Back to Journals » OncoTargets and Therapy » Volume 13
Histamine H3 Receptor Promotes Cell Survival via Regulating PKA/CREB/CDKN1A Signal Pathway in Hepatocellular Carcinoma
Authors Zhang C, Yu Y, Ma L, Fu P
Received 20 February 2020
Accepted for publication 15 April 2020
Published 4 May 2020 Volume 2020:13 Pages 3765—3776
DOI https://doi.org/10.2147/OTT.S250655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Chunle Zhang,1 Yang Yu,2 Liang Ma,1 Ping Fu1,2
1Kidney Research Laboratory, Division of Nephrology and National Clinical Research Center for Geriatrics, West China Hospital of Sichuan University, Chengdu 610041, People’s Republic of China; 2Department of Nephrology, West China Hospital of Sichuan University, Chengdu 610041, People’s Republic of China
Correspondence: Ping Fu
Department of Nephrology, Kidney Research Laboratory, Division of Nephrology and National Clinical Research Center for Geriatrics, West China Hospital of Sichuan University, Chengdu 610041, People’s Republic of China
Tel/ Fax +86-028-85422286
Email [email protected]
Background: The histamine H3 receptor (HRH3) is mainly expressed in areas of the brain involved in the regulation of the release of various neurotransmitters. Recent studies have shown that HRH3 expression is increased in several types of carcinomas. However, the functional roles and underlying molecular mechanism by which HRH3 regulates cell survival in hepatocellular carcinoma (HCC) remain unknown.
Methods: The mRNA and protein expression level of target genes were evaluated by qRT-PCR, Western blot and immunohistochemistry, respectively. Cell viability and cell proliferation activity were assessed by MTS assay and EdU incorporation assay. Cell apoptosis and cell cycle were assessed by flow cytometry analysis. A xenograft mouse model was constructed to investigate the effect of HRH3 on tumor growth in vivo.
Results: Our results indicated that HRH3 was significantly upregulated in HCC, which promoted cell survival by accelerating cell proliferation and inhibiting cell apoptosis. Our results also showed that HRH3 in HCC downregulated the expression of cyclin-dependent kinase inhibitor p21 (CDKN1A) to promote G1-S phase transition by inactivating the cAMP/PKA/CREB pathway, which finally contributed to the malignant growth of HCC.
Conclusion: Our findings indicated that HRH3 functioned in promoting HCC survival by inactivating the cAMP/PKA/CREB pathway to downregulate CDKN1A expression. Thus, HRH3 might serve as a potential therapeutic target in HCC treatment.
Keywords: HRH3, CDKN1A, cell survival, hepatocellular carcinoma
Introduction
Despite significant research advances, cancer remains a worldwide health problem with high mortality.1 Liver cancer is predicted to be the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide in 2018, with about 841,000 new cases and 782,000 deaths annually.1 Hepatocellular carcinoma (HCC) comprises 75%-85% of primary liver cancer.1 In the last decades, studies confirmed that the tumor microenvironment functioned in cancer initiation, progression, and invasion.2 Tumor microenvironment factors are comprised of pro-inflammatory cytokines, growth factors, angiogenesis-inducing factors, and immune factors, which contribute to carcinoma initiation and progression.2 Data accumulated over the years strongly suggested that histamine, one kind of immune factor, is involved in cancer initiation, growth, and chemotherapy-resistant response.3,4 For instance, histamine significantly increased the radiosensitivity of MDA-MB-231 and MCF-7 cells.3 Histamine deficiency promoted inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells.4
Histamine functions via its four receptors (HRH1, HRH2, HRH3, and HRH4), which belong to the G-protein-coupled receptors.5 Abnormal expression levels of histamine receptors have been reported in different types of human cancers.5–9 Zhao et al reported that upregulation of HRH1 promoted tumor progression and contributed to poor prognosis in hepatocellular carcinoma.8 Tanaka demonstrated that both HRH2 antagonist cimetidine and HRH3 antagonist clobenpropit attenuated inflammation-associated colorectal carcinogenesis in male ICR mice.6 Meng et al reported that activation of HRH4 suppressed human cholangiocarcinoma progression by disruption of epithelial mesenchymal transition and tumor metastasis.9 Moreover, the data of Cricco et al suggested that HRH3 was involved in pancreatic carcinoma cell growth.5 However, the results of Francis et al indicated that the upregulated HRH3 inhibited the growth of cholangiocarcinoma through mediating the activation of protein kinase Cα.7 Recently, it was reported that HRH3 was significantly upregulated in HCC tissues, and HRH3 agonist facilitated the growth and metastasis of HCC cells by inducing the formation of lamellipodia.10 However, the functional roles and underlying mechanism by which HRH3 promotes cell survival need to be further investigated in cancer cells, especially in HCC.
In the present study, we systematically investigated the expression and functional roles of HRH3 in HCC. Our findings facilitate an understanding of the roles of HRH3 and provide experimental evidences for the application of HRH3 as a potential therapeutic target in HCC treatment.
Materials and Methods
Antibodies and Reagents
Primary antibodies used in this study were listed in Table S1. The detailed information of the reagents were described in the Supplementary files.
Cell Culture and Tissue Collection
The HepG2, BEL-7402, Huh-7, PLC/PRF/5 and QSG-7701 cell lines purchased from Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China) were routinely cultured in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS). 86 HCC cancer tissues and adjacent normal liver tissues were obtained from HCC patients at the West China Hospital, Sichuan University in Chengdu, China. The inclusion criteria for HCC patients were set as follows: (1) histologically-confirmed HCC; (2) receiving surgical resection; (3) no preoperative anticancer therapy; (4) no other malignancy tumor. The time from surgery to HCC specific death was defined as overall survival. Our study was approved by the Ethics Committee of Sichuan University, and written informed consents were obtained from all the patients who participated in this study. Our study was conducted in accordance with the Declaration of Helsinki.
qRT-PCR, Western Blotting, and Immunohistochemistry (IHC)
RNA extraction, first cDNA synthesis, and qRT-PCR reactions were conducted according to the manufacturer’s instructions. Primer sequences used in this study were listed in Table S2. Western blotting and IHC were conducted as previously described.11,12
Plasmid and siRNA
All siRNAs used in this study were synthesized by GenePharm Corporation (Shanghai, China). Sequences of siRNAs and primers were listed in Table S2. HRH3 knockdown and overexpression plasmids were constructed by GenePharm Corporation.
Cell Viability and Proliferation Assay
Cell viability was determined using the MTS reagent according to the manufacturer’s instructions. Briefly, cells seeded into a 96-well plate were cultured normally. Then, MTS reagent (20 μL) was added into the well and incubated at 37 °C for 2 h in the dark. Read the plate at 490 nm using a microplate reader (Bio-Rad, Hercules, CA, USA). Cell proliferation activity was measured using an Ethynyl deoxyuridine (EdU) incorporation assay kit according to the manufacturer’s instructions. Briefly, cells were incubated with EdU (5 μM) reagent at 37 °C for 2 h, followed by fixing with 4% formaldehyde for 30 min and permeated with 0.5% Triton X-100 for 20 min. Then, cells were incubated with Apollo reaction cocktail for 30 min, followed by staining with Hoechst 33,342 for 30 min. Cells were visualized under a fluorescent microscope (DM5000B; Leica, Heerbrugg, Switzerland).
Cell Cycle Assay
Cell cycle was conducted as previously described.13 The cell cycle was detected using the propidium iodide staining kit according to the manufacturer’s instructions. Briefly, the cells were collected and fixed with 70% ethanol at 4 °C for 24 h. Then, the cells were stained with propidium iodide (1 mg/mL) for 30 min in the dark after digesting with RNase. The cell cycle was analyzed by flow cytometry (Beckman Coulter, Fullerton, CA, USA).
Cell Apoptosis Assays
The cell apoptosis was detected using the Annexin V-FITC detection kit according to the manufacturer’s instructions. Briefly, cells were collected and resuspended with 400 μL binding buffer and 5 μL Annexin V at 4 °C for 10 min in the dark. After adding 5 μL propidium iodide, cells were incubated at 4 °C for 10 min in the dark. The cell samples were analyzed using flow cytometry (Beckman Coulter). The apoptosis in the xenograft tissues was detected using the TUNEL kit according to the manufacturer’s instructions. Briefly, tissue sections were incubated with Proteinase K at 37 °C for 30 min, followed by incubating with TUNEL reaction buffer at 37 °C for 1 h in the dark and staining with DAPI. Images of the tissue sections were obtained using a fluorescence microscope (DM5000B; Leica).
cAMP Level and PKA Activity Assay
The cAMP level was detected using the cAMP direct immunoassay kit according to the manufacturer’s instructions. Briefly, cells cultured in the cell plate were lysed with HCl (0.1 M) for 20 min, followed by harvesting and centrifuging at 21 000 g for 10 min. Then, the supernatant extract was further analyzed for cAMP level according to the manufacturer’s protocol. Read the plate at 450 nm using a microplate reader (Bio-Rad). The cAMP concentration was quantified according to the standard curve. The PKA activity was detected using the PKA kinase activity kit. Briefly, cells were lysed with lysis buffer for 10 min on ice, followed by collecting and centrifuging at 21 000 g for 15 min. Then, the supernatant extract was further analyzed for PKA kinase activity according to the manufacturer’s protocol. Finally, the microplate was read at 450 nm using a microplate reader (Bio-Rad).
Nude Mice Xenograft Model
All animal procedures were approved by the Institutional Animal Care and Use Committee of West China Hospital of Sichuan University. Experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publications) and according to the institutional ethical guidelines for animal experiments. Fifteen male BALB/c mice (5 weeks old; body weight, 18–22 g) were randomly divided into five groups. Xenografts were initiated by subcutaneous injection of HCC cells into the back of mice on the right side (n = 3 per group). One week later, 1.5 mg/kg cholesterol-conjugated siCDKN1A was administered thrice per week for four weeks by intratumoral injection. The mice in other groups were similarly injected with the same volume of dimethyl sulfoxide (DMSO). The mice were euthanized by injection of an overdose of pentobarbital sodium, and tumor nodules were photographed and weights calculated. A tumor growth curve was plotted according to the data of tumor volume. The tumor volume (mm3) was calculated by the formula (length × width2)/2.
Statistical Analysis
Independent experiments were performed thrice where appropriate. SPSS 17.0 (SPSS, Chicago, IL) was used for all statistical analyses and P < 0.05 was considered statistically significant. The unpaired t-test was used for comparisons between two groups where appropriate. Correlations between measured variables were tested using Spearman rank correlation analysis. For prognosis analysis, variables (the IHC score of HRH3) were divided as high or low level by the median value for further analysis. The Log rank test and Kaplan–Meier survival curve were used to distinguish subgroups of patients with different survival. For every figure, the statistical tests are justified as appropriate and the data meet the assumptions of the tests.
Results
HRH3 Is Upregulated in HCC Tissues and Contributes to Tumor Progression and a Poor Prognosis
To explore the functional roles of HRH3 in HCC, both mRNA and protein expression of HRH3 were detected using qRT-PCR and Western blotting in 15 pairs of HCC tissues. Our results showed that both mRNA and protein expression of HRH3 were upregulated in HCC tissues compared with peritumor tissues (P < 0.01, P < 0.01, respectively) (Figure 1A and B). Similarly, IHC staining results also showed that HRH3 protein expression was remarkably increased in HCC tissues when compared with that in the peritumor tissues (P < 0.01) (Figure 1C). Moreover, we analyzed the relationship between the HRH3 expression level and the pathological characteristic of patients with HCC (Table S3) and found that HRH3 expression level in patients with low differentiation HCC was remarkably higher than that in patients with high and medium differentiation HCC (P = 0.008). Kaplan–Meier survival analysis revealed that HCC patients with high HRH3 expression had significantly shorter overall survival when compared with those in HCC patients with low HRH3 expression (Figure 1D). Taken together, these data indicate that HRH3 is upregulated in HCC, which contributes to the progression and poor prognosis of HCC.
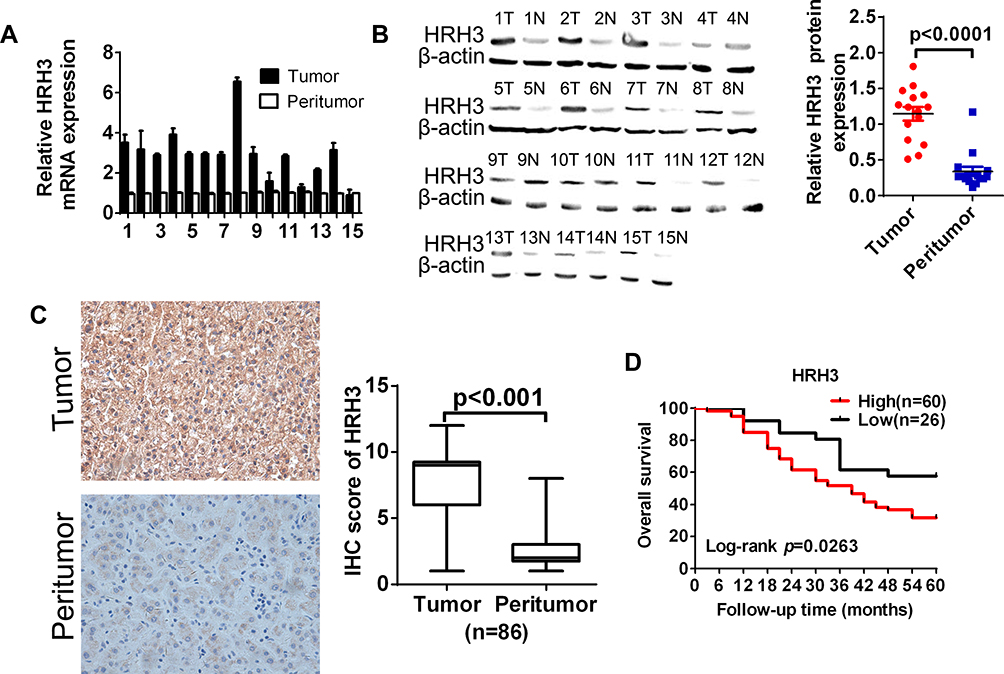 |
Figure 1 HRH3 was upregulated and associated with clinical prognosis in HCC. (A) qRT-PCR and (B) Western blot analyses of the relative mRNA and protein expression level of HRH3 in 15 paired tumor and peritumor tissues, respectively. β-actin was used as the internal control. (C) Representative immunohistochemistry (IHC)staining images of HRH3 in paired HCC tissues (n = 86). (D) Kaplan–Meier curve analysis of overall survival (OS) in patients with HCC by the expression of HRH3 in HCC tissues. Total number of patients in each subgroup is presented. Data are presented as mean ± SEM from three independent experiments. |
HRH3 Promotes HCC Cell Growth in vitro
To assess the potential effects of HRH3 on cell growth, a series of biological experiments were performed with gain-of-function or loss-of-function of HRH3.We used siRNA to silence HRH3 expression in HepG2 and PLC/PRF/5 cells, and verified the silencing efficiency by qRT-PCR (Supplementary Figure 1A) and Western blot (Figure 2A). MTS assay showed that HRH3 knockdown in HepG2 and PLC/PRF/5 cells significantly reduced cell growth in comparison with the control (Figure 2B). As supported, EdU incorporation assay also demonstrated that knockdown of HRH3 in HepG2 and PLC/PRF/5 cells significantly attenuated cell proliferation activity compared to the control (Figure 2C). Moreover, we also tested the effect of HRH3 knockdown on the apoptosis of HCC cells. As shown in Figure 2D and supplementary Figure 1B, knockdown of HRH3 in HepG2 and PLC/PRF/5 cells remarkably induced cell apoptosis compared to the control, whereas forced expression of HRH3 in Huh-7 and BEL-7402 cells dramatically promoted cell growth (Figure 2E and F). Similarly, EdU incorporation assay also showed that forced expression of HRH3 in Huh-7 and BEL-7402 cells significantly increased cell proliferation activity compared to the control (Figure 2G). Taken together, our results indicate that HRH3 promotes HCC cell survival by accelerating cell proliferation and inhibiting cell apoptosis.
 |
Figure 2 HRH3 promotes HCC cell survival in vitro. (A) Western blot analysis for the protein expression of HRH3 in HepG2 and PLC/PRF/5 cells 48 h after transfection with treatment as indicated. siNC, negative control siRNA, siHRH3, siRNA against HRH3. (B) MTS cell viability assay in HepG2 and PLC/PRF/5 cells treated as indicated. (C) Cell proliferation ability was evaluated using EdU incorporation assay 48 h after transfection with treatment as indicated. (D) Cell apoptosis analysis by flow cytometry 24 h after treatment as indicated in HepG2 and PLC/PRF/5 cells. (E) Western blot analysis for the protein expression of HRH3 in Huh-7 and BEL-7402 cells 48 h after transfection with treatment as indicated. EV, empty vector; HRH3, HRH3 forced expression vector. (F) MTS cell viability assay in Huh-7 and BEL-7402 cells treated as indicated. (G) Cell proliferation ability was evaluated using EdU incorporation assay 48 h after transfection with treatment as indicated. Data are presented as mean ± SEM from three independent experiments. Scale bar: 50μm. **P < 0.01. |
HRH3 Accelerates HCC Cell G1-S Phase Transition
The effect of HRH3 on cell cycle was further investigated in HCC cells. As shown in Figure 3A, HRH3 knockdown significantly elevated the percentage of HepG2 and PLC/PRF/5 cells in the G1 phase, but reduced the percentage S phase compared with the control. In contrast, forced expression of HRH3 significantly reduced the percentage of Huh-7 and BEL-7402 cells in the G1 phase, but elevated the percentage S phase compared with the control. The above results indicate that HRH3 promotes HCC cell growth through accelerating G1-S phase transition (Figure 3B).
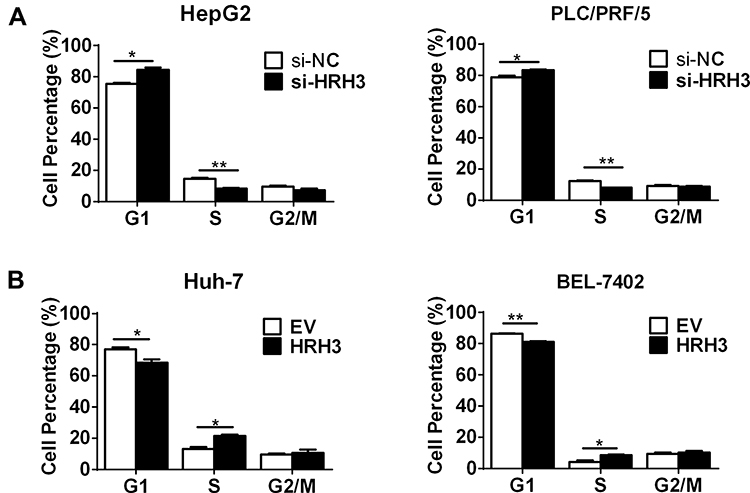 |
Figure 3 HRH3 promotes G1-S phase transition in HCC cells. (A) Cell cycle analyses by flow cytometry in HepG2 and PLC/PRF/5 cells 48 h after treatment as indicated. (B) Cell cycle analyses by flow cytometry in Huh-7 and BEL-7402 cells 48 h after treatment as indicated. Data are presented as mean ± SEM from three independent experiments. *P <0 0.05, **P <0 0.01. |
HRH3 Promotes Malignant Growth of HCC Cell Through Activating PKA
To reveal the molecular mechanism underlying the tumorigenic role of HRH3 in HCC cells, we firstly evaluated the effect of HRH3 on the intracellular level of cAMP. As shown in Figure 4A, HRH3 knockdown in HepG2 and PLC/PRF/5 cells resulted to a significant upregulated of cAMP content in comparison with the control. Based on the above results, we speculated that HRH3 might promote malignant growth of HCC cells by altering intracellular cAMP content. To prove this, we treated HepG2 and PLC/PRF/5 cells with 1 mM SQ22536, a highly inhibitor of adenylyl cyclase, which functioned to inhibit the formation of cAMP for 96 h in cell viability assay and for 48 h in EdU incorporation assay. Our results showed that the inhibitory effect of HRH3 depletion on cell growth was effectively reversed upon SQ22536 treatment (Figure 4B and supplementary Figure 2A and 2B), as also supported by EdU incorporation assay (Figure 4C).
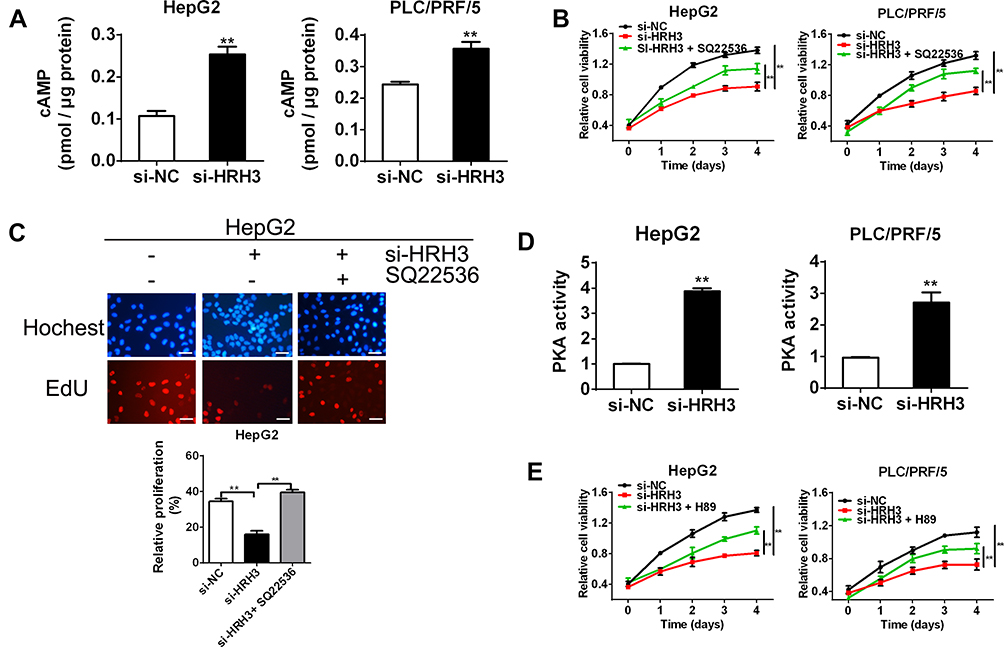 |
Figure 4 HRH3 promotes HCC cell survival through altering intracellular cAMP content and PKA activity. (A) cAMP level was detected by cAMP direct immunoassay in HepG2 and PLC/PRF/5 cells 48 h after treatment as indicated. (B) MTS cell viability assay in HepG2 and PLC/PRF/5 cells treated as indicated. Q22536, 1 mM. (C) Cell proliferation ability was evaluated using EdU incorporation assay 48 h after transfection with treatment as indicated. Q22536, 1 mM. (D) Activity of intracellular PKA was determined by enzyme assays in HepG2 and PLC/PRF/5 cells 48 h after treatment as indicated. H89, 10 μM. (E) MTS cell viability assay in HepG2 and PLC/PRF/5 cells treated as indicated. H89, 10 μM. Data are presented as mean ± SEM from three independent experiments. Scale bar: 50μm. **P < 0.01. |
PKA is involved in cAMP signaling in cells, and believed to function in the pathological process of tumorigenesis. Thus, we speculated that HRH3 might regulate the activity of cAMP-dependent PKA through altering intracellular cAMP content. To prove this, we used PKA Activity Assay Kit to investigate the effect of HRH3 knockdown on PKA activity in HepG2 and PLC/PRF/5 cells. As shown in Figure 4D, knockdown of HRH3 dramatically enhanced the PKA activity in HepG2 and PLC/PRF/5 cells relative to the control. Next, we treated the above cells with 10 μM H89, a highly inhibitor of PKA, for 96 h in cell viability assay and for 48 h in EdU incorporation assay, and reasonably found that the inhibitory effect of HRH3 depletion on cell growth was reversed upon H89 treatment (Figure 4E and supplementary Figure 2C and 2D). All of the above inhibitors did not show significant toxicity effect on the cell viability in normal hepatic QSG-7701 cells at concentrations used (P>0.05) (supplementary Figure 2A and 2C). Altogether, these results indicate that HRH3 promotes malignant growth in HCC cells through altering intracellular cAMP content and PKA activity.
HRH3 Downregulates the Expression of CDKN1A by Regulating Intracellular cAMP Content and PKA Activity
PKA is a kind of protein kinase with poor specificity and functions in many signal pathways through phosphorylation of different substrates. Thus, we speculated that HRH3/cAMP/PKA signaling axis might act on phosphorylation of some substrates and affect their activity, thus contributing to the malignant growth of HCC cells. Several studies have reported that cAMP responsive element-binding (CREB) protein could be phosphorylated by PKA, and the phosphorylated CREB, acting as a transcription factor, promotes the transcription of downstream molecules, including cell cycle related genes. To prove this, we tested the effect of HRH3 on the phosphorylation of CREB by Western blot. As shown in Figure 5A, forced expression of HRH3 significantly decreased the level of phosphorylated CREB in Huh-7 cell. In addition, HRH3 forced expression in Huh-7 cell clearly downregulated the mRNA and protein expression of CDKN1A, but not the mRNA expression of Cyclin D1, Cyclin E1, CDK2, CDK4, CDK6, and CDKN1B (Figure 5A and Supplementary Figure 3A). On the other hand, HRH3 knockdown in HepG2 cell significantly increased the level of phosphorylated CREB and the protein expression of CDKN1A, while these effects could be effectively reversed by SQ22536 treatment (Figure 5B). This was also supported by the results of qRT-PCR assay (Figure 5C). Similarly, H89 treatment also reversed the effects of HRH3 knockdown on upregulating the level of phosphorylated CREB and the expression of PKA in PLC/PRG/5 cells (Figure 5D and E). Next, we evaluated the potential role of CDKN1A in the process of HRH3-mediated tumorigenic growth in HCC cells. As shown in Figure 5F and Supplementary Figure 3B and 3C the promoting effect of HRH3 on HCC cell growth could be effectively reversed by the treatment of CDNK1A knockdown. Moreover, the inhibitory effect of HRH3 knockdown on G1-S phase transition was clearly reversed by the treatment of CDKN1A knockdown (Figure 5G). Taken together, our findings indicate that HRH3 activate CDKN1A signals through regulating intracellular cAMP content and PKA activity, thereby contributing to the malignant growth of HCC cells.
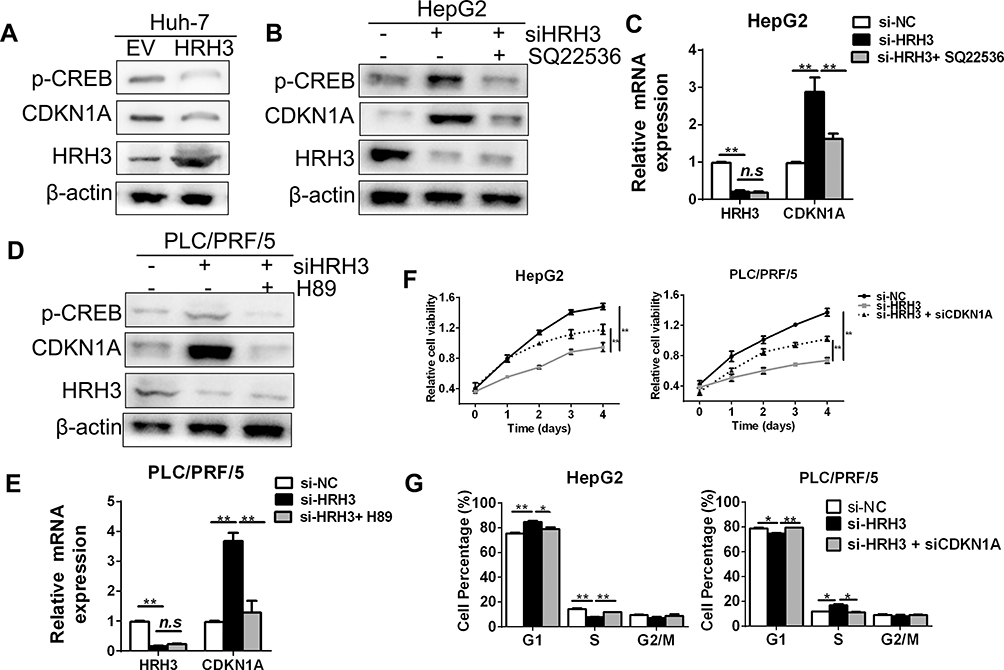 |
Figure 5 Transcriptional activation of CDKN1A by HRH3 in HCC cells through regulating intracellular cAMP content and PKA activity. (A) Western blot analysis of levels of the phosphorylated CREB and CDKN1A in Huh-7 cells 48 h after treatment as indicated. (B) Western blot analysis of the phosphorylated level CREB and CDKN1A in HepG2 cells 48 h after treatment as indicated. Q22536, 1 mM. (C) qRT-PCR analysis of the mRNA expressions of HRH3 and CDKN1A in HepG2 cells 48 h after treatment as indicated. (D) Western blot analysis of the level of phosphorylated CREB and CDKN1A in PLC/PRF/5 cells 48 h after treatment as indicated. H89, 10 μM. (E) qRT-PCR analysis of the mRNA expressions of HRH3 and CDKN1A in PLC/PRF/5 cells 48 h after treatment as indicated. (F) MTS cell viability assay in HepG2 and PLC/PRF/5 cells treated as indicated. siCDKN1A, siRNA against CDKN1A. (G) Cell cycle analyses by flow cytometry in HepG2 and PLC/PRF/5 cells 48 h after treatment as indicated. Data are presented as mean ± SEM from three independent experiments. n.s, not significant, *P <0 0.05, **P <0 0.01. |
HRH3 Enhances Tumorigenic Potential in Nude Mice
Next, we determined the tumorigenic potential of HRH3 in nude mice. Our results showed that the HRH3-forced expression tumors showed a much faster rate and bigger tumor volumes compared to the control (Figure 6A and B). Conversely, the HRH3-knockdown tumors showed a much slower rate and smaller tumor volumes compared to the control, while this inhibitory effect of HRH3 knockdown on tumor growth could be partially reversed by the treatment of siCDKN1A intratumoral injection (Figure 6A and B). In addition, we determined the effect of HRH3 on CREB phosphorylation and the protein expression of CDKN1A in xenograft tumors. As shown in Figure 6C, HRH3 expression was clearly downregulated in the HRH3-knockdown tumors relative to the control tumors, whereas the HRH3 expression was clearly upregulated in the HRH3-forced expression tumors relative to the control tumors. Meanwhile, the level of phosphorylated CREB and protein expression of CDKN1A were clearly downregulated in HRH3-forced expression tumors compared to the control tumors (Figure 6C). Moreover, the level of phosphorylated CREB and protein expression of CDKN1A were clearly upregulated in HRH3-knockdown tumors compared to the control tumors, and the effect of HRH3 knockdown on upregulating the expression of CDKN1A was effectively reversed by the treatment of CDKN1A knockdown (Figure 6C). We further investigated the effect of HRH3 on cell apoptosis in vivo using the TUNEL staining assay. Our data showed that HRH3 forced expression inhibited tumor cell apoptosis, whereas HRH3 knockdown increased the apoptosis rate of tumor cells in vivo, and the effect of HRH3 knockdown on promoting cell apoptosis was effectively reversed by the treatment of CKDN1A knockdown (Figure 6D).
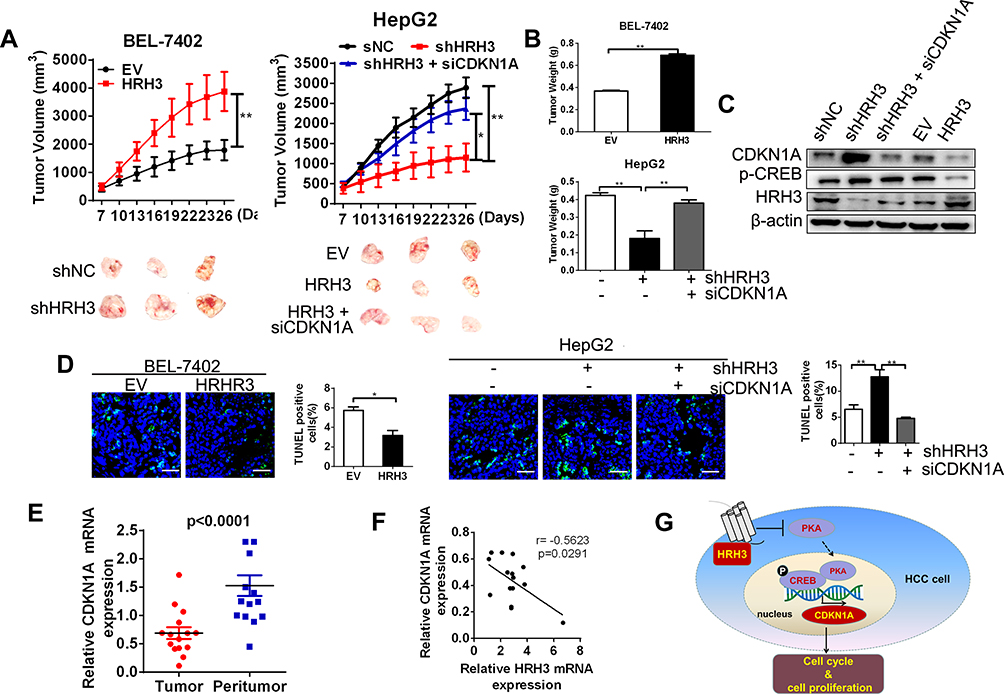 |
Figure 6 HRH3 promotes tumor growth in vivo. (A) Upper, tumor growth curves of subcutaneous xenograft tumor model developed from stable cell lines. Lower, Dissected tumors from sacrificed mice are shown. (B) Tumor weights were calculated and shown. (C) Western blot analysis of the levels of phosphorylated CREB, CDKN1A and HRH3 in tumors from representative mice. (D) TUNEL staining in tumor sections of xenograft mice model with treatment as indicated. Blue, DAPI; Green, TUNEL-positive nucleus. (E) qRT-PCR analyses of the relative mRNA expression level of CDKN1A in 15 paired tumor and peritumor tissues. (F) Correlation analysis between HRH3 and CDKN1A expression in 15 paired tumor and peritumor tissues. (G) Schematic depicting the regulation of cell growth by HRH3 in HCC. Data are presented as mean ± SEM from three independent experiments. Scale bar: 50μm. *P <0 0.05, **P < 0.01. |
We then analyzed the correlational relationship between the expression level of HRH3 and CDKN1A in 15 pairs of HCC tumor and peritumor tissues through qRT-PCR. Our data showed that the expression of CDKN1A was significant decreased in HCC tumor tissues compared with peritumor tissues (Figure 6E). In addition, Pearson r correlation analysis indicated a significant negative correlation between HRH3 expression and CDKN1A expression in tumor tissues from 15 HCC patients (r = −0.5623, P = 0.0291), which provided further supporting evidence (Figure 6F). Based on the above findings, we propose a model to illustrate the mechanism underlying the tumorigenic role of HRH3 in HCC (Figure 6G). Collectively, our data support tumorigenic role of HRH3 by regulating PKA/CREB/CDKN1A signaling axis in HCC.
Discussion
Liver cancer is one of the most common digestive system carcinomas, of which both the incidence and mortality are increasing.1 Numerous studies have shown that the tumor microenvironment is closely related with the occurrence and progression of HCC.6,7,14 Infiltrated macrophages and mast cells in the tumor microenvironment promote malignant tumor growth through secreting inflammatory mediators.6 Histamine, a key mediator in allergic inflammatory responses, not only modulates the immune system, but also acts as a critical mediator in the occurrence and progression of malignancy tumors.7,14 The results from Nabil et al indicated that high serum histamine level in early stages of HCC might contributed to the tumor growth.15 Moreover, previous studies demonstrated that histamine functioned in cell proliferation, migration, and invasion via four G-protein-coupled receptors (HRH1, HRH2, HRH3, and HRH4).6,7,16 Zhao et al reported that upregulated HRH1 promoted tumor progression and contributed to poor prognosis in HCC.8 Histamine and histamine receptors have been identified as critical molecules during inflammation and carcinogenesis, which might be potential molecular target for cancer therapeutics.6,17
Abnormal expression of HRH3 has been reported in different types of cancer, including HCC, cholangiocarcinoma, melanoma, pancreatic, and breast cancer.7,10 Yu et al reported that HRH3 was upregulated in HCC tissues.10 Consistently, our present study demonstrated that HRH3 was significantly increased in HCC tissues and contributed to tumor progression, which indicated that upregulation of HRH3 may serve as a potential prognostic biomarker for early detection and diagnosis of HCC. The results of Francis et al indicated that HRH3 was upregulated in cholangiocarcinoma cells and tissues.7 Chen et al reported that HRH3 was overexpressed in prostate cancer.18 Taken together, all these results indicate that HRH3 is upregulated in malignant carcinomas, strongly suggesting that it may act as a novel malignancy therapeutic target.
Previous studies have reported that HRH3 has dual effects on malignancy occurrence and progression. For instance, Tanaka et al indicated that feeding with the diets containing HRH3 antagonist clobenpropit significantly lowered the multiplicity of colonic adenocarcinoma in male ICR mice, suggesting that HRH3 may be involved in inflammation-related colorectal carcinogenesis.6 Furthermore, inhibition of HRH3 suppressed glioblastoma tumor growth, invasion, and epithelial-to-mesenchymal transition.19 HRH3 antagonist OUP-186 attenuated the proliferation activity of human breast cancer cells.20 Cricco et al reported that HRH3 promoted pancreatic carcinoma cell growth.5 A similar effect was also verified in non-tumor cells. For instance, HRH3 agonist promoted cell proliferation throughout the rat gastrointestinal tract.21 In contrast, Francis et al reported that HRH3 inhibited the growth of cholangiocarcinoma in vitro and in vivo.7 In addition, HRH3 agonists inhibited the biliary growth of BDL rats.22 The results from Davenas et al also indicated that HRH3 antagonists increased McA-RH7777 hepatoma cell proliferation.23 Consistent with the previous studies, the results in the present study demonstrated that upregulated HRH3 increased the proliferation activity, and inhibited cell apoptosis in HCC cells, strongly indicating that HRH3 facilitated cell survival in HCC. The controversial effects of HRH3 on malignancy cell growth may be cell-type specific, which was well demonstrated by the results of Lampiasi N et al, indicating that histamine reduced cell viability and proliferation in HuH-6 cells, whereas in HA22T/VGH cells histamine induced a significant increase in cell growth, suggesting histamine may play different roles depending on the tumor cell features.24
Thus far, the mechanism by which HRH3 promotes tumor growth remains unclear. HRH3 belongs to the family of Gi-coupled G protein coupled receptors.25 Interaction of histamine with HRH3 activates a member of the Gi protein family.26 The detached α-subunit of the Gi protein suppresses the activation of adenylyl cyclase (AC) to inhibit the generation of cAMP. cAMP activates PKA, which in turn promotes the phosphorylation of CREB.26 The detached βγ-subunits of the Gi protein are involved in the activation of phospholipase Cβ (PLC-β), which hydrolyzes the phospholipid phosphatidylinositol (4,5)-bisphosphate to the second messengers inositol trisphosphate (IP3) and diacylglycerol (DAG).26 The present study demonstrated that the level of cAMP was significantly reduced, the activity of PKA was remarkably decreased, and the phosphorylation level of CREB was markedly reduced in HCC cells after forced expression of HRH3. Together, all our results indicate that HRH3 facilitates cell growth through inactivating the cAMP/PKA/CREB signaling pathway in HCC.
Previous studies revealed a cluster of PKA, protein kinase C (PKC), and casein kinase II consensus recognition sites near the N terminus of CREB, which indicated the possibility of interaction in a positive or negative fashion to regulate CREB bioactivity.27 Transcription was stimulated on binding to the cAMP response element of a phosphorylated CREB dimer. Activity of the transcription factor CREB was disrupted in malignancy tumors.28 Xia et al reported that blocking CREB activation completely abolished the development of small cell lung cancer, strongly suggesting that targeting CREB is a promising therapeutic strategy against this cancer.28 Similarly, Srinivasan et al reported that tobacco carcinogen-induced production of GM-CSF activated CREB to promote pancreatic ductal adenocarcinoma growth.29 Notably, repressing CREB inhibited the CREB/EZH2 axis that in turn suppressed prostate cancer growth.30 However, in the present study, our data indicated that forced expression of HRH3 inhibited the phosphorylation of CREB, whereas knockdown of HRH3 increased the phosphorylation of CREB, which indicated that HRH3 promoted cell growth by attenuating the phosphorylation of CREB in HCC.
The transcriptional coactivators p300 and CREB binding protein (CBP) are important regulators of the cell cycle, and tumorigenesis.31 Activated CREB permits its binding to the CREB binding protein at the cAMP response elements in the promoters of target genes, including cell-cycle negative regulator p21/cip1, cyclin A1, and Bcl-2 family members.32,33 p21(CDKN1A), a cyclin-dependent kinase inhibitor, acts as an antioncogene. CDKN1A binds to and inhibits the activity of CDK2 or CDK4 complexes, and functions as a negative regulator of cell cycle progression at the G1 phase. The results of Sun et al demonstrated that CREB reduced smooth muscle cell proliferation via a CDKN1A-dependent mechanism.32 In addition, Cui et al reported that androgen-induced bZIP, belonging to the CREB/ATF family, facilitated proliferation of prostate cancer cells through downregulation of CDKN1A expression.34 Our present study demonstrated that the expression of CDKN1A was regulated by CREB in HCC cells. Furthermore, HRH3 downregulated the expression of CDKN1A via inhibiting the phosphorylation of CREB in HCC cells. Together, our present study indicated that HRH3 downregulated the expression of CDKN1A, and facilitated G1-S phase transition, which finally led to uncontrolled growth of HCC cells.
Several studies also indicated that HRH3-mediated activation of Gi protein stimulates the mitogen-activated protein kinase (MAPK) signaling pathway. The results of Francis et al indicated that HRH3 inhibited cholangiocarcinoma growth by protein kinase Cα-dependent ERK1/2 dephosphorylation.7 Heather et al demonstrated that HRH3 agonist inhibited biliary growth of BDL rats by downregulation of the cAMP-dependent PKA/ERK1/2/ELK-1 pathway.22 The results of Lin et al demonstrated that HRH3 promoted invasion and epithelial-to-mesenchymal transition by activating the PI3K/Akt and MEK/ERK pathways in gliomas.19 However, whether the MAPK signal pathway is activated by HRH3 in HCC cells requires further investigation.
Conclusion
In summary, the results of this study indicate that upregulated HRH3 promotes the growth of HCC by accelerating G1-S phase transition, suppressing cell apoptosis, and inactivating cAMP/PKA/CREB signal pathway to downregulate the expression of CDKN1A. These findings suggest that HRH3 plays a vital oncogenic role in HCC and might be a potential novel therapeutic target for HCC treatment in the future.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Wang M, Zhao J, Zhang L, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–773. doi:10.7150/jca.17648
3. Maggiorella L, Deutsch E, Frascogna V, et al. Enhancement of radiation response by roscovitine in human breast carcinoma in vitro and in vivo. Cancer Res. 2003;63(10):2513–2517.
4. Yang XD, Ai W, Asfaha S, et al. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nat Med. 2011;17(1):87–95. doi:10.1038/nm.2278
5. Cricco GP, Mohamad NA, Sambuco LA, et al. Histamine regulates pancreatic carcinoma cell growth through H 3 and H 4 receptors. Inflamm Res. 2008;57(S1):23–24. doi:10.1007/s00011-007-0611-5
6. Tanaka T, Kochi T, Shirakami Y, et al. Cimetidine and clobenpropit attenuate inflammation-associated colorectal carcinogenesis in male ICR mice. Cancers (Basel). 2016;8(2):25. doi:10.3390/cancers8020025
7. Francis H, Onori P, Gaudio E, et al. H3 histamine receptor-mediated activation of protein kinase Calpha inhibits the growth of cholangiocarcinoma in vitro and in vivo. Mol Cancer Res. 2009;7(10):1704–1713. doi:10.1158/1541-7786.MCR-09-0261
8. Zhao J, Hou Y, Yin C, et al. Upregulation of histamine receptor H1 promotes tumor progression and contributes to poor prognosis in hepatocellular carcinoma. Oncogene. 2020;39(8):1724–1738. doi:10.1038/s41388-019-1093-y
9. Meng F, Han Y, Staloch D, Francis T, Stokes A, Francis H. The H4 histamine receptor agonist, clobenpropit, suppresses human cholangiocarcinoma progression by disruption of epithelial mesenchymal transition and tumor metastasis. Hepatology. 2011;54(5):1718–1728. doi:10.1002/hep.24573
10. Yu D, Zhao J, Wang Y, et al. Upregulated histamine receptor H3 promotes tumor growth and metastasis in hepatocellular carcinoma. Oncol Rep. 2019;41(6):3347–3354. doi:10.3892/or.2019.7119
11. Ahmadian E, Eftekhari A, Babaei H, et al. Anti-cancer effects of citalopram on hepatocellular carcinoma cells occur via cytochrome c release and the activation of NF-kB. Anticancer Agents Med Chem. 2017;17(11):1570–1577. doi:10.2174/1871520617666170327155930
12. Ahmadian E, Khosroushahi AY, Eftekhari A, Farajnia S, Babaei H, Eghbal MA. Novel angiotensin receptor blocker, azilsartan induces oxidative stress and NFkB-mediated apoptosis in hepatocellular carcinoma cell line HepG2. Biomed Pharmacother. 2018;99:939–946. doi:10.1016/j.biopha.2018.01.117
13. Kattan SW, Nafie MS, Elmgeed GA, Alelwani W, Badar M, Tantawy MA. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: implication of PI3K/AKT/mTOR pathway [published online ahead of print, 2020 Jan 23]. J Steroid Biochem Mol Biol. 2020;198:105604. doi:10.1016/j.jsbmb.2020.105604
14. Schneider EH, Detlef N, Roland S. Modulation of behavior by the histaminergic system: lessons from HDC-, H3R- and H4R-deficient mice. Neurosci Biobehav Rev. 2014;47:101–121. doi:10.1016/j.neubiorev.2014.07.020
15. Abdel-Hamid NM, Abdullah AH. Serum histamine and acetylcholine variations as new noninvasive biochemical markers in staging of experimental hepatocellular carcinoma. Clin Exp Med. 2019;19(1):115–120. doi:10.1007/s10238-018-0537-y
16. Wang Y, Jiang Y, Ikeda J, et al. Roles of histamine on the expression of aldehyde dehydrogenase 1 in endometrioid adenocarcinoma cell line. Cancer Med. 2015;3(5):1126–1135. doi:10.1002/cam4.296
17. Nicoud MB, Formoso K, Medina VA. Pathophysiological role of histamine h4 receptor in cancer: therapeutic implications. Front Pharmacol. 2019;10:556. doi:10.3389/fphar.2019.00556
18. Chen J, Hu XY. Inhibition of histamine receptor H3R suppresses prostate cancer growth, invasion and increases apoptosis via the AR pathway. Oncol Lett. 2018;16(4):4921–4928. doi:10.3892/ol.2018.9310
19. Lin JJ, Zhao TZ, Cai WK, et al. Inhibition of histamine receptor 3 suppresses glioblastoma tumor growth, invasion, and epithelial-to-mesenchymal transition. Oncotarget. 2015;6(19):17107–17120. doi:10.18632/oncotarget.3672
20. Tanaka S, Sakaguchi M, Yoneyama H, et al. Histamine H 3 receptor antagonist OUP-186 attenuates the proliferation of cultured human breast cancer cell lines. Biochem Biophys Res Commun. 2016;480(3):479–485. doi:10.1016/j.bbrc.2016.10.077
21. Grandi D, Schunack W, Morini G. Epithelial cell proliferation is promoted by the histamine H3 receptor agonist (R)-α-methylhistamine throughout the rat gastrointestinal tract. Eur J Pharmacol. 2006;538(1–3):141–147. doi:10.1016/j.ejphar.2006.03.049
22. Francis H, Franchitto A, Ueno Y, et al. H3 histamine receptor agonist inhibits biliary growth of BDL rats by downregulation of the cAMP-dependent PKA/ERK1/2/ELK-1 pathway. Lab Invest. 2007;87(5):473–487. doi:10.1038/labinvest.3700533
23. Davenas E, Rouleau A, Morisset S, et al. Autoregulation of McA-RH7777 hepatoma cell proliferation by histamine H 3 receptors. J Pharmacol Exp Ther. 2008;326(2):406–413. doi:10.1124/jpet.107.135368
24. Lampiasi N, Azzolina A, Montalto G, et al. Histamine and spontaneously released mast cell granules affect the cell growth of human hepatocellular carcinoma cells. Exp Mol Med. 2007;39(3):284–294. doi:10.1038/emm.2007.32
25. Baker JG. Antagonist affinity measurements at the Gi-coupled human histamine H3 receptor expressed in CHO cells. BMC Pharmacol. 2008;8(1):9. doi:10.1186/1471-2210-8-9
26. De Esch IJ, Thurmond RL, Jongejan A, Leurs R. The histamine H 4 receptor as a new therapeutic target for inflammation. Trends Pharmacol Sci. 2005;26(9):462–469. doi:10.1016/j.tips.2005.07.002
27. Quinn PG, Granner DK. Cyclic AMP-dependent protein kinase regulates transcription of the phosphoenolpyruvate carboxykinase gene but not binding of nuclear factors to the cyclic AMP regulatory element. Mol Cell Biol. 1990;10(7):3357–3364. doi:10.1128/MCB.10.7.3357
28. Xia Y, Zhan C, Feng M, et al. Targeting CREB pathway suppresses small cell lung cancer. Mol Cancer Res. 2018;16(5):825–832. doi:10.1158/1541-7786.MCR-17-0576
29. Srinivasan S, Totiger T, Shi C, et al. Tobacco carcinogen-induced production of GM-CSF activates CREB to promote pancreatic cancer. Cancer Res. 2018;78(21):6146–6158. doi:10.1158/0008-5472.CAN-18-0579
30. Zhang Y, Zheng D, Zhou T, et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun. 2018;9(1):4080. doi:10.1038/s41467-018-06177-2
31. Snowden AW, Anderson LA, Webster GA, et al. A novel transcriptional repression domain mediates p21(WAF1/CIP1) induction of p300 transactivation. Mol Cell Biol. 2000;20(8):2676–2686. doi:10.1128/MCB.20.8.2676-2686.2000
32. Sun L, Zhao R, Bai Y, et al. CREB is activated in smooth muscle cells isolated from atherosclerotic plaques and reduces smooth muscle cell proliferation via p21-dependent mechanism. Int J Cardiol. 2014;174(3):764–767. doi:10.1016/j.ijcard.2014.04.078
33. Park MH, Lee HS, Lee CS, et al. p21-Activated kinase 4 promotes prostate cancer progression through CREB. Oncogene. 2013;32(19):2475–2482. doi:10.1038/onc.2012.255
34. Cui X, Cui M, Asada R, et al. The androgen-induced protein AIbZIP facilitates proliferation of prostate cancer cells through downregulation of p21 expression. Sci Rep. 2016;6(1):37310. doi:10.1038/srep37310
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.