Back to Archived Journals » Open Access Surgery » Volume 9
Hirschsprung disease: current perspectives
Authors Moore SW
Received 20 November 2015
Accepted for publication 11 February 2016
Published 27 May 2016 Volume 2016:9 Pages 39—50
DOI https://doi.org/10.2147/OAS.S81552
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Cataldo Doria
SW Moore
Division of Paediatric Surgery, Faculty of Medicine, University of Stellenbosch, Cape Town, South Africa
Abstract: Hirschsprung disease is a complex congenital condition of the intestine, which is recognized as being of genetic origin and results from a disturbance of the normal development of the enteric nervous system. As a result, aganglionosis of the distal bowel occurs. It is the most common cause of a low intestinal obstruction in the neonate as well as older children. Occurring as an isolated condition in 70% of cases, it may be associated with other associated congenital abnormalities as well as a number of syndromic phenotypes. A number of distinct genetic sites have been identified in these syndromic phenotypes, which identify potential underlying genetic associations of the disease and indicate the probable gene–gene interaction in its pathogenesis. This review looks at the prevalence, congenital associations, and possible genetic factors influencing the development of Hirschsprung disease. Diagnostic dilemmas, surgical management, potential postsurgical complications, and outcomes are also explored.
Keywords: Hirschsprung disease, children, diagnosis, management, complications and outcome
Introduction
Hirschsprung disease (HSCR) is defined as a functional intestinal obstruction that results from the congenital deficiency of the normal myenteric plexus parasympathetic ganglion cells in the distal portion of the large intestine.
The initial description of congenital megacolon is attributed to Frederick Ruysch in 1691. It went mostly unnoticed in the medical world until it was reported by Harald Hirschsprung in 18871 which started the scientific attention on this unique condition. Nevertheless, it was 50 years until the essential role of the distal aganglionic bowel was identified.2,3 Successful treatment then followed based on the surgical excision of the aganglionic segment and reanastomosis with ganglionated bowel.
The widespread practice and success of this surgery have made HSCR one of the successful surgical treatments of modern times. Nevertheless, over 120 years later, “misunderstandings and diagnostic challenges still make it one of the most challenging diagnoses in certain cases”.4 This is mainly due to the interpretation of the findings and difficulties in separating it from allied congenital neurodysplastic conditions causing functional disturbances of the gastrointestinal tract (GIT).
Demographics
HSCR remains the most common condition to cause low functional intestinal obstruction in children. It is mostly identifiable at birth, but often presents later, particularly in resource-challenged environments. Although the major condition in this spectrum is HSCR, there are a number of other HSCR-like conditions collectively known as “variant Hirschsprung disease”.5–7
The worldwide incidence of HSCR approximates 1 in every 5,000 live births. This has been consistently reported in many countries, including South Africa. A reported large series found a prevalence of 1 per 5,726 live births, and included all ethnic groups.8 An approximate 4:1 male-to-female ratio is substantiated worldwide, but appeared in our previous study to vary between the various South African ethnic groups, being 3.6:1 in Caucasians and 3.9:1 in black African patients, but only 2.5:1 in those of mixed descent, because of a greater proportion of females in this ethnic group.8
The length of the aganglionic segment also affects the male-to-female ratio and differs from the accepted 4:1 ratio to approach a 1:1 ratio in those with a total colonic aganglionosis (TCA). This suggests an incomplete gene penetrance with a variable phenotype in certain patients.
Approximately 92% of mothers with HSCR babies have a normal antenatal history, and the average birth weight of the babies in our series was 3,129 g.8 HSCR has previously been recognized as being rare or absent in premature babies, although it is not excluded in this group. In our earlier study, although 12.2% of babies weighed under 2.5 kg at birth, a scored gestational age of <37 weeks could only be established in 1.9%, illustrating its low prevalence in this group. More recent reports9,10 indicate a rising prevalence in premature babies.
HSCR is accepted as being a sex-linked heterogonous disorder with differing phenotypic expressions and degrees of severity. It has a clear inheritance risk and recurs in certain families in autosomal dominant, recessive, and multigenic transmission patterns.
Associated anomalies
From a molecular perspective, HSCR remains a complex congenital condition resulting from interference of the normal developmental enteric nerve cell signaling mechanisms. This results in the lack of a normal enteric nervous system (ENS) and aganglionosis of the distal bowel segment. As a result, the nature and scope of associated anomalies remain to be of considerable interest.
HSCR-associated congenital anomalies are reported in between 5% and 32% of patients (mean, 21.1%). These include 12% with associated chromosomal anomalies. Apart from the known syndromic phenotypic expressions, there is no reported link between these reported congenital anomalies and familial HSCR recurrence.
Congenital anomalies of the GIT (8.05%), as well as the genitourinary (6.05%) and central nervous system (6.79%) have been reported. In addition anomalies in the musculoskeletal system (5.12%) and those affecting the cardiovascular system (4.99%), craniofacial area (3%), and skin (ectodermal dysplasia) (Figure 1). Syndromic Hirschsprung associations include Down syndrome, dominant sensorineural deafness, Waardenburg syndrome, neurofibromatosis, neuroblastoma, pheochromocytoma, the multiple endocrine neoplasia Type II syndromes, as well as other abnormalities such as syndromes related to cholesterol and fat metabolism. In addition to associations with neuroblastoma and tumors related to MEN2B, HSCR may also be associated with tumors of neural origin such as ganglioneuroma, ganglioneuroblastoma, retinoblastoma, and tumors associated with neurofibromatosis and other autonomic nervous system disturbances.
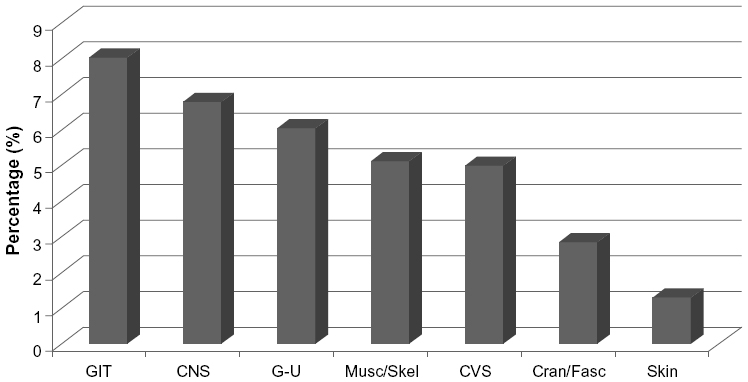 | Figure 1 Bar graph of relative incidence of associated anomalies of Hirschsprung disease |
Some of the syndromic expressions have been linked to different genes or genetic sites, indicating probable gene–gene interaction in the pathogenesis of HSCR. These genetic variations lead to the syndromes being transmitted within families.
Major susceptibility genes on chromosome 10 (RET) and chromosome 13 (EDNRB) have been shown to affect the phenotypic expression of HSCR. Haploinsufficiency of these major RET mutations has been shown to cause HSCR in approximately 20%–25% of cases, particularly in syndromic phenotypes. However, the etiology and pathogenesis of the majority of sporadic HSCR are not yet clear and appears to be caused by the cumulative effects of a number of susceptibility areas on genes actively involved in the control of the mechanisms of neuronal cell proliferation, differentiation, and maturation during ENS development. In addition, there are potential “modifying” associations with loci on chromosomes 2, 9, 20, 21, and 22. In this context, the role of certain flanking genes of critical areas of these genes in the final phenotypic expression of HSCR remains unclear.
Familial transmission
Family history should always be a part of history taking in HSCR evaluation, as a familial recurrence of 2.4%–9% and has been reported. Reports include recurrence in both mono- and dizygotic twins. As a result, a greater than 200 times increased risk of recurrence exists for families with HSCR. No significant difference is noted between male and female probands, but longer phenotypic expressions carry a higher risk. In our previous series study, 50% of male patients with TCA had a family history, occasionally transmitted through a female sibling.
However, genetic counseling via pedigree analysis remains complex, and the significance of specific genetic variations in terms of risk is still unclear. However, an association between long-segment aganglionosis and an increased risk of recurrence exists, with 15%–21% of families with long-segment aganglionosis in our previous study showing an increased risk of recurrence, particularly in patients with TCA (P<0.01)11 This risk of recurrence can achieve >50% prevalence in patients with ultralong-segment aganglionosis (Zuelzer’s disease). In addition, a progression of the length of segment was noted in succeeding generations in a few affected families.11
Long-segment HSCR along with several known associated syndromes demonstrates an autosomal dominant inheritance pattern with incomplete penetrance and is often related to mutations in the RET proto-oncogene. Short-segment HSCR, on the other hand, appears to be transmitted in an autosomal recessive manner or due to multiplicative effects of a number of involved genes.
Etiology/pathophysiology
Developmental etiology of HSCR
One of the areas of advancing knowledge is in our understanding of how the ENS forms during fetal development. HSCR occurs due to a disturbance of the normal ENS development, resulting in the lack the normal ganglion cells in the myenteric plexuses. As a result, there is a functional loss of coordinated peristalsis, leading to intestinal obstruction, as occurs in HSCR.
The ENS originates from neural crest-derived cells that undergo aboral migration in the gut following the vagal pathways. The role of a second sacral inflow of neural cells12 appears to be uncertain.
Early studies showed that the migration of neuroblasts in the vagal trunk begins in about week 5 of embryonic life and progresses down the gut and reaches the rectum by week 12.13 The more recent work of Fu et al14 showed an earlier migration of vagal neuroblasts that colonize the entire gut in a rostrocaudal manner between weeks 4 and 7.
As they migrate, the enteric neuroblasts undergo key developmental processes such as migration, proliferation, survival, and differentiation, differentiating into at least 14 different classes of cells that make up the ENS.15,16 These neuroblasts then form definable intramural ganglia collections by perpendicular migrations within the gut wall.12 The ENS then develops into the complex interconnecting network of neurons and glial cells that regulate motility, sensory response, secretion, and blood flow within the GIT.17
This development of the ENS relies on coordinated interactions between neural crest-derived neuroblasts, the controlling signaling pathways, as well as developmental processes (eg, maturation, survival) to coordinate a normal ENS development. They are under the molecular control of a number of interdependent signaling pathways and transcription and neurotrophic factors that interact with the extracellular matrix components to form a functional ENS. Defective molecular signaling during this developmental process results in maldevelopment and functional conditions such as HSCR.
Genetic associations of HSCR
HSCR is known to be a congenitally determined, sex-modified multifactorial condition, which arises largely from a multigenetic malfunction of ENS development.18–20 The reason for the incomplete migration and development of ganglion cells is as yet not fully understood. Early estimates were that genetic variations could be identified in at least 12% of HSCR cases, which is higher than the expected in the normal population. It is now understood that the major controlling signaling pathway of the developing enteric neuroblasts appears to be the RET proto-oncogene, with up to 80% of patients with HSCR harboring alteration of at least one RET allele.20 Most of these are noncoding mutations/polymorphisms within the gene. The RET intronic SNP rs2435357 has been shown to demonstrate significant frequency differences by affecting RET transcription regulation affecting sex, segment length of aganglionosis, and family recurrence.18 Mutations affecting the coding of RET mRNA are less common overall and appear more frequently in cases of extended aganglionosis (eg, TCA).
Ongoing research has identified a number of susceptibility/ modifying genes related to the RET and EDNRB signaling pathways. These include the EDNRB ligand EDN 3, GDNF situated at chromosome 5p12–13, and its related receptor GFRα. In addition, the association with PHOX2B and the SOX-10 gene on chromosome 22q13 appears to synergize with the endothelin system in very long aganglionic segments.19 Along with the other major susceptibility pathways (eg, the EDNRB), these genetic variations also appear to play a critical role in the etiology of HSCR.
However, the disorder is complex, as shown by the number of affected genes identified (at least 12 have been identified to date and are continually being added to), and gene interaction and crosstalk are understood to modify the genetic expression, resulting in the eventual phenotype. The best example to date lies in the additional susceptibility gene for semaphorin 3A, which results in signaling pathway dysfunction in conjunction with a variant in Ret, leading to malfunction and disease.20 It is probable that this could function as a viable hypothesis for other identified genetic variations, in keeping with the current concept of a multigenetic etiology for HSCR. Some then lead to syndromic phenotypic expressions of genes identified in associated syndromes (eg, chromosome 21 and ZEB2 genetic variations associated with the Mowat–Wilson syndrome).
In addition to all these, local cell adhesion molecules and Rho GTPases, as well as possible somatic mutations,21 resulting from alterations in the local tissue microenvironment are also involved in the development of the clinical phenotype.
Pathology
Macroscopically, the bowel in patients with HSCR can be seen to have a narrow aganglionic segment, a proximal transitional zone progressing from a narrow to a dilated lumen, and a dilated proximal portion of bowel (megacolon) The bowel wall is thickened as a result of hypertrophy of the muscular wall.
HSCR varies considerably in aganglionic length and classically affects the rectum and sigmoid in >70% of cases. The aganglionosis extends proximally for short or long segments depending on the phenotypic expression. A long colonic segment is present in approximately 20% and TCA occurrs in 8%–10%. Ultralong segments of aganglionosis involving extensive small bowel segments (>50 cm) are uncommon, (<2%) and may even involve the entire small intestine (Zuelzers disease).
The functional abnormality results from the uncoordinated function of the aganglionic bowel and its failure to relax to allow forward propulsion of the intestinal contents. The functional obstruction always includes internal anal sphincter dysfunction and failure to relax, which can be demonstrated manometrically.
Diagnosis
HSCR, although often regarded as a relatively easy diagnosis, may be “one of the most difficult diagnoses in Paediatric Surgery”,4 mainly due to variations and difficulties in interpreting the relevant clinical, histological, and radiological findings.
Clinical presentation of HSCR
The clinical evaluation of the patient remains the most important diagnostic step in the diagnosis of potential HSCR.
History
The clinical presentation depends not only on the aganglionosis length but also the age of presentation. More than 90% of the cases can be diagnosed in the neonatal period but are frequently overlooked in poorly resourced health situations, with late presentation is therefore common.
A delay in passage of meconium is the most pertinent observation in the neonate suspected of having HSCR (>80%). Whereas normal babies pass meconium within 24 hours, and even up to 48 hours, any baby who passes no or little meconium even after 24 hours should be investigated for HSCR.
Intestinal obstruction presents with bile-stained vomit, and abdominal distention is often present by day 2. In developing countries, presentation may be significantly late, and the main symptoms include abdominal distention, constipation, or diarrhea, with failure to thrive and developmental delay. These children often have a history of early onset of mild constipation followed by acute low intestinal obstruction. The early onset of chronic constipation (often since birth) is an indication to exclude HSCR. Stools when passed are irregular and passed with great difficulty. Abdominal distention occurs in almost 100% of the cases and may be marked. The child does not develop normally and is often thin and malnourished. The presence of a megacolon of the proximal colon, although being a classic sign of HSCR, may be present in other variations of functional intestinal obstruction.
A family history of HSCR or severe constipation is not infrequent. Other associated anomalies may be present in approximately 16% of HSCR cases.8
In addition to the signs of intestinal obstruction on physical examination, the rectum is often empty on examination in HSCR. However, there may be an explosive discharge of soft stool and bowel emptying if the examining finger reaches beyond the aganglionic segment.
The real danger in HSCR is that of Hirschsprung-associated enterocolitis (HAEC), which carries a significant morbidity and mortality risk. Occurring in approximately 16% of patients, it contributes to >50% of the mortality from this condition, presenting clinically with bloody diarrhea and mucus associated with abdominal distention and possible vomiting.8 Although the majority of cases present during the first 2–4 weeks postnatally, it can also occur in the older patient or following surgical HSCR pull through. The diagnosis of HAEC is important as it requires urgent bowel decompression and relief of obstruction combined with a gentle nursing back to health. Hirschsprung-associated enterocolitis may leads to toxic megacolon and bowel perforation.
Urogenital problems may also present secondary to chronic obstruction (eg, vesicoureteric reflux, hydronephrosis).
Radiological investigation
Because HSCR results in a decrease in bowel segment motility, there is a functional obstruction with decreased peristaltic activity into the aganglionic segment with an abnormal or absent relaxation of the aganglionic segment. These features can often be demonstrated radiologically but varies, and as such, it is not sensitive enough to exclude HSCR. However, contrast studies may give an indication of the transition zone level. Diagnosis is then confirmed by other tests (eg, histology of rectal biopsies).
Abdominal X-rays
A diagnostic evaluation should begin with abdominal X-rays (diagnostic accuracy: 52%), but normal practice would be to proceed to a contrast enema to evaluate the diagnosis further.
Essentially, it is necessary to look for signs of low intestinal obstruction and distended bowel loops of different calibers on the abdominal radiographs. Erect abdominal views can demonstrate air–fluid levels, indicating intestinal obstruction. There would be absence of rectal air, and the lateral view may occasionally demonstrate a narrow rectum (Figure 2). If small bowel obstruction is prominent, a longer aganglionic segment must be considered.
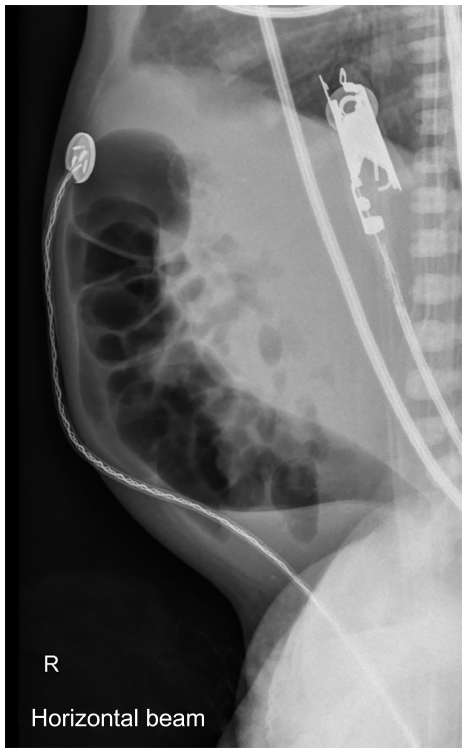 | Figure 2 Lateral abdominal X-ray in a Hirschsprung disease patient showing transition zone. |
Contrast enema
A reduced size of rectum or rectosigmoid, with transition to a dilated, proximal colon on contrast enema, is typical of short-segment (rectosigmoid) aganglionosis.
The radiologic study should show variation in lumen size (the so-called transition zone). In addition, an irregular or “sawtooth” appearance may be present (Figure 3).
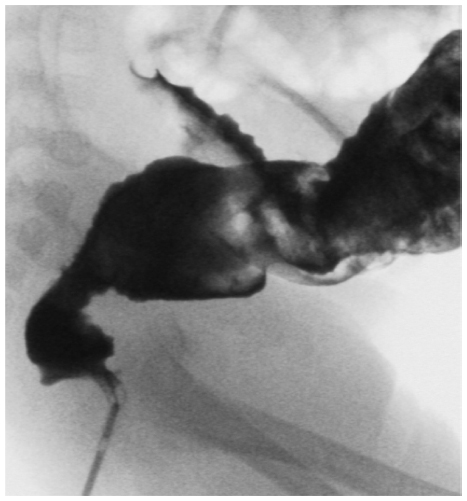 | Figure 3 Contrast enema showing the transition zone in a Hirschsprung disease patient and abnormal rectosigmoid ratio. |
These findings may vary, particularly in neonates, as the transition zone may not have developed sufficiently, or in patients with extended aganglionic segments (TCA).
A contrast enema is reported to be diagnostic in approximately two-thirds of patients with HSCR, particularly if low ion pressures are employed in neonates as the bowel can easily be distended. Colonic irrigation or washout of the colon should not be performed prior to the contrast as it may result in possible decompression and false interpretations.
The essential diagnostic feature on contrast enema is demonstration of the narrow aganglionic segment with dilatation of the proximal bowel segment, a reversed rectosigmoid ratio, and a demonstrable transitional segment. The aganglionic segment may be irregular, demonstrating a sawtooth mucosal appearance, probably as a result of mucosal edema and muscular fasciculations.
A further delay in the clearing of contrast (barium sulfate) within 24 hours is also a reliable sign, and a follow-up X-ray should be performed the following day.
In rectosigmoid aganglionosis, the rectosigmoid ratio (ratio of the diameter of the rectum to the sigmoid colon; normal ratio, 1:1) may prove a useful guide, but considerable variation may exist at different ages and aganglionic lengths. In one study,22 the rectosigmoid index and radiological transitional zone supported the histopathologic diagnosis in 79%–87% of the cases. Positive predictive values were not significant in this study, but the negative predictive values reached clinical significance in smaller children but not in neonates. Occasionally, additional ultrasonographic findings may also suggest the diagnosis.
Radiologic features of HAEC
The features of HAEC on plain abdominal X-ray are very nonspecific and difficult to interpret and must be interpreted in light of the clinical findings.23 The so-called Delphi analysis24 includes signs such as a rectosigmoid “cutoff” without any distal air, dilated proximal loops of bowel (including small bowel), air fluid levels on erect films, possible pneumatosis intestinalis, or a “sawtooth” appearance with an irregular mucosal or intestinal wall. In addition, free intra-abdominal or subdiaphragmatic air represents an intestinal perforation. One review25 reported colonic dilatation as having a 90% sensitivity but low specificity (24%) on plain radiography Care should be taken in contrast enemas, and this technique probably should be avoided in the acute phase because of the perforation risk. The findings on computerized tomography scan have also been reported.26
Histologic diagnosis
The absence of ganglion cells in intramuscular myenteric (Auerbach’s) plexus and the submucosal Meissner’s plexus are the recognized classic histological picture of HSCR as identified on rectal biopsy. There is usually an additional proliferation of peripheral nerves and neurofibrils present.
Biopsies are obtained from sites 2 and 4 cm from the dentate line (3 and 5 cm in older children) to avoid the normal aganglionic zone in the first centimeter proximal to the dentate line. The fresh biopsy specimens are then frozen and sectioned (slices of ±15 μm), stained, and submitted to histopathologic examination. Issues surrounding rectal biopsy techniques are discussed in further detail in the “Dilemmas and controversies in HSCR” section.
Despite innovations in the biopsy forceps over the years and the development of a superior tool with disposable capsules, these problems remain. As a result, procedure to full-thickness biopsy is often required. Full-thickness rectal biopsy, however, results in problems being encountered during subsequent surgical procedures that require mucosal stripping (eg, endorectal pull-through [ERP] procedures).
Demonstration of aberrant neurofibrils
Acetylcholinesterase (AChE) staining techniques27 are useful for demonstrating the HSCR-related increase in neurofibrils in the lamina propria and muscularis mucosa. As such, it remains the investigation of choice in many units to evaluate suction biopsies since it demonstrates the HSCR increase in parasympathetic nerves in the affected zone as well as the abnormal lamina propria and muscularis mucosal neurofibril activity.
Because certain staining techniques (eg, AChE) are mostly performed in specialized centers and require frozen sections, many histopathology services experience logistical problems and rely on the use of alternative special histochemical stains for evaluation. Where diagnostic difficulties are experienced on hematoxylin and eosin staining, further histochemical and immunocytochemical staining may be required28 However, a clear understanding of the capabilities of these staining methods and limitations are required for interpretation of the results. Immunohistochemical stains such as dehydrogenase stains, neuron specific enolase (NSE), S100 and PGP9.5, calretinin,29 and synaptophysin30 are currently being used.
Therefore, the histopathologic diagnosis of HSCR essentially rests on observing the absence of ganglion cells in the intermyenteric plexuses, as well as observation of the presence of enlarged peripheral nerve trunks in the submucosa and the observed increased proliferation of neurofibrils in the lamina propria and the muscularis mucosa on special stains (absent in normally innervated intestinal wall). These findings may vary, particularly in neonates and in long-segment aganglionosis.31
Anorectal manometry
The absence of the rectosphincteric reflex (demonstrated manometrically in normal bowel following rectal distention) in patients with HSCR has been previously used in diagnosis, but it is currently in limited use, except in the diagnosis of sphincter achalasia (ultrashort-segment Hirschsprung).
Differential diagnosis
Differential diagnosis of HSCR includes the small left colon syndrome (particularly in diabetic mothers), a meconium plug in neonates, as well as other dysganglionosis of the GIT. In addition, visceral hollow muscular myopathies and chronic idiopathic intestinal pseudo-obstruction should also be borne in mind as differentials.32
In the older child (generally at >1 year of age), the differential includes conditions such as acquired megacolon, mostly result from anal fissure or anal or rectal strictures or anorectal malformations, local tumors, or other psychogenic pathologies.
Controversies and diagnostic dilemmas in HSCR
The first area of discussion of dilemmas associated with HSCR is that of the diagnosis and rectal biopsy in patients with Hirschsprung disease is unclear. It must be stressed that the final diagnosis rests on a combination of clinical suspicion, radiographic findings, as well as rectal biopsy results.
First, the criteria used for selecting patients for biopsy require further clarification. In this regard, the NICE guidelines for biopsy in children with chronic constipation (published in 2010),33 although a helpful guide to the selection of patients, are not infallible and are currently being reevaluated.
The second issue relates to interpretation of the biopsy results. This requires an understanding of the type of rectal biopsy and the level at which the biopsy was taken. The method of suction biopsy initially described by Noblett34 is widely used as it allows specimens of the submucosal and mucosal layers to be obtained with minimal discomfort and without anesthesia. However, biopsy by this technique is often inadequate, particularly in the older patient, due to tissue thickness and possible mucosal edema. A number of innovations to the biopsy forceps over the years have culminated in the current availability of a superior tool with disposable capsules for specimen taking which yields more reliable results. However, problems remain, and as a result, full-thickness biopsy is frequently recommended in cases of unclear diagnosis. However, full-thickness biopsy may result in problems being encountered during subsequent surgical procedures that require mucosal stripping (eg, ERP procedures).
Further limitations of this technique include the fact that that the biopsy specimen often does not show the deeper plexuses, and, in many centers, relies on specialized staining methods (eg, AChE or immunostaining) to demonstrate the neurofibrils in the lamina propria and muscularis mucosa when ganglion cells cannot be visualized.
Issues surrounding the method and type of histological staining remain controversial. Many units in the USA rely on hematoxylin and eosin staining, with the occasional use of calretinin staining in difficult cases. AChE staining, as well as the use of more specific stains, for neural tissues is commonly used in much of Europe and the rest of the world to enhance diagnosis.
HSCR is fairly well understood as a functional obstruction due to the congenital absence of ganglion cells in the distal bowel. Further controversy has been the occasional interpretation of histological results in the presence of clinical and radiological evidence of a localized functional rectal obstruction. The status of other conditions involving the ENS which are sometimes included in the Hirschsprung spectrum of conditions and loosely termed “variant Hirschsprung disease” is not yet clear.5 It has become increasingly clear that a number of congenital neurodysplastic conditions exist that cause a functional disturbance of the GIT. These include both HSCR and other HSCR-like conditions. These patients have signs and symptoms of a functional intestinal obstruction, but some ganglion cells may be evident on the rectal biopsy specimen. These are a problem group and are among the most difficult to treat.6 Therefore, there is a considerable lack of consensus as to their significance.
Further areas of controversy include postnatal ENS plasticity, which may contribute to and possibly explain the histological differences observed in “Skip” aganglionic segments and acquired aganglionosis in Hirchsprung disease. Occasional reports supported these concepts as being uncommon variants sometimes seen in association with HSCR.35–37 These are of considerable interest to surgeons, both from an etiological as well as a management point of view.
It has become clear over time that there is a spectrum of histopathological findings in the ENS of Hirschsprung patients. Recognition of the histologically identified transitional zone is important as it may result in postoperative obstructive symptoms39,40 following pull-through surgery for HSCR. The importance of an adequate HSCR diagnosis as well as an intraoperative pathological evaluation of the intended pull-through segment at operation is therefore important in order to avoid postoperative obstructive dysfunction.
Issues surrounding the best methods of surgical correction and the special issue of TCA and extended aganglionosis will be discussed. In general, the outcomes for Hirschsprung surgery are not always as good as surgeons think, and long term follow-up is important.
Management
The standard surgical management of HSCR varies from center to center.
However, the initial aim of management is to resuscitate the patient (some children with enterocolitis have severe fluid deficits). Once the diagnosis has been confirmed by the previously described methodology.
The next aim is to relieve the intestinal obstruction. In many patients, this can be achieved by regular adequate rectal washouts with warm saline every 6 hours. If relief from obstruction is obtained by this means, then the washouts may be continued until early corrective surgery can be performed (preferentially in the neonatal period). Should complete decompression of the distended bowel not be obtained by this means, a surgical defunctioning stoma may still be necessary in patients not decompressing fully or when nursing care is defective. The stoma must be based on the normally enervated bowel level.
The current practice of managing the obstruction by bowel irrigation techniques allows for early one-stage surgery to be performed within the neonatal period. This allows for early treatment and restoring of normal gastrointestinal function.
Principles of surgical management and results
The correction of the problem by means of definitive surgery is aimed at resecting the abnormal aganglionic bowel and ensuring that normally ganglionated bowel is anastomosed to the rectum without affecting continence.
Pull-through surgical procedures for HSCR have been subject to numerous modifications since the original description by Swenson and Bill in 1948.40 Most of these modifications still adhere to the principle of removing the aganglionic obstructive segment and reanastomosis of healthy bowel to the rectum. The popularity of the Duhamel41 and ERP described by Soave42 have waned in recent years but remain in the surgeons armamentarium for procedures performed by open surgery.
Neonatal pull-through
The current trend toward a primary procedure by the transanal technique (described in the “Transanal pull-through technique” section) versus a two-stage or laparoscopically assisted technique represents a major change in surgical practice. Arguments for the laparoscopic-assisted modification are that it permits histological mapping of intestinal biopsies, thus permitting identification of the level of the transitional zone.
In general therefore, there is evidence to support the view that the current neonatal pull-through is a safe and feasible method of treatment of HSCR, particularly in patients diagnosed in the neonatal period. Patients who present later with grossly distended megacolon are probably best managed by surgical diversion of the fecal stream by a colostomy, allowing the chronically distended bowel to return to a normal caliber lumen before definitive surgical correction.44
Transanal pull-through techniques
The transanal pull-through approach is currently in vogue in many parts of the world as it avoids abdominal scars, promotes earlier feeding, and simplifies postoperative recovery. It is particularly suited to a short aganglionic segment. The anastomosis is usually by a modified Soave or Swenson technique adapted for anal approach (technique performed in reverse).
Prognosis and outcome of Hirschsprung surgery
HSCR is regarded as a surgically correctable condition in the majority of cases. Patients who are appropriately treated may live as productive members of society with a good quality of life. Growth and development are mostly within the normal population parameters and intellectual function approximates normal.44 In general, most patients achieve socially acceptable anorectal function, provided that sufficient time is allowed for adjustment in the beginning.
Poor outcomes are reported in approximately 15%–30% of patients, particularly in those with syndromic expressions. However, fecal continence improves with time in most children. However, this may be delayed into adolescence. In a 47-year follow-up study, evaluation via an evacuation score was graded as “excellent” in 66.7%, “good” in 19.0%, and “fair” in 11.9%, Incontinence occurred in 16.7%, and soiling was present in 19.0%.45
The functional outcome depends on a number of factors, including the length of the aganglionic segment, the surgical procedure performed, the presence or absence of surgical complications, as well as the social circumstances, family support, and associated anomalies of the patient among other factors. Psychological problems be magnified in those with poor support systems.
Genetic counseling is important but should be handled with care. Cognizance of potential ethical issues is of importance, particularly in cases of very long aganglionic segments or syndromic phenotypic expressions, as these may include a number of disabling associated anomalies.
Postoperative complications of Hirschsprung surgery
Early postoperative complications appear to be consistent, despite varying ages of surgical correction. Similarly, the published results of single-stage repair show little effect on the complication rate if the repair was performed prior to or later than the neonatal period or in patients weighing less than 40 kg.46 Recognized early, postoperative complications of Hirschsprung surgery include anastomotic insufficiency, rectal stenosis, prolonged ileus, intestinal adhesive obstruction, and neorectal retraction. Wound or other septic complications may occur with intrapelvic or presacral abscesses related to anastomotic complications. Excessive fluid and electrolyte losses may result in metabolic derangements, particularly in long-segment disease and enterocolitis associated with HSCR.
Late complications
The overall long-term development of function (continence, sexual, and psychological) remains largely unevaluated. In one large study,45 a 6% incidence of postoperative enterocolitis and a 4% stricture rate were reported. The complications associated with transanal surgery techniques have been reported in a multicenter study as being <1%–2%.45
In a further long-term evaluation of 194 of 259 consecutive patients, bowel function was normal in 68%, whereas 10.3% had soiling and 21.7% required laxatives or enemas for resistant constipation.48 The majority of patients with HSCR continue to have disturbances of bowel function for many years before attaining normal continence. This is particularly true of patients with associated Down syndrome, where 34% achieved normal continence.
The causes of persistent constipation and obstructive symptoms may be related to a possible residual aganglionic segment, sphincter achalasia, associated dysganglionosis of the ENS, as well as acquired aganglionosis, and other functional causes. Postoperative causes include surgical complications such as strictures, restrictive cuffs following ERP (Soave), or a retained spur following a Duhamel anastomosis,
Persistent postoperative sphincter achalasia may give rise to postoperative obstructive symptoms and be difficult to manage. Previous attempts at repeated anal dilatation have resulted in mixed success. A topical application of a glyceryltrinitrate paste is fairly effective, as a cheap alternative. The alternative is the injection of botulin toxin, which is advocated by some.
Genitourinary symptoms such as enuresis, incontinence of urine, dysuria, and impotence are not infrequent and occur in 0%–9.7% of patients postoperatively. The frequency with which these occur may be related to the surgical technique used, possibly due to damage to pelvic nerves (especially with techniques involving extensive pelvic dissection).44 It has also been suggested that these symptoms may be related to psychological disturbances associated with long periods of hospitalization and trauma.
HAEC remains a significant cause of morbidity and mortality.24 The etiology of HAEC remains obscure, despite considerable research,24,25 but current evidence suggests a complex interaction of genetic factors with underlying factors, suggesting impairment of local defense mechanisms. Whether environmental factors also play a part is not yet clear.
Mortality is uncommon, being reported in 1.5%–2.8% of cases and depends on the surgical procedure.45 Many of these cases may be influenced by a higher incidence in patients with HAEC and Down syndrome (mortality rate of 26%).
Management of special problems
Enterocolitis management
Management of HAEC includes resuscitation, antibiotics, decompression (6-hourly), and colostomy when stabilized. Surgery, if performed in the acute stage of HAEC, may result in an increased frequency of severe complications.
Total colonic aganglionosis (TCA)
TCA or long-segment aganglionosis may lead to a number of special problems due to the length of the bowel involved. It occurs in approximately 2%–13% of cases and appears to represent a different clinical spectrum of disease in terms of presentation and diagnostic difficulties.31 TCA can be divided into the more frequent TCA type, where the aganglionosis extends from the anus to <50 cm beyond the ileocecal valve into the small bowel. A second type exists where total colonic and small bowel aganglionosis (TCSA) exist, which may involve a very long segment of aganglionosis (sometimes referred to as Zuelzer’s disease).2
Initial surgical management usually involves intestinal defunctioning in ganglionated bowel by means of an ileostomy. Subsequent anal pull-through procedure may necessitate anastomotic modification depending on the length of intestinal involvement.
It is not yet clear whether TCA merely represents a long form of HSCR or a different expression of the disease, but its nature suggests a different pathophysiology from the more common forms of HSCR.48
It has long been recognized as presenting particular problems in diagnosis and management.48–50 Many different surgical techniques have been utilized for TCA,49–52 with outcomes mostly related to the type of surgical technique performed. Many surgeons now accept a modification of the Duhamel procedure as the best option in TCA in terms of long-term function.51,52
Genetic counseling and prevention
Genetic factors are known to be implicated in HSCR pathogenesis. However, the pattern of inheritance remains unclear, and variable Mendelian inheritance with autosomal dominant, recessive, and multigenic patterns being reported. This is not entirely surprising as HSCR is a complex condition involving a multifactorial and multigenic etiology.
Pedigree analysis alone is a particularly difficult basis for genetic counseling in a multifactorial condition, such as HSCR, and may depend on a number of unrelated factors such as small family size, poor history, and the possibility of adoption into a family. Despite recent advances in understanding its molecular background, practical therapeutic interventions are still very limited.11
Should a major genetic defect be detected in a particular family, the door is open for fetal genetic testing and evaluation of risk.
Acknowledgments
This work is based on the research supported in part by the National Research Foundation and the Medical Research Council of South Africa.
Disclosure
The author reports no conflicts of interest in this work.
References
Hirschsprung H. Stulträgheit Neugeborener in Folge von Diltation und Hypertrophie des Colons. Jb Kinderheilk. 1887;27:1–7. | |
Zuelzer WW, Wilson JL. Functional intestinal obstruction on a congenital neurogenic basis in infancy. Am J Dis Child. 1948;75(1):40–64. | |
Whitehouse FR, Kernohan JW. Myenteric plexus in congenital megacolon. Study of eleven cases. Arch Int Med. 1948;82(1):75–111. | |
Martucciello G. Hirschsprung’s disease, one of the most difficult diagnoses in pediatric surgery: a review of the problems from clinical practice to the bench. Eur J Pediatr Surg. 2008;18(3):140–149. | |
Puri P. Variant Hirschsprung’s disease. J Pediatr Surg. 1997;32(2):149–157. | |
Puri P, Gosemann JH. Variants of Hirschsprung disease. Semin Pediatr Surg. 2012;21(4):310–318. | |
Obermayr F, Hotta R, Enomoto H, Young HM. Development and developmental disorders of the enteric nervous system. Nat Rev Gastroenterol Hepatol. 2013;10(1):43–57. | |
Moore SW. The Study of the Etiology of Post-Surgical Obstruction in Patients with Hirschsprungs Disease [doctoral MD thesis]. Cape Town, South Africa: University of Cape Town; 1993. | |
Sharp NE, Pettiford-Cunningham J, Shah SR, et al. The prevalence of Hirschsprung disease in premature infants after suction rectal biopsy. J Surg Res. 2013;184(1):374–377. | |
Duess JW, Hofmann AD, Puri P. Prevalence of Hirschsprung’s disease in premature infants: a systematic review. Pediatr Surg Int. 2014; 30(8):791–795. | |
Moore SW, Zaahl MG. A review of genetic mutation in familial Hirschsprung’s disease in South Africa: towards genetic counseling. J Pediatr Surg. 2008;43(2):325–329. | |
Burns AJ, Thapar N. Advances in ontogeny of the enteric nervous system. Neurogastroenterol Motil. 2006;18(10):876–887. | |
Okamoto E, Ueda T. Embryogenesis of intramural ganglia of the gut and its relationship to Hirschsprungs disease. J Pediatr Surg. 1967;2:437–443. | |
Fu M, Lui VC, Sham MH, Cheung AN, Tam PK. HOXB5 expression is spatially and temporarily regulated in human embryonic gut during neural crest cell colonization and differentiation of enteric neuroblasts. Dev Dyn. 2003;228(1):1–10. | |
Le Douarin N, Teillet MA. [Origin of intramural ganglionic system cells of the digestive tract of bird embryos]. C R Acad Sci Hebd Seances Acad Sci D. 1971;273(16):1411–1414. French. | |
Le Douarin N. Migration and differentiation of neural crest cells. Curr Top Dev Biol. 1980;16:31–85. | |
Anderson RB, Newgreen DF, Young HM. Neural crest and the development of the enteric nervous system. Adv Exp Med Biol. 2006;589:181–196. | |
Kapoor A, Jiang Q, Chatterjee S, et al. Population variation in total genetic risk of Hirschsprung disease from common RET, SEMA3 and NRG1 susceptibility polymorphisms. Hum Mol Genet. 2015;24(10):2997–3003. | |
Sasselli V, Pachnis V, Burns AJ. The enteric nervous system. Dev Biol. 2012;366(1):64–73. | |
Jiang Q, Arnold S, Heanue T, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am J Hum Genet. 2015;96(4):581–596 | |
Moore SW, Zaahl M. Intronic RET gene variants and somatic mutations in Down syndrome associated Hirschsprungs disease (ds-hscr) in an African population. J Pediatr Surg. 2012;47(2):299–302. | |
Garcia R, Arcement C, Hormaza L, et al. Use of the recto-sigmoid index to diagnose Hirschsprung’s disease. Clin Pediatr (Phila). 2007;46(1):59–63. | |
Frykman PK, Short SS. Hirschsprung-associated enterocolitis: prevention and therapy. Semin Pediatr Surg. 2012;21(4):328–335. | |
Pastor AC, Osman F, Teitelbaum DH, Caty MG, Langer JC. Development of a standardized definition for Hirschsprung’s-associated enterocolitis: a Delphi analysis. J Pediatr Surg. 2009;44(1):251–256. | |
Elhalaby EA, Teitelbaum DH, Coran AG, Heidelberger KP. Enterocolitis associated with Hirschsprung’s disease: a clinical histopathological correlative study. J Pediatr Surg. 1995;30(7):1023–1026; discussion 1026–1027. | |
Sheth J, Nour S, Dickinson F. CT images of Hirschsprung’s associated enterocolitis: a rare finding. Arch Dis Child. 2009;94(10):816. | |
Meier-Ruge W, Lutterbeck PM, Herzog B, Morger R, Moser R, Schärli A. Acetylcholinesterase activity in suction biopsies of the rectum in the diagnosis of Hirschsprung’s disease. J Pediatr Surg. 1972;7(1):11–17. | |
Lake BD, Malone MT, Risdon RA. The use of acetylcholinesterase (AChE) in the diagnosis of Hirschsprung’s disease and intestinal neuronal dysplasia. Pediatr Pathol. 1989;9(3):351–354. | |
Kapur RP, Reed RC, Finn LS, Patterson K, Johanson J, Rutledge JC. Calretinin immunohistochemistry versus acetylcholinesterase histochemistry in the evaluation of suction rectal biopsies for Hirschsprung disease. Pediatr Dev Pathol. 2009;12(1):6–15. | |
Kobayashi H, Miyano T, Yamataka A, Lane GJ, Fujimoto T, Puri P. Use of synaptophysin polyclonal antibody for the rapid intraoperative immunohistochemical evaluation of functional bowel disorders. J Pediatr Surg. 1997;32(1):38–40. | |
Moore SW, Zaahl M. Clinical and genetic differences in total colonic aganglionosis in Hirschsprung’s disease. J Pediatr Surg. 2009;44(10):1899–1903. | |
Moore SW, Schneider JW, Kaschula RD. Non familial visceral myopathy clinical and pathologic features of African degenerative leiomyopathy. Pediatr Surg Int. 2002;18(1):6–12. | |
Bardisa-Ezcurra L, Ullman R, Gordon J. Diagnosis and management of idiopathic childhood constipation: summary of NICE guidance. BMJ. 2010;340:c2585. | |
Noblett HR. A rectal suction biopsy tube for use in the diagnosis of Hirschsprung’s disease. J Pediatr Surg. 1969;4(4):406–409. | |
Kapur RP, deSa DJ, Luquette M, Jaffe R. Hypothesis: pathogenesis of skip areas in long-segment Hirschsprung’s disease. Pediatr Pathol Lab Med. 1995;15(1):23–37. | |
Moore SW, Sidler D, Schubert PA. Segmental aganglionosis (zonal aganglionosis or “skip” lesions) in Hirschsprungs disease: a report of 2 unusual cases. Pediatr Surg Int. 2013;29(5):495–500. | |
Cohen MC, Moore SW, Neveling U, Kaschula RO. Acquired aganglionosis following surgery for Hirschsprung’s disease: a report of 5 cases during a 33-year experience with pull-through procedures. Histopathology. 1993;22(2):163–168. | |
Moore SW, Millar AJ, Cywes S. Long term clinical, manometric, and histologic evaluation of obstructive symptoms in the postoperative Hirschsprungs patient. J Paediatr Surg. 1994;29(1):106–111. | |
Kapur RP, Kennedy AJ. Transitional zone pull through: surgical pathology considerations. Semin Pediatr Surg. 2012;21(4):291–301. | |
Swenson O, Bill AH Jr. Resection of rectum and rectosigmoid with preservation of the sphincter for benign spastic lesions producing megacolon; an experimental study. Surgery. 1948;24(2):212–220. | |
Duhamel B. A new operation for the treatment of Hirschsprungs disease. Arch Dis Child. 1960;35(179):38–39. | |
Soave F. New surgical technique for the treatment of Hirschsprung’s disease. Surgery. 1960;56:1007–1014. | |
Moore SW, Albertyn R, Cywes S. Clinical outcome and long term quality of life after surgical correction of Hirschsprungs disease. J Pediatr Surg. 1996;31(11):1496–1502. | |
Ieiri S, Nakatsuji T, Akiyoshi J, et al. Long-term outcomes and the quality of life of Hirschsprung disease in adolescents who have reached 18 years or older – a 47-year single-institute experience. J Pediatr Surg. 2010;45(12):2398–2402. | |
Langer JC, Durrant AC, de la Torre L, et al. One-stage transanal Soave pullthrough for Hirschsprung disease: a multicenter experience with 141 children. Ann Surg. 2003;238(4):569–583; discussion 583–585. | |
Elhalaby EA, Hashish A, Elbarbary MM, Soliman HA, Wishahy MK, Elkholy A, et al. Transanal one-stage endorectal pull-through for Hirschsprung’s disease: a multicenter study. J Pediatr Surg. 2004; 39(3):345–351. | |
Menezes M, Puri P. Long-term outcome of patients with enterocolitis complicating Hirschsprung’s disease. Pediatr Surg Int. 2006; 22[4], 316–318. | |
Moore SW. Total colonic aganglionosis and Hirschsprung’s disease: shades of the same or different? Pediatr Surg Int.2009;25(8):659–666. | |
Martin LW. Surgical Management of total colonic aganglionosis. Ann Surg. 176, 343–346. 1972. | |
Tsuji H, Spitz L, Kiely EM, Drake DP, Pierro A. Management and long-term follow-up of infants with total colonic aganglionosis. J Pediatr Surg. 1999;34(1):158–161. | |
Goto S, Gunter M, Scherer LR, Bloch T, Grosfeld JL. Surgical treatment of total colonic aganglionosis: efficacy of aganglionic patch enteroplasty in the rat. J Pediatr Surg. 1986;21(7):601–607. | |
Escobar MA, Grosfeld JL, West KW, Scherer LR, Rouse TM, Engum SA, et al. Long-term outcomes in total colonic aganglionosis: a 32-year experience. J Pediatr Surg. 2005;40(6):955–961. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.