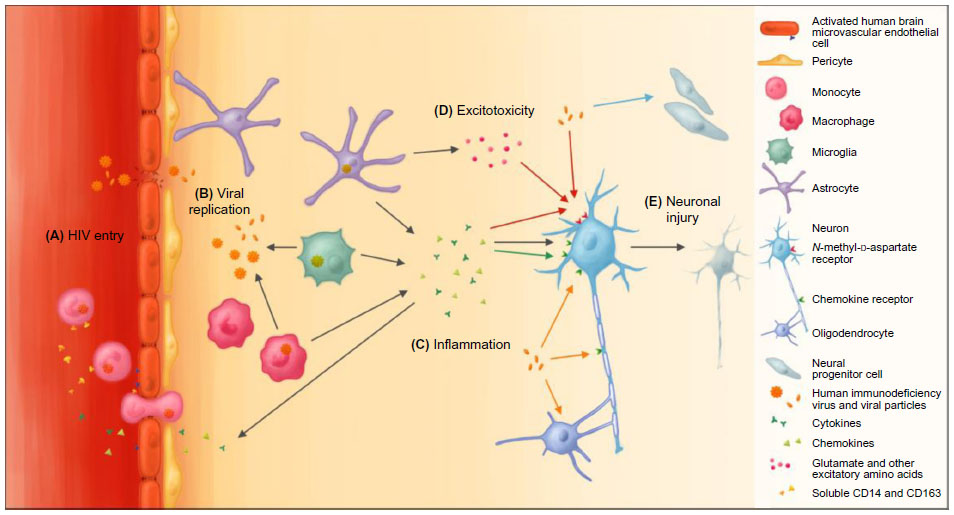 | Figure 1 The effects of HIV on the cells of the central nervous system. |
HIV affects most CNS cell types, but primarily targets microglia and monocyte-derived macrophages in the perivascular space as the primary site of HIV infection and production in the brain (Figure 1B). These infected sites correspond with the highest concentrations of HIV in histopathological studies.56 HIV-infected and activated microglia and macrophages are major sources for viral products, chemokines, proinflammatory cytokines, and small molecules, such as platelet-activating factor and quinolinic acid or derivatives thereof, that lead to further immune activation and cerebral injury (Figure 1C).55 Furthermore, fusion of these cells is thought to give rise to multinucleated giant cells, considered a hallmark of HIV encephalopathy.55
Astrocytes are highly abundant in the CNS, and regulate the environment of neurons, integrity of the BBB, and response to injury. Similar to microglia, when activated by HIV, they proliferate (termed reactive astrocytosis) and release several inflammatory mediators.55 While HIV DNA has been detected within astrocytes, this may be the result of nonspecific – thus CD4 receptor-independent – uptake and release of the virus, and sustained infection leading to viral DNA integration and production of viral proteins remains doubtful.56,57 The same is true for oligodendrocytes, which produce the myelin sheath that insulates neuronal axons.58 HIV can also affect neural progenitor cells, which reside in select regions of the brain and play a critical role in brain growth and repair after injury, as they are capable of forming new neurons, astrocytes, and oligodendrocytes.59 Exposure to HIV reduces proliferation of these cells, which may contribute to cognitive impairment, consistent with the finding of fewer hippocampal neural progenitor cells in adults with HAD.60
HIV-induced neuronal injury
Interestingly, neurons also lack the CD4 receptor necessary for HIV infection, and intraneuronal HIV is not consistently detected in adults or children with HIV-related CNS disease.56,61 However, the receptor-expression profile of neurons, which includes the HIV coreceptors CCR5 and CXCR4, renders them sensitive to injury caused by viral proteins as well as inflammatory mediators triggered by HIV infection.62
Direct exposure to structural viral proteins has been shown to be neurotoxic in vitro, although their exact contributions in vivo remain uncertain. Several viral proteins, including gp120, Tat, and Vpr, can directly induce neuronal apoptosis via chemokine receptors and intracellular pathways.62–65 Additionally, gp120 increases glutamate (Glu) release from microglia and impairs Glu uptake by astrocytes, leading to overstimulation of N-methyl-d-aspartate-receptors (Figure 1D) and subsequent lethal Ca2+ influx (Figure 1E).66,67 Both Tat and gp120 can further contribute to this mechanism by direct binding to N-methyl-d-aspartate-receptors.68,69 Local axonal degeneration may also occur through gp120-induced caspase activation of CCR5 or CXCR4.70
Viral proteins also cause astrocyte dysfunction, which may lead to additional neuronal injury. Tat promotes expression of inducible nitric oxide synthase and production of nitric oxide by infected astrocytes, causing oxidative injury to neurons.71 Oligodendrocyte injury induced by gp120 or Tat could reduce myelination, which subsequently increases axonal vulnerability.72,73
Neuroinflammation
The CNS has a predominantly innate immune response to injury, depending on activated glia and monocyte-derived cells, whereas in contrast with the periphery, the role of T cells is much less clear.58 Monocyte- and macrophage-activation markers, such as plasma and CSF neopterin, soluble CD14 (sCD14) and plasma sCD163, are elevated in HIV-infected adults, correlating with presence or severity of neurocognitive impairment.2,46,74,75 In cognitively impaired HIV-infected adults, CSF sCD14 and sCD163 are also associated with CSF NFL, a marker of neurodegeneration, underpinning the role of persistent immune activation in HIV neuropathogenesis.76 Furthermore, cognitively impaired HIV-infected adults have higher proportions of CD14+ CD16+ monocytes in the blood. This subpopulation is associated with chronic inflammation and a higher susceptibility to HIV infection and MCP-1 chemoattraction in comparison to their CD14+ CD16− counterparts.49,77,78 In PHIV-infected children, only neopterin and sCD14 levels have been evaluated, and were found to be systemically elevated.79,80 Potential associations with other measures of HIV-related cerebral injury have not yet been examined.
The ongoing immune activation leads to sustained release of cytokines and chemokines, consistent with increased levels of inflammatory mediators in the CSF of HIV-infected adults and children despite cART.46,81,82 Many HIV-induced proinflammatory cytokines, including TNFα, IL-1β, and IL-6, amplify the effects of HIV through the induction of viral replication and further immune activation, as well as being directly neurotoxic.55 In vitro, TNFα and IL-1β interfere with Glu uptake, contributing to excitotoxicity.83 Increased TNFα also causes oligodendrocyte death, which subsequently could impair myelination.83,84 IL-6 causes cytoplasmic vacuoles in neurons, resulting in neuronal dysfunction. Further recruitment of immune cells and cytokine release is enhanced by various chemokines, such as IL-8, IP-10, MIP-1α, MIP-1β, MCP-1, and RANTES.85,86
Contrastingly, the chemokines MCP-1, MIP-1α, RANTES, SDF-1, and fractalkine can all in vitro protect neurons against apoptosis induced by gp120 or Tat, likely due to activation of pathways promoting neuronal survival.62,87,88 TNFα also prevents apoptosis of neurons expressing type 2 TNF receptors, indicating that expression of specific receptors is crucial in the distinction between neurotoxic and neurotrophic effects.89 As the net effect of many inflammatory mediators in vivo is unclear, their presence alone does not predict protection or injury. Indeed, while several inflammatory markers were detectable in the CSF of PHIV-infected children on cART, they did not correlate with systemic inflammation, HIV disease markers, or neurological symptoms.82 However, it does imply sustained brain inflammation in chronic HIV infection, warranting further research to identify inflammatory mechanisms that may serve as predictors and possible therapeutic targets for cerebral injury in PHIV-infected children.
Evidence of cerebral injury
Typical findings of HIV encephalopathy on conventional computed tomography or magnetic resonance imaging (MRI) sequences are cerebral atrophy, basal ganglia calcifications, and white-matter abnormalities. In the cART era, advanced imaging modalities to evaluate white-matter integrity, cerebral metabolites, and cerebral blood flow (CBF) show increasing evidence for microstructural injury in PHIV-infected children, even in the absence of neurological symptoms or macrostructural lesions.90 These imaging modalities and the type of cerebral injury they capture are detailed in this section.
It is important to interpret neuroimaging findings within the context of a developing brain, as many physiological changes occur throughout childhood and adolescence. The majority of brain growth and myelin deposition occurs in infancy. In childhood and adolescence, cerebral volume continues to increase at a slower rate until adulthood, with a relative increase in white matter due to ongoing myelination and thinning of cortical gray matter as a consequence of synaptic pruning. This maturation occurs in a posterior–anterior direction, corresponding to early development of visuomotor areas, while higher-order cognitive functions utilizing prefrontal and parietal cortical regions mature in late childhood and adolescence.6,21
Macrostructural injury
Cerebral atrophy, manifesting as cortical and subcortical volume reduction, sulcal widening, and ventriculomegaly (Figure 2A), occurs in many clinically and immunologically stable PHIV-infected children and correlates with poorer cognitive functioning.91–93 Gray-matter atrophy is generally considered to represent loss of neuronal cells, but could also be a manifestation of impaired brain growth in HIV-infected children.90 In a longitudinal study, gray-matter volume increased over time, suggesting that in the setting of adequate treatment, the physiological development of the CNS may at least partially compensate for neuronal loss in these children.91
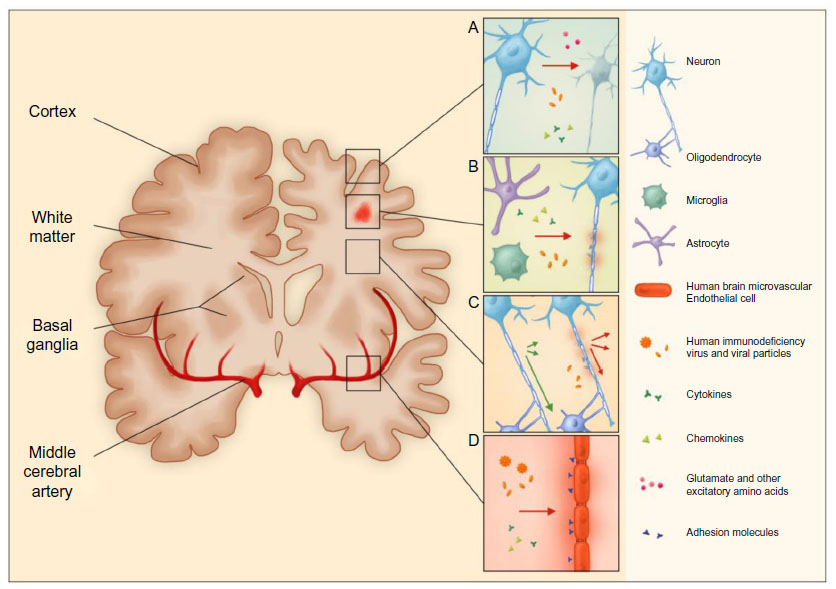 | Figure 2 HIV-induced cerebral injury. |
White-matter lesions (WML) are best seen on fluid-attenuated inversion-recovery MRI images as hyperintense areas diffusely spread throughout periventricular, deep, and juxtacortical white matter.91,92,94 WML are thought to represent sites of inflammation with glial proliferation and myelin injury (Figure 2B), while the presence of lesions in the well-perfused juxtacortical regions makes an exclusively vascular origin less likely.95,96 WML have been detected in PHIV-infected infants initiated on cART within the 1st months of life, and remained stable over time in another study, suggesting a very early onset of pathogenesis.91,94 However, neither the presence, volume, nor distribution of WML clearly relate to neurodevelopmental outcomes in PHIV-infected children on cART.91,92,94
Microstructural injury
Even in the absence of structural imaging abnormalities, such as WML, white-matter damage measured by changes in diffusion values with diffusion-tensor imaging is apparent in stable PHIV-infected children.38,92 These changes are widespread throughout the white matter, indicating profuse myelin and axonal injury (Figure 2C). Reduced white-matter integrity was found to correlate with higher HIV load, lower CD4+ T-cell nadir, and poorer cognitive functioning, and may therefore serve as a potentially early marker of HIV-related developmental delay.38,92,97 However, longitudinal studies studying such decay are lacking.
Several CSF biomarkers that are frequently reported in other degenerative and inflammatory CNS diseases, such as dementia and multiple sclerosis, may also be useful in the evaluation of HIV-related cerebral injury. Elevated CSF NFL, indicative of damage to myelinated axons in the white matter, has repeatedly been reported in HIV-infected adults, most prominently in association with HAD or low CD4+ T-cell nadirs.74,98 Higher total or phosphorylated Tau levels, indicating cortical neurodegeneration, are seen most prominently in HIV-infected adults with HAD.98,99 Altered markers of amyloid processing have also been reported, possibly corresponding with increased intraneuronal amyloid-β accumulation.74,98–100 Various other pediatric infectious, inflammatory, and demyelinating CNS disorders show distinctive patterns of biomarkers indicative of neuronal injury, including total Tau and NFL.101 Evaluation of these markers in PHIV-infected children may thus provide new insights into the underlying mechanisms of cerebral injury, and may help distinguish neurodegeneration from impaired brain growth due to HIV.
Magnetic resonance spectroscopy can examine the metabolic integrity and density of neurons noninvasively through detection of changes in cerebral metabolites, even before structural changes occur or clinical symptoms become manifest.102 These metabolites are discussed here as a concentration ratio to creatine (Cre). Acute HIV infection in adults is associated with a rise in Glu (Glu:Cre) to potentially excitotoxic levels, accompanied by increases of choline (Cho:Cre) and myo-inositol (MI:Cre), representing increased membrane turnover and glial proliferation, respectively.2,74,103 Initiation of cART partially reverses some of these changes.103 Chronically HIV-infected adults and children show persistent elevations in MI:Cre and Cho:Cre, suggestive of ongoing inflammation, while Glu:Cre and N-acetylaspartate (NAA:Cre) decrease, indicating neuronal dysfunction and degeneration.103–105 Contrastingly, decreased Cho:Cre in basal ganglia and frontal white matter has also been reported in immunologically stable PHIV-infected children.106 As the developing brain of children has a higher requirement for Cho, which is a component of cell membranes and myelin, this decrease may reflect impaired myelination and brain growth.107 In PHIV-infected children, an age-related increase in NAA – as normally seen in healthy controls – was lacking, again suggesting impaired brain growth in HIV infection.106
Metabolite changes in HIV-infected adults correlate with the presence and severity of cognitive impairment,108 and although pediatric studies relating cerebral metabolites to neurodevelopmental outcomes are limited, reports include associations between hippocampal Cho:Cre and spatial memory deficits and between overall NAA:Cho ratio and verbal IQ.104,106 Furthermore, while adult metabolite alterations correlate with several other indicators of HIV-related cerebral injury, such as cortical atrophy, elevated NFL, and increased systemic or intrathecal inflammation, such relationships have not yet been studied in PHIV-infected children.2,74,81,109
Cerebral blood flow and vascular disease
A higher risk of cerebrovascular disease is well established in chronic HIV-infected adults, and is increasingly recognized in PHIV-infected children. Neuroimaging studies predating cART displayed calcifications of basal ganglia, the thalamus, and deep white matter in the majority of children with CNS HIV disease.95 Histopathological studies correspondingly revealed vasculitis with calcifications within cerebral blood vessels.95,110 Other cerebrovascular abnormalities have been reported in PHIV-infected children, including ischemic stroke, aneurysms, and moyamoya malformations.111 However, their relation to neurodevelopmental outcomes is unclear.
Even with optimal cART, there are many likely contributors to vasculopathy in PHIV-infected children, including chronic low-grade inflammation, endothelial dysfunction, hypercoagulability, and disturbed glucose and lipid metabolism (Figure 2D). Indeed, HIV infection in children stable on therapy has been associated with higher levels of high-sensitivity CRP, soluble adhesion molecules, and fibrinogen, as well as an unfavorable lipid profile.44,45,112 Elevations in P-selectin and fibrinogen were linked to poorer neurocognitive performance.45 Additionally, findings of increased carotid intima-media thickness and decreased flow-mediated distension reveal structural and functional vascular changes, respectively. These measures also correlate with increased inflammation and an unfavorable lipid profile, and are most pronounced in children receiving a protease inhibitor.113 These findings allude to an early onset of vasculopathy accelerated by HIV-related inflammation and influenced by cART, rendering it a potential source of long-term comorbidity in PHIV-infected children surviving into adulthood. This underlines the relevance of further exploration of vascular health in this population.114
CBF has not been extensively researched in PHIV-infected children, as many of the neuroimaging modalities require exposure to radioactivity or contrast agents. A small positron-emission tomography study, predating cART, showed cortical hypometabolism in PHIV-infected children with and without encephalopathy, as well as basal ganglia hypermetabolism in those with encephalopathy.115 In HIV-infected adults, increased flow or metabolism is predominantly found in the basal ganglia and in early stages of infection, whereas reduced CBF is associated with chronic infection and localized in both cortical and subcortical structures.116,117 Flow alterations are more pronounced in cognitively impaired individuals, but also occur in neuroasymptomatic patients, suggesting CBF may prove useful as an early marker of cerebral injury in HIV.117,118 In theory, higher flow may reveal areas of compensatory increased usage of brain reserve after HIV injury, as suggested by altered task-based activation patterns on functional MRI.119,120 It may also reflect ongoing inflammation or vasodilatation due to increased release of nitrous oxide.116 Lower flow may be related to loss of cells, although it is currently unclear whether flow changes are a cause or effect of volume changes.118 Furthermore, the relation between CBF and the vascular and coagulation effects of HIV is unclear. A promising opportunity to evaluate CBF in children is with the use of arterial spin labeling, a noninvasive MRI technique that yields results comparable to positron-emission tomography in HIV-infected adults,120 and was successfully used to assess resting-state flow and the relation to cognitive performance in healthy children and children with sickle-cell anemia (Table 1).121
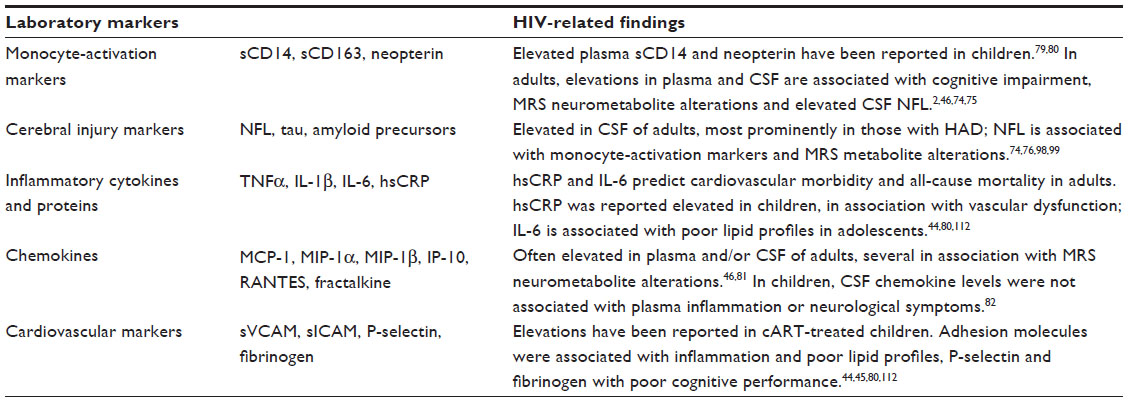 | Table 1 Selected laboratory biomarkers in HIV neuropathogenesis |
Treatment effects
While cART has been shown to reduce HIV load and inflammatory markers effectively in CSF,39 it evidently does not fully prevent or reverse accrued CNS injury. Long-term effects of cART on cerebral health in HIV-infected children are unclear; most studies evaluating the effect of treatment initiation or optimization report on improvement of neurodevelopmental outcomes, but some do not.9,15,22 While lack of improvement during treatment may reflect the severity of immunocompromise and injury prior to cART initiation, it also raises concern about cART neurotoxicity.
Timing of cART initiation
Neurodevelopmental delay may result from accrued CNS involvement, after the virus has already entered and injured the CNS and prior to initiation of cART. Reported effects of early versus deferred initiation of cART – ie, at study inclusion versus when criteria of immunosuppression or symptomatic disease are met – are not consistent. Better neurodevelopmental scores were seen in infants who initiated cART within 12 weeks of life versus deferral to a mean age of 31 weeks,13 in line with reports of better cognitive performance in school-age children with adequate viral suppression early in life or cART initiation before an AIDS-defining event.122,123 Another study found similar poor outcomes in both early and deferred treatment groups, although the mean age of initiating cART was significantly higher, at 6 years.9 This suggests the window for benefit of early cART initiation in PHIV-infected children lies in infancy, and is in line with the theory that the younger age-group has an increased vulnerability to the neurological sequelae of HIV.
Challenges of cART in the CNS
While peripheral viral suppression with cART may reduce the amount of HIV entering the CNS, effective treatment within the CNS compartment requires antiretroviral agents to penetrate the BBB. If HIV is exposed to subtherapeutic cART levels in the CNS, viral replication and mutation may occur despite adequate systemic viral suppression, potentially leading to ongoing damage and cART resistance.39,40 This is generally referred to as compartmentalization, and has been associated with the presence and severity of cognitive impairment in adults.124 The CNS-penetration effectiveness score is often used to categorize cART regimens based on CSF pharmacokinetics of the different antiretroviral agents, but it has never been validated in children and was indeed not associated with IQ in two recent studies.122,123 Pediatric studies comparing plasma and CSF levels of antiretroviral agents are lacking. Additionally, it is unknown how well pharmacokinetics of cART in the CSF represent those in brain tissue.
The toxicity of antiretroviral agents has long been speculated to play a part in HIV-related neurocognitive impairments. Particularly concerning are reports of children experiencing significant neurological decline, despite immunological recovery after treatment initiation, and the occurrence of neuropsychiatric symptoms with use of the non-nucleoside reverse-transcriptase inhibitor efavirenz. In these reports, symptoms were reversible by treatment alteration or interruption.125–127 Little is known about the potential effects of cART on the different nervous system cell types and the exact mechanisms by which cART causes neurotoxicity. Several antiretroviral agents were indeed reported toxic to neurons in vitro, mostly causing dendrite pruning and altered responses to Glu.47 However, minimal cell death occurred, and only a few agents were toxic at concentrations typically achieved in the CSF of cART-treated HIV-infected adults. Additionally, nucleoside reverse-transcriptase inhibitors may interfere with mitochondrial DNA γ-polymerase, resulting in mitochondrial dysfunction, although this has not been proven in children.128 Protease inhibitors have been associated with disturbed lipid and glucose metabolism and increased carotid intima-media thickness in children, all risk factors for vascular disease.113 Such changes may affect the CNS and thereby influence neurodevelopment, especially with the prolonged exposure to cART that occurs as PHIV-infected children survive into adulthood.
Adjuvant treatment options
Considering the increasing evidence for inflammatory influences in HIV neuropathogenesis, adjuvant treatment with anti-inflammatory agents may reduce HIV-related cerebral injury. So far, the drugs studied in adults have failed to elicit an effective response and thus have not been evaluated in children. Various nonpharmaceutical interventions, such as caregiver-training programs, may be effective in improving neurocognitive functioning of PHIV-infected children, and should be explored further.21
Conclusion and future directions
Neurodevelopmental delay affects a large proportion of PHIV-infected children, with deficits spanning multiple motor, language, and cognitive domains. The vulnerability of children with a young age at primary HIV infection or a more severe disease history stresses the importance of prevention of mother-to-child transmission, as well as early diagnosis and intervention. While cART is effective in reducing cerebral injury by suppressing HIV load and subsequent inflammation, it evidently does not fully protect against HIV-induced damage in the brain. Future studies should examine the underlying mechanisms of pediatric neurological HIV disease, focusing on age-related host immune responses, pharmacokinetics, and toxicity of cART in the pediatric CNS. Neuroimaging of white-matter integrity, cerebral metabolites, and CBF are especially promising for the pediatric population, as they can detect subtle abnormalities before major structural damage and clinical symptoms occur or become apparent. When combined with laboratory evaluation of neurodegeneration and inflammation, this could significantly contribute to increasing our understanding of the neuropathogenesis of HIV infection in the pediatric brain, and possibly expand the window for optimal treatment or prevention. As PHIV-infected children on cART now survive into adulthood, longitudinal research is warranted to observe the long-term effects of HIV infection and cART exposure.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Global Update on the Health Sector Response to HIV, 2014. Geneva: WHO; 2014. | |
Valcour V, Chalermchai T, Sailasuta N, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–282. | |
Patel K, Ming X, Williams PL, Robertson KR, Oleske JM, Seage GR. Impact of HAART and CNS-penetrating antiretroviral regimens on HIV encephalopathy among perinatally infected children and adolescents. AIDS. 2009;23(14):1893–1901. | |
Tardieu M, Le Chenadec J, Persoz A, Meyer L, Blanche S, Mayaux MJ. HIV-1-related encephalopathy in infants compared with children and adults. French Pediatric HIV Infection Study and the SEROCO Group. Neurology. 2000;54(5):1089–1095. | |
Cohen S, Ter Stege JA, Geurtsen GJ, et al. Poorer cognitive performance in perinatally HIV-infected children versus healthy socioeconomically matched controls. Clin Infect Dis. 2015;60(7):1111–1119. | |
Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology. 2010;35(1):147–168. | |
Spudich S, González-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med. 2012;2(6):a007120. | |
Van Rie A, Harrington PR, Dow A, Robertson K. Neurologic and neurodevelopmental manifestations of pediatric HIV/AIDS: a global perspective. Eur J Paediatr Neurol. 2007;11(1):1–9. | |
Puthanakit T, Ananworanich J, Vonthanak S, et al. Cognitive function and neurodevelopmental outcomes in HIV-infected children older than 1 year of age randomized to early versus deferred antiretroviral therapy: the PREDICT neurodevelopmental study. Pediatr Infect Dis J. 2013;32(5):501–508. | |
Le Doaré K, Bland R, Newell ML. Neurodevelopment in children born to HIV-infected mothers by infection and treatment status. Pediatrics. 2012;130(5):e1326–e1344. | |
Lindsey JC, Malee KM, Brouwers P, Hughes MD. Neurodevelopmental functioning in HIV-infected infants and young children before and after the introduction of protease inhibitor-based highly active antiretroviral therapy. Pediatrics. 2007;119(3):e681–e693. | |
Ferguson G, Jelsma J. The prevalence of motor delay among HIV infected children living in Cape Town, South Africa. Int J Rehabil Res. 2009;32(2):108–114. | |
Laughton B, Cornell M, Grove D, et al. Early antiretroviral therapy improves neurodevelopmental outcomes in infants. AIDS. 2012;26(13):1685–1690. | |
Chase C, Ware J, Hittelman J, et al. Early cognitive and motor development among infants born to women infected with human immunodeficiency virus. Women and Infants Transmission Study Group. Pediatrics. 2000;106(2):E25. | |
Jeremy RJ, Kim S, Nozyce M, et al. Neuropsychological functioning and viral load in stable antiretroviral therapy-experienced HIV-infected children. Pediatrics. 2005;115(2):380–387. | |
Ruel TD, Boivin MJ, Boal HE, et al. Neurocognitive and motor deficits in HIV-infected Ugandan children with high CD4 cell counts. Clin Infect Dis. 2012;54(7):1001–1009. | |
Lowick S, Sawry S, Meyers T. Neurodevelopmental delay among HIV-infected preschool children receiving antiretroviral therapy and healthy preschool children in Soweto, South Africa. Psychol Health Med. 2012;17(5):599–610. | |
Kandawasvika GQ, Kuona P, Chandiwana P, et al. The burden and predictors of cognitive impairment among 6- to 8-year-old children infected and uninfected with HIV from Harare, Zimbabwe: a cross-sectional study. Child Neuropsychol. 2015;21(1):106–120. | |
Fishkin PE. Brief report: Relationship between HIV infection and WPPSI-R performance in preschool-age children. J Pediatr Psychol. 2000;25(5):347–351. | |
Koekkoek S, de Sonneville LM, Wolfs TF, Licht R, Geelen SP. Neurocognitive function profile in HIV-infected school-age children. Eur J Paediatr Neurol. 2008;12(4):290–297. | |
Laughton B, Cornell M, Boivin M, Van Rie A. Neurodevelopment in perinatally HIV-infected children: a concern for adolescence. J Int AIDS Soc. 2013;16(1):18603. | |
Whitehead N, Potterton J, Coovadia A. The neurodevelopment of HIV-infected infants on HAART compared to HIV-exposed but uninfected infants. AIDS Care. 2014;26(4):497–504. | |
Brahmbhatt H, Boivin M, Ssempijja V, et al. Neurodevelopmental benefits of antiretroviral therapy in Ugandan children aged 0–6 years with HIV. J Acquir Immune Defic Syndr. 2014;67(3):316–322. | |
Brackis-Cott E, Kang E, Dolezal C, Abrams EJ, Mellins CA. The impact of perinatal HIV infection on older school-aged children’s and adolescents’ receptive language and word recognition skills. AIDS Patient Care STDS. 2009;23(6):415–421. | |
Van Rie A, Mupuala A, Dow A. Impact of the HIV/AIDS epidemic on the neurodevelopment of preschool-aged children in Kinshasa, Democratic Republic of the Congo. Pediatrics. 2008;122(1):e123–e128. | |
Rice ML, Buchanan AL, Siberry GK, et al. Language impairment in children perinatally infected with HIV compared to children who were HIV-exposed and uninfected. J Dev Behav Pediatr. 2012;33(2):112–123. | |
Souza E, Santos N, Valentini S, Silva G, Falbo A. Long-term follow-up outcomes of perinatally HIV-infected adolescents: infection control but school failure. J Trop Pediatr. 2010;56(6):421–426. | |
Wood SM, Shah SS, Steenhoff AP, Rutstein RM. The impact of AIDS diagnoses on long-term neurocognitive and psychiatric outcomes of surviving adolescents with perinatally acquired HIV. AIDS. 2009; 23(14):1859–1865. | |
Kerr SJ, Puthanakit T, Vibol U, et al. Neurodevelopmental outcomes in HIV-exposed-uninfected children versus those not exposed to HIV. AIDS Care. 2014;26(11):1–9. | |
Lyman WD, Kress Y, Kure K, Rashbaum WK, Rubinstein A, Soeiro R. Detection of HIV in fetal central nervous system tissue. AIDS. 1990; 4(9):917–920. | |
Williams PL, Marino M, Malee K, Brogly S, Hughes MD, Mofenson LM. Neurodevelopment and in utero antiretroviral exposure of HIV-exposed uninfected infants. Pediatrics. 2010;125(2):e250–e260. | |
Crowell CS, Malee KM, Yogev R, Muller WJ. Neurologic disease in HIV-infected children and the impact of combination antiretroviral therapy. Rev Med Virol. 2014;24(5):316–331. | |
Chiriboga CA, Fleishman S, Champion S, Gaye-Robinson L, Abrams EJ. Incidence and prevalence of HIV encephalopathy in children with HIV infection receiving highly active anti-retroviral therapy (HAART). J Pediatr. 2005;146(3):402–407. | |
Smith R, Chernoff M, Williams PL, et al. Impact of HIV severity on cognitive and adaptive functioning during childhood and adolescence. Pediatr Infect Dis J. 2012;31(6):592–598. | |
Nachman S, Chernoff M, Williams P, Hodge J, Heston J, Gadow KD. Human immunodeficiency virus disease severity, psychiatric symptoms, and functional outcomes in perinatally infected youth. Arch Pediatr Adolesc Med. 2012;166(6):528–535. | |
Kandawasvika GQ, Ogundipe E, Gumbo FZ, Kurewa EN, Mapingure MP, Stray-Pedersen B. Neurodevelopmental impairment among infants born to mothers infected with human immunodeficiency virus and uninfected mothers from three peri-urban primary care clinics in Harare, Zimbabwe. Dev Med Child Neurol. 2011;53(11):1046–1052. | |
Smith R, Malee K, Charurat M, et al. Timing of perinatal human immunodeficiency virus type 1 infection and rate of neurodevelopment. Pediatr Infect Dis J. 2000;19(9):862–871. | |
Hoare J, Fouche JP, Spottiswoode B, et al. A diffusion tensor imaging and neurocognitive study of HIV-positive children who are HAART-naïve “slow progressors”. J Neurovirol. 2012;18(3):205–212. | |
McCoig C, Castrejón MM, Castaño E, et al. Effect of combination antiretroviral therapy on cerebrospinal fluid HIV RNA, HIV resistance, and clinical manifestations of encephalopathy. J Pediatr. 2002;141(1):36–44. | |
Nightingale S, Winston A, Letendre S, et al. Controversies in HIV-associated neurocognitive disorders. Lancet Neurol. 2014;13(11):1139–1151. | |
Chan P, Brew BJ. HIV associated neurocognitive disorders in the modern antiviral treatment era: prevalence, characteristics, biomarkers, and effects of treatment. Curr HIV/AIDS Rep. 2014;11(3):317–324. | |
Edén A, Fuchs D, Hagberg L, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis. 2010;202(12):1819–1825. | |
Canestri A, Lescure FX, Jaureguiberry S, et al. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis. 2010;50(5):773–778. | |
Ross AC, O’Riordan MA, Storer N, Dogra V, McComsey GA. Heightened inflammation is linked to carotid intima-media thickness and endothelial activation in HIV-infected children. Atherosclerosis. 2010;211(2):492–498. | |
Kapetanovic S, Leister E, Nichols S, et al. Relationships between markers of vascular dysfunction and neurodevelopmental outcomes in perinatally HIV-infected youth. AIDS. 2010;24(10):1481–1491. | |
Kamat A, Lyons J, Misra V. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr. 2012;60(3):234–243. | |
Robertson K, Liner J, Meeker RB. Antiretroviral neurotoxicity. J Neurovirol. 2012;18(5):388–399. | |
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011; 12(12):723–738. | |
Hong S, Banks WA. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun. 2015;45:1–12. | |
Chaudhuri A, Duan F, Morsey B, Persidsky Y, Kanmogne GD. HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: putative mechanisms of blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 2008;28(4):697–711. | |
Stins MF, Shen Y, Huang SH, Gilles F, Kalra VK, Kim KS. Gp120 activates children’s brain endothelial cells via CD4. J Neurovirol. 2001;7(2):125–134. | |
Muratori C, Mangino G, Affabris E, Federico M. Astrocytes contacting HIV-1-infected macrophages increase the release of CCL2 in response to the HIV-1-dependent enhancement of membrane-associated TNFα in macrophages. Glia. 2010;58(16):1893–1904. | |
Dohgu S, Banks WA. Brain pericytes increase the lipopolysaccharide-enhanced transcytosis of HIV-1 free virus across the in vitro blood-brain barrier: evidence for cytokine-mediated pericyte-endothelial cell crosstalk. Fluids Barriers CNS. 2013;10(1):23. | |
Louboutin JP, Agrawal L, Reyes BA, Van Bockstaele EJ, Strayer DS. HIV-1 gp120-induced injury to the blood-brain barrier: role of metalloproteinases 2 and 9 and relationship to oxidative stress. J Neuropathol Exp Neurol. 2010;69(8):801–816. | |
Rao VR, Ruiz AP, Prasad VR. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res Ther. 2014;11(1):13. | |
Trillo-Pazos G, Diamanturos A, Rislove L, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol. 2003;13(1):144–154. | |
Sabri F, Tresoldi E, Di Stefano M, et al. Nonproductive human immunodeficiency virus type 1 infection of human fetal astrocytes: independence from CD4 and major chemokine receptors. Virology. 1999;264(2):370–384. | |
Kramer-Hämmerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111(2):194–213. | |
Schwartz L, Major EO. Neural progenitors and HIV-1-associated central nervous system disease in adults and children. Curr HIV Res. 2006;4(3):319–327. | |
Krathwohl MD, Kaiser JL. HIV-1 promotes quiescence in human neural progenitor cells. J Infect Dis. 2004;190(2):216–226. | |
An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58(11):1156–1162. | |
Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95(24):14500–14505. | |
Bachis A, Biggio F, Major EO, Mocchetti I. M- and T-tropic HIVs promote apoptosis in rat neurons. J Neuroimmune Pharmacol. 2009; 4(1):150–160. | |
Eugenin EA, King JE, Nath A, et al. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104(9):3438–3443. | |
Jones GJ, Barsby NL, Cohen EA, et al. HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration. J Neurosci. 2007;27(14):3703–3711. | |
Huang Y, Zhao L, Jia B, et al. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J Neurosci. 2011;31(42):15195–15204. | |
Wang Z, Pekarskaya O, Bencheikh M, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312(1):60–73. | |
Self RL, Mulholland PJ, Nath A, Harris BR, Prendergast MA. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-d-aspartate receptor polyamine-sensitive site. Brain Res. 2004;995(1):39–45. | |
Gemignani A, Paudice P, Pittaluga A, Raiteri M. The HIV-1 coat protein gp120 and some of its fragments potently activate native cerebral NMDA receptors mediating neuropeptide release. Eur J Neurosci. 2000;12(8):2839–2846. | |
Melli G, Keswani SC, Fischer A, Chen W, Höke A. Spatially distinct and functionally independent mechanisms of axonal degeneration in a model of HIV-associated sensory neuropathy. Brain. 2006;129(Pt 5):1330–1338. | |
Liu X, Jana M, Dasgupta S, et al. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277(42):39312–39319. | |
Bernardo A, Agresti C, Levi G. HIV-gp120 affects the functional activity of oligodendrocytes and their susceptibility to complement. J Neurosci Res. 1997;50(6):946–957. | |
Hauser KF, Hahn YK, Adjan VV, et al. HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia. 2009;57(2):194–206. | |
Peluso MJ, Meyerhoff DJ, Price RW, et al. Cerebrospinal fluid and neuroimaging biomarker abnormalities suggest early neurological injury in a subset of individuals during primary HIV infection. J Infect Dis. 2013;207(11):1703–1712. | |
Burdo TH, Weiffenbach A, Woods SP, Letendre S, Ellis RJ, Williams KC. Elevated sCD163 in plasma but not cerebrospinal fluid is a marker of neurocognitive impairment in HIV infection. AIDS. 2013;27(9):1387–1395. | |
McGuire J, Gill A, Douglas S, Kolson D. Central and peripheral markers of neurodegeneration and monocyte activation in HIV-associated neurocognitive disorders. J Neurovirol. 2015;21(4):439–448. | |
Fischer-Smith T, Croul S, Sverstiuk AE, et al. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. 2001;7(6):528–541. | |
Williams DW, Calderon TM, Lopez L, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One. 2013;8(7):e69270. | |
Syed SS, Balluz RS, Kabagambe EK, et al. Assessment of biomarkers of cardiovascular risk among HIV-1 infected adolescents: role of soluble vascular cell adhesion molecule (sVCAM) as an early indicator of endothelial inflammation. AIDS Res Hum Retroviruses. 2013;29(3):493–500. | |
Sainz T, Diaz L, Navarro ML, et al. Cardiovascular biomarkers in vertically HIV-infected children without metabolic abnormalities. Atherosclerosis. 2014;233(2):410–414. | |
Anderson AM, Harezlak J, Bharti A, et al. Plasma and cerebrospinal fluid biomarkers predict cerebral injury in HIV-infected individuals on stable combination antiretroviral therapy. J Acquir Immune Defic Syndr. 2015;69(1):29–35. | |
McCoig C, Castrejón MM, Saavedra-Lozano J, et al. Cerebrospinal fluid and plasma concentrations of proinflammatory mediators in human immunodeficiency virus-infected children. Pediatr Infect Dis J. 2004;23(2):114–118. | |
Ye L, Huang Y, Zhao L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125(6):897–908. | |
Wilt SG, Milward E, Zhou JM, et al. In vitro evidence for a dual role of tumor necrosis factor-α in human immunodeficiency virus type 1 encephalopathy. Ann Neurol. 1995;37(3):381–394. | |
Yang B, Akhter S, Chaudhuri A, Kanmogne GD. HIV-1 gp120 induces cytokine expression, leukocyte adhesion, and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. Microvasc Res. 2009;77(2):212–219. | |
González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81. | |
Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96(14):8212–8216. | |
Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85(5):1299–1311. | |
Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel ULM. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation: essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-κB pathway. J Biol Chem. 2004;279(31):32869–32881. | |
Hoare J, Ransford GL, Phillips N, Amos T, Donald K, Stein DJ. Systematic review of neuroimaging studies in vertically transmitted HIV positive children and adolescents. Metab Brain Dis. 2014;29(2):221–229. | |
van Arnhem LA, Bunders MJ, Scherpbier HJ, et al. Neurologic abnormalities in HIV-1 infected children in the era of combination antiretroviral therapy. PLoS One. 2013;8(5):e64398. | |
Cohen S, Caan M, Mutsaerts H, et al. Cerebral injury in perinatally HIV-infected children compared to matched healthy controls. Neurology. In press 2015. | |
Martin SC, Wolters PL, Toledo-Tamula MA, Zeichner SL, Hazra R, Civitello L. Cognitive functioning in school-aged children with vertically acquired HIV infection being treated with highly active antiretroviral therapy (HAART). Dev Neuropsychol. 2006;30(2):633–657. | |
Ackermann C, Andronikou S, Laughton B, et al. White matter signal abnormalities in children with suspected HIV-related neurologic disease on early combination antiretroviral therapy. Pediatr Infect Dis J. 2014;33(8):e207–e212. | |
Kure K, Llena JF, Lyman WD, et al. Human immunodeficiency virus-1 infection of the nervous system: an autopsy study of 268 adult, pediatric, and fetal brains. Hum Pathol. 1991;22(7):700–710. | |
Kim KW, MacFall JR, Payne ME. Classification of white matter lesions on magnetic resonance imaging in elderly persons. Biol Psychiatry. 2008;64(4):273–280. | |
Hoare J, Fouche JP, Phillips N, et al. Clinical associations of white matter damage in cART-treated HIV-positive children in South Africa. J Neurovirol. 2015;21(2):120–128. | |
Peterson J, Gisslen M, Zetterberg H, et al. Cerebrospinal fluid (CSF) neuronal biomarkers across the spectrum of HIV infection: hierarchy of injury and detection. PLoS One. 2014;9(12):e116081. | |
Calcagno A, Atzori C, Romito A, et al. Cerebrospinal fluid biomarkers in patients with plasma HIV RNA below 20 copies/mL. J Int AIDS Soc. 2014;17(4 Suppl 3):19719. | |
Achim CL, Adame A, Dumaop W, Everall IP, Masliah E. Increased accumulation of intraneuronal amyloid β in HIV-infected patients. J Neuroimmune Pharmacol. 2009;4(2):190–199. | |
Shahim P, Darin N, Andreasson U, et al. Cerebrospinal fluid brain injury biomarkers in children: a multicenter study. Pediatr Neurol. 2013;49(1):31–39. e2. | |
Masters MC, Ances BM. Role of neuroimaging in HIV-associated neurocognitive disorders. Semin Neurol. 2014;34(1):89–102. | |
Young AC, Yiannoutsos CT, Hegde M, et al. Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology. 2014;83(18):1592–1600. | |
Gabis L, Belman A, Huang W, Milazzo M, Nachman S. Clinical and imaging study of human immunodeficiency virus-1-infected youth receiving highly active antiretroviral therapy: pilot study using magnetic resonance spectroscopy. J Child Neurol. 2006;21(6):486–490. | |
Prado PT, Escorsi-Rosset S, Cervi MC, Santos AC. Image evaluation of HIV encephalopathy: a multimodal approach using quantitative MR techniques. Neuroradiology. 2011;53(11):899–908. | |
Keller MA, Venkatraman TN, Thomas A, et al. Altered neurometabolite development in HIV-infected children: correlation with neuropsychological tests. Neurology. 2004;62(10):1810–1817. | |
Rae CD. A guide to the metabolic pathways and function of metabolites observed in human brain 1H magnetic resonance spectra. Neurochem Res. 2014;39(1):1–36. | |
Mohamed MA, Barker PB, Skolasky RL, et al. Brain metabolism and cognitive impairment in HIV infection: a 3-T magnetic resonance spectroscopy study. Magn Reson Imaging. 2010;28(9):1251–1257. | |
Cohen RA, Harezlak J, Gongvatana A, et al. Cerebral metabolite abnormalities in human immunodeficiency virus are associated with cortical and subcortical volumes. J Neurovirol. 2010;16(6):435–444. | |
Shah SS, Zimmerman RA, Rorke LB, Vezina LG. Cerebrovascular complications of HIV in children. Am J Neuroradiol. 1996;17(10):1913–1917. | |
Wilmshurst JM, Donald KA, Eley B. Update on the key developments of the neurologic complications in children infected with HIV. Curr Opin HIV AIDS. 2014;9(6):533–538. | |
Miller TL, Borkowsky W, Dimeglio LA, et al. Metabolic abnormalities and viral replication are associated with biomarkers of vascular dysfunction in HIV-infected children. HIV Med. 2012;13(5):264–275. | |
Charakida M, Donald AE, Green H, et al. Early structural and functional changes of the vasculature in HIV-infected children: impact of disease and antiretroviral therapy. Circulation. 2005;112(1):103–109. | |
Idris N, Grobbee D, Burgner D, et al. Cardiovascular manifestations of HIV infection in children. Eur J Prev Cardiol. 2015;22(11):1452–1461. | |
Depas G, Chiron C, Tardieu M, et al. Functional brain imaging in HIV-1-infected children born to seropositive mothers. J Nucl Med. 1995;36(12):2169–2174. | |
Tracey I, Hamberg LM, Guimaraes AR, et al. Increased cerebral blood volume in HIV-positive patients detected by functional MRI. Neurology. 1998;50(6):1821–1826. | |
Ances BM, Sisti D, Vaida F, et al. Resting cerebral blood flow: a potential biomarker of the effects of HIV in the brain. Neurology. 2009;73(9):702–708. | |
Ances BM, Roc AC, Wang J, et al. Caudate blood flow and volume are reduced in HIV+ neurocognitively impaired patients. Neurology. 2006;66(6):862–866. | |
Melrose RJ, Tinaz S, Castelo JM, Courtney MG, Stern CE. Compromised fronto-striatal functioning in HIV: an fMRI investigation of semantic event sequencing. Behav Brain Res. 2008;188(2):337–347. | |
Towgood KJ, Pitkanen M, Kulasegaram R, et al. Regional cerebral blood flow and FDG uptake in asymptomatic HIV-1 men. Hum Brain Mapp. 2013;34(10):2484–2493. | |
Kilroy E, Liu CY, Yan L, et al. Relationships between cerebral blood flow and IQ in typically developing children and adolescents. J Cogn Sci (Seoul). 2011;12(2):151–170. | |
Lazarus JR, Rutstein RM, Lowenthal ED. Treatment initiation factors and cognitive outcome in youth with perinatally acquired HIV infection. HIV Med. 2015;16(6):355–361. | |
Crowell CS, Huo Y, Tassiopoulos K, et al. Early viral suppression improves neurocognitive outcomes in HIV-infected children. AIDS. 2015;29(3):295–304. | |
Svicher V, Ceccherini-Silberstein F, Antinori A, Aquaro S, Perno CF. Understanding HIV compartments and reservoirs. Curr HIV/AIDS Rep. 2014;11(2):186–194. | |
Tamula MA, Wolters PL, Walsek C, Zeichner S, Civitello L. Cognitive decline with immunologic and virologic stability in four children with human immunodeficiency virus disease. Pediatrics. 2003;112(3):679–684. | |
Price RW, Spudich S. Antiretroviral therapy and central nervous system HIV type 1 infection. J Infect Dis. 2008;197(Suppl 3):S294–S306. | |
Church JA, Mitchell WG, Gonzalez-Gomez I, et al. Mitochondrial DNA depletion, near-fatal metabolic acidosis, and liver failure in an HIV-infected child treated with combination antiretroviral therapy. J Pediatr. 2001;138(5):748–751. | |
Alimenti A, Forbes JC, Oberlander TF, et al. A prospective controlled study of neurodevelopment in HIV-uninfected children exposed to combination antiretroviral drugs in pregnancy. Pediatrics. 2006;118(4):e1139–e1145. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.