Back to Archived Journals » Oncolytic Virotherapy » Volume 5
High-throughput screening to enhance oncolytic virus immunotherapy
Authors Allan K, Stojdl D, Swift S
Received 3 October 2015
Accepted for publication 14 January 2016
Published 5 April 2016 Volume 2016:5 Pages 15—25
DOI https://doi.org/10.2147/OV.S66217
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
KJ Allan,1,2 David F Stojdl,1–3 SL Swift1
1Children’s Hospital of Eastern Ontario (CHEO) Research Institute, 2Department of Biology, Microbiology and Immunology, 3Department of Pediatrics, University of Ottawa, Ottawa, ON, Canada
Abstract: High-throughput screens can rapidly scan and capture large amounts of information across multiple biological parameters. Although many screens have been designed to uncover potential new therapeutic targets capable of crippling viruses that cause disease, there have been relatively few directed at improving the efficacy of viruses that are used to treat disease. Oncolytic viruses (OVs) are biotherapeutic agents with an inherent specificity for treating malignant disease. Certain OV platforms – including those based on herpes simplex virus, reovirus, and vaccinia virus – have shown success against solid tumors in advanced clinical trials. Yet, many of these OVs have only undergone minimal engineering to solidify tumor specificity, with few extra modifications to manipulate additional factors. Several aspects of the interaction between an OV and a tumor-bearing host have clear value as targets to improve therapeutic outcomes. At the virus level, these include delivery to the tumor, infectivity, productivity, oncolysis, bystander killing, spread, and persistence. At the host level, these include engaging the immune system and manipulating the tumor microenvironment. Here, we review the chemical- and genome-based high-throughput screens that have been performed to manipulate such parameters during OV infection and analyze their impact on therapeutic efficacy. We further explore emerging themes that represent key areas of focus for future research.
Keywords: oncolytic, virus, screen, high-throughput, cancer, chemical, genomic, immunotherapy
Introduction
High-throughput screens can rapidly and efficiently capture large amounts of data across multiple biological parameters. Depending on the screen design, the ability to collect unbiased information can identify novel ways of impacting pathways of interest that may be overlooked by rationally designed approaches.
Screening technologies have considerable power to systematically probe novel targets, across multiple stages of virus infection. Genome-wide RNA interference (RNAi) screens have focused on short interfering RNA (siRNA) and short hairpin RNA (shRNA) technologies.1 Genome-editing screens have employed clustered, regularly interspaced, short palindromic repeats (CRISPR)–Cas9 technology.2 Pharmaceutical screens have also explored a variety of chemical compound libraries, including bioactive and small molecules, chemotherapeutic compounds, and clinical drug panels.
Typically, a primary screen is performed to find a number of hits above a certain statistical threshold, followed by a smaller secondary screen to validate and replicate these hits (Figure 1). Finally, independent assays are performed to confirm activity and therapeutic relevance (Figure 1). Such approaches have not only yielded biological insights in diverse fields that include cancer biology, developmental biology, cellular metabolism, and virology but have also discovered biological and biotherapeutic targets for disease. However, the vast potential of these powerful new technologies to uncover targets in the oncolytic virus immunotherapy (OVIT) field has yet to be fully realized.
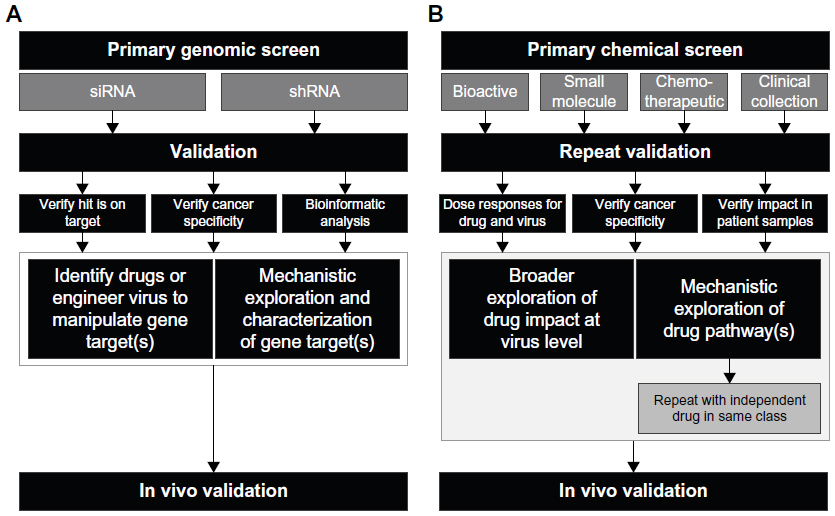 | Figure 1 Flow charts showing typical experimental approaches for high-throughput screening. |
A huge network of highly complex biological interactions occur between an oncolytic virus (OV) and an infected host. OVs have multimodal capabilities, including the direct lysis of infected cells, the establishment of an inflammatory environment, and the amplification of immunostimulatory signals as viral replication proceeds.3–5 OVs can also infect endothelial cells, which can lead to the collapse of tumor vasculature.6 Perhaps, most importantly, OVs can activate antitumor immune responses that can control, regress, and prevent the relapse of tumors.7,8 This occurs, at least in part, through the immunogenic release of tumor-associated antigens (TAAs) following oncolysis of infected tumor cells.9–11 Such TAAs are processed and presented by dendritic cells (DCs) to drive T-cell engagement against a broad repertoire of tumor targets in a phenomenon known as “antigen spreading.”12,13 This capacity to generate TAA-specific T-cell responses can also be artificially enhanced during OVIT by encoding one or multiple TAAs into the viral vector. For example, rhabdoviruses expressing the melanoma TAA, human dopachrome tautomerase, can drive large curative CD8+ T-cell responses against subcutaneous B16.F10 tumors in vivo.14,15 In addition to directly stimulating CD8+ T-cell responses, OVs can selectively minimize immunosuppressive cell populations within the tumor microenvironment through the depletion of regulatory T-cells (Tregs)16 or myeloid-derived suppressor cells (MDSCs).17,18
Several key areas of the interaction between an OV and an infected host represent potential biological targets that can be manipulated to impact therapeutic outcomes. From a virological perspective, these parameters may include delivery to the tumor site, infectivity, oncolysis, bystander killing, replication/productivity/burst size, spread, and persistence (Figure 2). These distinct temporally regulated stages of the OV infection process can be assessed by performing screens at early, intermediate, or late time points following virus inoculation to uncover targets that are therapeutically relevant at multiple stages of infection, following single or multiple rounds of replication. From a host perspective, extra levels of complexity that can potentially be modified to enhance OVIT outcomes include immune engagement and manipulation of the tumor microenvironment (Figure 2).
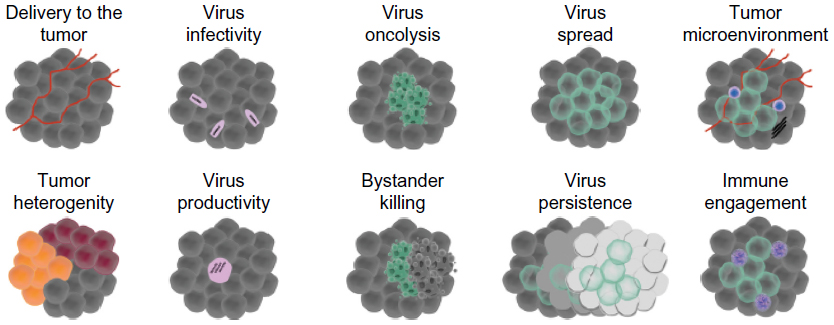 | Figure 2 Ten key parameters affecting oncolytic virus therapeutic efficacy that can be probed by high-throughput in vitro or in vivo screens. |
Here, we review the application of high-throughput genomic and chemical screens to manipulate such parameters during OVIT to improve therapeutic outcomes. While relatively few screens have been performed, of particular interest are those that have explored viruses that are currently undergoing clinical testing, or are already clinically approved, including herpes simplex virus (HSV), vesicular stomatitis virus (VSV), vaccinia virus (VV), and Maraba (MRB) virus.
Modulating OV infectivity and productivity at the tumor site
There are currently two concepts that address the desirability of viral replication and persistence at the tumor site. One concept aims to maximize the replicative capacity and burst size of the OV to enhance oncolysis at the tumor site. This potentially enables a greater magnitude of endogenous TAA spreading since a larger number of heterogeneous tumor cells, presumably across a wider antigenic spectrum, are lysed in a highly inflammatory context. An alternative concept aims to minimize the persistence of the virus at the tumor site to ensure immune responses are focused on lysed tumor antigens rather than viral targets.
Several RNAi screen have identified host genes that can be knocked down to reduce viral infection.19–21 Two of these screens assessed genes that had an early impact on VSV or VV infection by analyzing outcomes at 7–8 hours following inoculation,20,21 while a third screen analyzed outcomes at 18 hours following VSV inoculation to capture genes involved at an intermediate stage of the infection cycle (Table 1).19 Such genes could be knocked down during OVIT to minimize viral persistence at the tumour site, or, alternatively, could be encoded directly into an OV to achieve local overexpression at the tumour site with the aim of enhancing viral replication. For example, SMAD family member 2 (Smad2), which can be knocked down to inhibit VSV replication,19 functions as a tumor suppressor,22 and loss of Smad2 and other Smad proteins correlates with advanced disease and poor prognosis for colorectal cancer.23 Yet, overexpressing Smad2 in subcutaneously implanted Mv1Lu cells can inhibit cellular growth.24
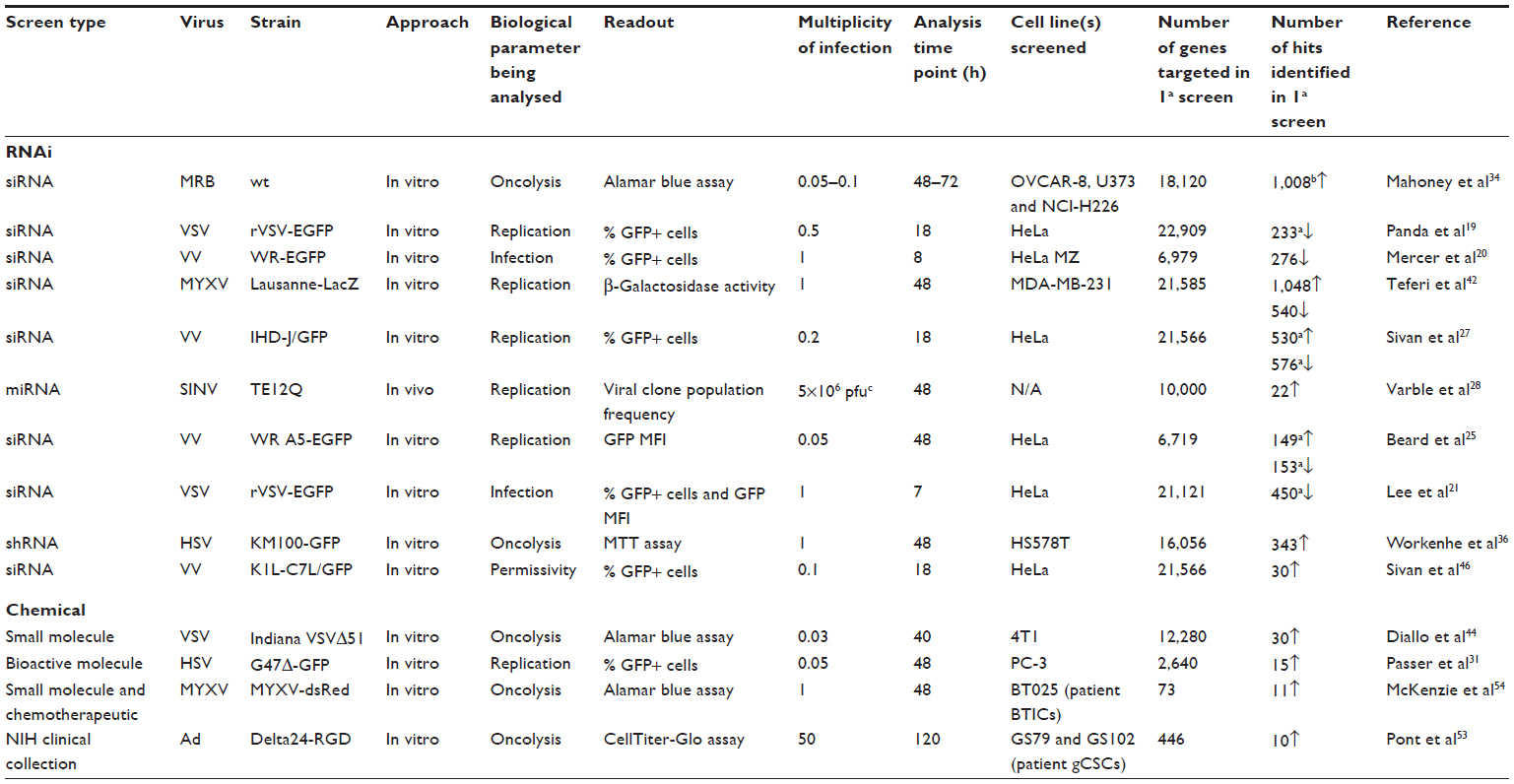 | Table 1 Overview of key high-throughput screens involving oncolytic viruses |
Conversely, screens have also identified host genes that, when knocked down, can improve virus replication. Beard et al25 conducted a screen in HeLa cells with a druggable siRNA human genome library targeting 6,719 genes during infection with recombinant VV-A5-EGFP, a VV strain expressing enhanced green fluorescent protein fused to the A5 core protein in the Western Reserve backbone (Table 1). The primary screen identified 153 proviral host factors (factors that inhibited replication following knockdown) and 149 antiviral factors (factors that increased replication upon knockdown). Seventy-two of these genes were selected for secondary validation using a deconvoluted set of four individual siRNAs targeting each gene. Tumor necrosis factor receptor-associated factor 2 was among their confirmed proviral hits. Further mechanistic exploration determined that this gene precipitated rapid entry of VV into the host cell.26 The nuclear pore protein, nucleoporin 98, was also a confirmed proviral hit. Since VV replication and morphogenesis take place in the cytoplasm, such nuclear-localized hits were unexpected; yet, other RNAi screens have previously shown that nuclear pore proteins have an important role in VV replication.20,27 Among the antiviral hits, only seven genes could be validated, but no further assessment was performed to decipher their role in VV replication.
Using a powerful in vivo screening approach, Varble et al28 identified host gene targets capable of improving viral replication in the context of an in vivo infection with the OV candidate, Sindbis virus (SINV) (Table 1). A library of miR30-based microRNAs (miRNAs) was encoded into a large cohort of SINVs such that each individual virus encoded a unique miRNA targeting a single open-reading frame (ORF). After infecting naïve mice with this library, viral clones equipped with replication-enhancing miRNAs outpaced poorly replicating clones to become the dominant circulating viral populations. This process of natural selection in a host animal identified ~20 viral clones in the first of four parallel screens. Of those 20 clones, eight were shared between two parallel screens, three were shared between three parallel screens, and a single clone was shared between all four parallel screens. The viral clones that provided the greatest replicative advantage across the parallel screens targeted genes encoding zinc finger protein X or MAX gene-associated protein. Both proteins (or their close homologs) have previously been shown to have a role in cellular self-renewal, in addition to cell cycle and cell death pathways.29,30 In this screen, functional analysis suggested that both genes had a role in maintaining the host antiviral response to SINV replication, with a reduction in interferon (IFN)-stimulated gene expression following knockdown.28 From a therapeutic perspective, it is eminently conceivable to perform similar in vivo screens with RNAi library-encoding OVs in tumor-bearing animals to allow the natural selection and subsequent identification of high-value viral clones capable of replicating to high titers at tumor sites.
Finally, in a chemical screen, Passer et al31 analyzed the impact of 2,640 bioactive compounds across three pharmacologically active libraries on the replication index of the oncolytic HSV-1, G47Δ (Table 1). From this library, 15 drugs were identified that amplified viral replication. Two drugs within the same class of ENT1 inhibitors, dipyridamole and dilazep, were further explored. Both dipyridamole and dilazep increased viral productivity across multiple tumor cell lines and in several independent human patient prostate tumor explants. Overall, this correlated with an increase in the killing capacity of the virus on tumor cells following combination therapy; yet, some tumor cell lines remained resistant to the additive activity of the drug and virus combination, despite expressing the target ENT1 protein by Western blot. The combination therapy also improved tumor control in xenograft mice bearing subcutaneous DU145 tumors. Interestingly, the ability of HSV vectors to achieve synergistic outcomes following ENT1 inhibition was fully dependent on the infected cell protein 6 (ICP6)-null status of the HSV vector used. ICP6 encodes for the large subunit of ribonucleotide reductase (RR1) in HSV-1,32 which is essential for viral DNA replication; the removal of ICP6 from oncolytic HSV-1 solidifies its cancer specificity by restricting infection to replicating cells. Indeed, it was subsequently demonstrated that ENT1 inhibition promoted the activity of host ribonucleotide reductase to enhance viral productivity in infected cells.31
Enhancing oncolytic death
Oncolysis is a key step in the viroimmunotherapeutic process that not only directly kills malignant cells to ablate the tumor mass but also releases TAAs to drive inflammatory immune engagement against tumor targets. The level of oncolysis and the type of tumor cell death elicited are critical for establishing immunogenic responses.33
To our knowledge, our laboratory was the first to perform a high-throughput siRNA screen to analyze the impact of host gene manipulation on viral oncolysis to uncover new therapeutic targets (Table 1).34 We carried out two independent genome-wide screens with a siRNA library targeting 18,120 genes across three distinct tumor cell lines to identify host genes that mediated MRB virus-induced oncolysis. We observed 1,008 synthetic lethal hits that were common to at least two of the three malignant cell lines. Following gene cluster analysis, we observed several hits in expected biological pathways, including metabolism, cellular mitosis, and adenylate cyclase activity. However, we also uncovered an unexpected and critical role for endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) during MRB infection. Following further mechanistic exploration, we subsequently demonstrated that specific inhibition of the inositol-requiring enzyme 1 alpha, an ER transmembrane protein kinase/endoribonuclease that acts as an UPR sensor, increased the apoptotic index following MRB infection in a tumor-specific manner. Such transient ER stress led to the accumulation of the caspase adaptor protein, RIP associated Ich-1/CED homologous protein with death domain, which prepared the cell to undergo caspase 2-dependent death following virus infection. Preconditioning tumor cells with a drug that inhibited ER stress sensitized patient glioblastoma cells to MRB oncolysis. Critically, pretreatment with this drug in vivo rendered resistant EMT6 syngeneic breast tumors sensitive to MRB oncolysis and extended survival in tumor-bearing animals. We observed that combining this drug with virus therapy did not simply achieve an additive effect but was in fact synthetic lethal. Essentially, the target cell was manipulated into allowing access to a cell death pathway that would otherwise have not been available to the virus. Strikingly, we found this to have a wider therapeutic application since inhibiting ER stress in combination with doxorubicin, a DNA-damaging chemotherapeutic agent that depends on caspase-2 signaling,35 also sensitized cancer cells to die.34 Linking such diverse biological pathways would have been highly improbable using traditional scientific approaches but was made possible through the unbiased nature of high-throughput screening. This study provided the first proof-of-concept that functional screening could be used to identify OVIT targets. It also highlighted how such knowledge could be used to design combination therapies not only for OVIT but also for broader cancer therapies.
Workenhe et al36 specifically tailored a genome-wide RNAi screen to identify host factors responsible for inhibiting HSV-1 KM100 oncolysis in a breast cancer cell line that was resistant to infection. KM100 is a HSV-1 mutant that lacks ICP0, making it hypersensitive to IFN.37 Using a pooled lentiviral shRNA library consisting of 78,432 shRNAs targeting 16,056 genes, 343 genes were identified that could be knocked down to improve KM100 oncolysis (Table 1). After selecting 24 genes for secondary validation and pathway analysis, the authors focused on the serine/arginine-rich splicing factor 2 (SRSF2) gene, which increased KM100-mediated oncolysis following knockdown in the HS578T breast cancer cell line but not in nonmalignant primary fibroblasts. SRSF2 belongs to a family of serine–arginine RNA-binding proteins that are critical for alternative and constitutive splicing of messenger RNA (mRNA) precursors, mRNA export, and translation38 and have been implicated in mammary gland tumorigenesis.39,40 Since SRSF2 is phosphorylated by DNA topoisomerase I,41 the authors explored the utility of combining KM100 with DNA topoisomerase I inhibitors. This combination synergistically enhanced the level of killing observed for in vitro breast cancer cell lines and increased survival in syngeneic TUBO tumor-bearing mice.
Improving virus spread across the tumor
Achieving virus coverage across the entire tumor is a rare occurrence during OVIT but has been documented on occasion in some patients in clinical trials (personal communication). Virus spread across the tumor site depends on at least two critical factors: how much virus makes it to the tumor site following inoculation and how well the virus replicates to produce progeny that can subsequently spread across the local (and distant) tumor mass.
The initial virus load at the tumor is often dependent on the route of delivery, with direct intratumoral injections typically depositing a larger functional viral load (Stojdl, 2015 unpublished data). Conversely, intravenously administered virus can accumulate at the tumor site but typically at lower levels than the input dose and with potentially lower levels of infectivity depending on the antibody serostatus of the treated host. Such route considerations represent physical constraints of therapy; yet, the ability of the virus to generate large numbers of infectious progeny that can spread across the tumor mass and the susceptibility of uninfected neighboring tumor cells to allow subsequent virus attachment and transduction are two key areas that can be leveraged by screening approaches.
Sivan et al27 conducted a genome-wide RNAi screen in HeLa cells with recombinant VV IHD-J/GFP, a strain with a point mutation that increases the release of extracellular enveloped virus from the cell surface (Table 1). This virus strain was deliberately employed to facilitate the identification of host genes that were capable of impacting virus spread. Taking a novel approach, two primary screens were conducted in parallel using different commercial siRNA libraries targeting the entire host genome. One library contained a pool of four siRNAs per well targeting 18,120 genes, while a second library comprised three nonpooled siRNAs targeting 21,566 genes. Both libraries shared 17,693 overlapping gene targets; 576 genes were identified that could be silenced to decrease replication, while 530 genes could be silenced to increase replication. Approximately 30% or 50% of these genes, respectively, were hits across both independent libraries from the primary screen. Intriguingly, several of the validated genes that decreased replication upon knockdown were nuclear pore proteins, including nucleoporin 62, which diminished infectious virus yield by ~100-fold in one-step growth curves and negatively impacted morphogenesis. No secondary validation was performed on the genes that improved viral replication upon knockdown since these were not the focus of the study but at least 27 of them enhanced spread with a minimum of three different siRNA reagents.
Teferi et al42 performed RNAi screens to identify host factors that impacted the replication of Myxoma virus (MYXV) using a recombinant Lausanne strain expressing LacZ from a late promoter (Table 1). A primary whole-genome screen was conducted using a siRNA library targeting 21,585 genes in MDA-MB-231 human breast cancer cells. This identified 1,048 antiviral genes and 540 proviral genes. After screening out toxic siRNAs and performing validation on hits from both the primary screen and an additional kinome screen, 711 genes were identified that could be knocked down to increase replication, while 333 genes could be knocked down to decrease virus replication. In follow-up studies, certain subsets of these genes were explored with a focus on glycolysis, mitogen-activated protein kinase signaling, DNA replication, and the cell cycle. Knockdown of genes that promoted glycolysis (eg, phosphofructokinase) was detrimental to virus growth, whereas knockdown of genes that interfered with glycolysis (eg, fructose-1,6-bisphosphatase 1) favored virus growth. This is particularly interesting since cancer cells typically rely on the glycolytic pathway for energy metabolism, a phenomenon described as the “Warburg effect.”43 This reliance on glycolytic gene function for MYXV replication may be at least partially responsible for the tumor tropism of the virus. This study highlights the utility of high-throughput screening for customizing OVIT by revealing genetic or metabolic signatures that could predict responses to therapy.
Dealing with tumor heterogeneity
Tumors are formed from a mixed population of subclones, each harboring a distinct mutational variance. OVs have several inherent natural features that can at least partially mitigate tumor heterogeneity, including the ability to induce antigen spreading. Yet, additional therapeutic value may be achieved by pairing OVs with complementary interventions that unlock their capacity to target a wider genetic complement of tumor subclones.
To identify small molecules capable of modulating OV activity in resistant tumor cells, Diallo et al44 screened a panel of 12,280 chemical compounds to identify small molecules capable of rendering the partially resistant 4T1 breast cancer cell line sensitive to oncolysis by the VSV mutant, VSVΔ51 (Table 1). This mutant harbors a single point mutation in the M protein that renders it unable to block nuclear export, thereby allowing the normal expression of host genes involved in the antiviral immune response, among others.45 A lead viral sensitizer compound, Vse1, rendered four malignant cell lines across a spectrum of resistance susceptible to VSVΔ51 activity, including those derived from breast, colon, kidney, and brain tumors. Nonmalignant cells remained unaffected. Virus replication capacity was also improved across patient-derived colon, vulvar, and bone tumor explants in the presence of Vse1. Finally, in syngeneic subcutaneous CT26 tumor-bearing animals, Vse1 improved the ability of intratumorally administered VSVΔ51 to control tumor growth. Subsequent mechanistic analysis demonstrated that Vse1 was capable of suppressing 96% of the cellular transcripts typically stimulated by virus infection.44 Importantly, when paired with the wild-type VSV virus, which has an intact M protein capable of blocking nuclear transport, Vse1 displayed no ability to improve viral replication. This suggested that the majority of Vse1 activity with VSVΔ51 may have been impacting the innate antiviral immune signaling response.
Sivan et al46 screened a siRNA library across 21,566 host genes to identify modulators of viral permissivity (Table 1). Using a VV mutant with deletions in K1L and C7L genes that block the infectious cycle prior to late-stage gene expression to prevent infection in most human cells, host genes were identified that allowed the virus to regain the capacity to replicate in the nonpermissive HeLa cell line. While only a small number of genes could be knocked down to improve permissivity beyond baseline, knockdown of sterile alpha motif domain-containing 9 (SAMD9) demonstrated a particularly noteworthy increase in infectivity from ~0% to 27%–47%, depending on the particular siRNA used. This represented an almost complete rescue since ~50% of cells were infected under the same conditions in the permissive BS-C-1 cell line. Further validation using CRISPR/Cas9 gene editing confirmed the role of SAMD9 in viral permissivity and replication for this particular virus strain. Mechanistic studies previously demonstrated that SAMD9 can be antagonized by poxvirus-encoded host range factors to prevent the formation of antiviral granules.47
Such screening approaches that focus on host genes capable of reversing resistance to infection have important therapeutic implications for heterogeneous tumor masses since manipulating such genes may achieve permissivity in otherwise resistant tumor subclones. While tumors represent genetically diverse populations, there are nevertheless several common cellular signaling pathways that sustain growth. These represent conserved targets that may offer therapeutic benefit across broad patient populations by allowing multiple stages of an entire signaling cascade to be targeted rather than focusing on a single gene or protein. As mentioned previously, our laboratory discovered that transient inhibition of components of the typically conserved ER stress response, in particular the activating transcription factor 6 and inositol-requiring enzyme 1 pathways, specifically sensitized tumor cells to caspase-2-dependent apoptosis induced by MRB virus infection.34 Inhibition of these ER components increased the potency of MRB virus up to 10,000-fold across several malignant cell lines, with no concomitant increase of cytotoxicity in normal cells, and substantially improved antitumor efficacy in vivo. Indeed, ER stress and UPR activation are considered crucial for oncogenic transformation, development, and growth48 and therefore represent prime targets for such approaches.
Directed targeting of specific tumor populations
In addition to screening for genomic or chemical interventions that increase replication within the bulk tumor mass, an alternative approach has been to perform screens on highly defined tumor populations. The tumor microenvironment contains a diverse number of cell types, including epithelial cells, stem cells, fibroblasts, stroma, endothelia, and immune populations. There is convincing evidence that cancer stem cells are involved in initiating and/or maintaining solid tumor growth49–51 and are similarly responsible for tumor relapse after cancer patients receive treatments that cannot effectively target and ablate this population.52 Thus, several chemical screens have focused on identifying drugs that can enhance viral replication and oncolysis specifically in cancer stem cell populations.
Pont et al53 screened >400 drugs to identify potential targets that could enhance the replicative and oncolytic ability of the oncolytic adenovirus (Ad), Delta24-RGD, in both resistant and partially resistant patient-derived glioblastoma cancer stem-like cell (gCSC) cultures (Table 1). Four viral sensitizer compounds – including anagrelide, ebselen, fluphenazine, and indirubin – were identified that synergistically enhanced oncolytic activity across gCSC samples. Each compound impacted one or more core biological mechanisms, including enhancing virus attachment to host cells by increasing CAR expression levels, boosting replication efficiency, or increasing cell death. Fluphenazine emerged as a lead candidate since it could improve Delta24-RGD oncolysis in an extended panel of 12 gCSC cultures across a spectrum of virus sensitivity. Interestingly, fluphenazine significantly decreased Ad titers but nevertheless improved the degree of both apoptotic and necrotic tumor cell death and the oncolytic index of the virus in gCSCs, suggesting an uncoupling between the number of infectious virus particles and the oncolytic effect. These four sensitizer compounds were further validated with Delta24-RGD across a broader panel of tumor cell lines derived from other tissues. Strikingly, the four lead compounds could also synergize with other OVs, including Newcastle Disease Virus and HSV-1-G47Δ,53 suggesting that these drugs impacted conserved pathways that were important to the life cycle of multiple viruses.
Similarly, McKenzie et al54 screened 73 chemotherapeutic compounds in preclinical or clinical development for their ability to sensitize the temozolomide-resistant patient-derived BT025 brain tumor-initiating cell (BTIC) culture to oncolytic MYXV (Table 1). A primary screen was performed at a drug dose of 1 μM to identify eight lead compounds, with a secondary screen at 100 nM to identify an additional three compounds, and a final screen at 10 nM, which identified no new candidate compounds. After further analysis, six of these eleven compounds achieved synergistic activity in combination with MYXV via Chou–Talalay analysis.54 Several compounds acted on the same similar signaling hub – namely, the PI3K/Akt/mTOR pathway – consistent with independent observations that rapamycin, which blocks mTOR, can increase rates of viral oncolysis.55,56 Pemetrexed, a folate antimetabolite drug, increased both viral gene expression and viral titers but was the only drug that positively impacted both these viral parameters. Axitinib, a tyrosine kinase inhibitor, was chosen for further validation across three additional BTIC cultures and demonstrated increased killing in combination with MYXV across all tested cells. Interestingly, this enhanced death index was not accompanied by any change in apoptotic markers. Although markers of immunogenic cell death were not explored, certain chemotherapeutic drugs can enhance the immunogenicity of tumor cell death57 to achieve synergistic anti-tumor efficacy during OVIT.58,59
Manipulating the tumor microenvironment and enhancing therapeutic immune responses
Despite the clear importance of OV-mediated activation of antitumor immune responses through either inherent or engineered mechanisms, very few screens have been published that focus on defining new approaches to solidifying immune engagement or manipulating immunosuppressive cell populations within the tumor microenvironment, such as M2 macrophages, MDSCs, and Tregs. Several aspects of OV biology have the potential to impact immune engagement, including the type and immunogenicity of tumor cell death induced by the OV (eg, apoptosis, necroptosis, necrosis, autophagy) and the level of proinflammatory cytokine activation in infected cells. These, in turn, impact the establishment of the chemotactic gradient that drives the infiltration and activation of innate immune cell populations and the subsequent presentation of immunogenic TAAs by DCs to activate tumor- (or virus-) specific T-cells.
One of the few immune-related screens performed using an OV assessed which genes encoded by Parapoxvirus ovis (Orf virus [ORFV]) were involved in the establishment of small DC-rich immune cell depots at sites of infection. This screen is unique among the currently published literature for its focus on viral rather than host genes. This elegant screen was carried out entirely in vivo, delivering full ORFV ORFs as linear expression elements at spatially separated dermal sites in mice using gene gun technology.60 Twenty-six pools of eight to ten ORFs were administered, and individual hits from pools that activated DC accumulation were rescreened. Ultimately, it was determined that the B2L gene from ORFV, a homolog of the VV F13L gene, was a direct driver behind the formation of these depots.
Given the importance of the immune response to the oncolytic effect,7,8,10 and the clear ability of TAA-expressing OVs to engage therapeutically relevant antitumor memory immune responses,61 in vivo screens have also been performed to identify optimal TAA targets during OVIT. Pulido et al created a VSV-expressed xenoantigen complementary DNA (cDNA) library mined from two human melanoma cell lines. Repeat intravenous treatment with this VSV library cured 60% of mice bearing syngeneic B16.F10 melanoma tumors. Individual VSV vectors within the cDNA library were screened for their ability to stimulate interleukin-17 production in splenocytes from treated mice restimulated with B16.F10 tumor lysates to identify the most potent encoded TAA targets. Three VSV vectors – encoding NRAS, TYRP1, and CYC1 – were identified that, when used in combination, achieved the same therapeutic outcome as the complete cDNA library.62 Importantly, using one or two of these vectors in combination was insufficient to achieve therapeutic efficacy, highlighting the importance of incorporating strategies designed to target multiple antigens. Indeed, this tallies with previous observations from the wider immunotherapeutic field.63,64 Intriguingly, treating intracranial B16.F10 tumors with the three VSV-encoded TAAs that demonstrated efficacy against subcutaneously implanted B16.F10 tumors failed to achieve efficacy,65 suggesting that the tissue in which the tumor resides has a significant role in selecting the nature of the TAAs that represent relevant therapeutic targets.
Future directions
It is widely accepted that therapeutic approaches that focus on more than one malignant target achieve better control of complex, genetically diverse tumor masses. High-throughput screening has begun to play an important role in characterizing interventions that can impact multiple independent steps of the host–OV response. In the future, high-throughput screening will undoubtedly have a key role in identifying stackable interventions capable of impacting several cancer-associated pathways at temporally distinct periods during OVIT that could run to thousands of potential configurations. For example, prior to OV inoculation, innate immune responses could be targeted to minimize IFN responses in residual IFN-competent tumor cells to prevent the suppression of viral replication. During OV therapy, a chemotherapeutic agent could be administered to deplete suppressive Treg populations, together with a viral synergizer to solidify IFN suppression. Post-OV therapy, compounds capable of promoting immune memory formation may be administered to solidify tumor control and protect against relapse.
In vitro screens have provided tantalizing insights into the utility of high-throughput screening to identify novel therapeutic targets during OVIT, especially when these targets have been validated across multiple cell lines to determine broader relevance and across patient samples to explore potential clinical utility. The establishment of miniaturized cultures that mimic the in vivo environment will consolidate the utility of screening technologies for OVIT. Ultimately, in vivo screens have perhaps the greatest potential to reveal the most clinically relevant insights for OVIT, and further efforts in this area are needed.
The published host–virus interactome screens performed with OV vectors have predominantly focused on targeting the host genome. Yet, targeting the virus genome could also beneficially modulate responses, especially for OVs encoded by large genomes that have not been fully characterized. For example, numerous VV genes have unknown functions66 and could be evaluated for their impact on oncolytic activity to generate a more potent OV.
Several other relatively new large-scale technological approaches have clear potential to provide insights for OVIT. Ribosome profiling is an emerging technology that provides a global snapshot of the mRNAs that are being translated in a cell at a given time. This is achieved by deep sequencing of all ribosome-protected mRNAs extracted from live cells, yielding mechanistic insights into viral and host gene expression and regulation during viral infection. In addition, genome-editing screens can employ CRISPR–Cas9 or transcription activator-like effector nuclease technology to completely remove or disrupt gene targets rather than knock them down, as in RNAi-based approaches. However, to our knowledge, none of these techniques have yet been applied in a primary OV screen.
Conclusion
High-throughput screening has achieved clear benefits in the field of infectious disease research; yet up to this point, there have been relatively few publications that have used this technology to improve OVIT. OV screens have identified genes or pathways that have rational impacts on virological outcomes at the level of antiviral immune responses, mRNA or DNA synthesis, and metabolism. Yet, many of these genes have affected these pathways in unexpected or indirect ways. In addition, several novel nonrational pathways and mechanisms have been uncovered that may have otherwise eluded hypothesis-driven approaches, such as the impact of ER stress on viral oncolysis and the significance of nuclear pore proteins during VV replication. Taking a similarly unbiased screening approach to focus on finding genes or pathways that consolidate immune system engagement is likely to provide important therapeutic insights to improve the long-term efficacy of OVIT and to guide the development and design of optimized clinical strategies.
Acknowledgments
This work was supported by a Vanier Canada Graduate Scholarship from the Government of Canada (to KJA) and a grant from the Ontario Institute for Cancer Research, the Canada Foundation for Innovation, Angels of Hope, the Ottawa Regional Cancer Foundation, and the Terry Fox Research Institute (to DFS). The author’s would also like to thank Scientific Illustration for the Research Scientist for providing the excellent open-source graphical resources that have been used in this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Mohr SE, Smith JA, Shamu CE, Neumüller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15(9):591–600. | |
Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16(5):299–311. | |
Stanford MM, Breitbach CJ, Bell JC, McFadden G. Innate immunity, tumor microenvironment and oncolytic virus therapy: friends or foes? Curr Opin Mol Ther. 2008;10(1):32–37. | |
Vacchelli E, Aranda F, Obrist F, et al. Trial watch: immunostimulatory cytokines in cancer therapy. Oncoimmunology. 2014;3:e29030. | |
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662. | |
Breitbach CJ, Arulanandam R, De Silva N, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73(4):1265–1275. | |
Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra232. | |
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3): 718–730. | |
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. | |
Pol J, Bloy N, Obrist F, et al. Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology. 2014;3:e28694. | |
Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2013;22(2):251–256. | |
Granot T, Yamanashi Y, Meruelo D. Sindbis viral vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8+ T-cell immunity. Mol Ther. 2014;22(1):112–122. | |
Janelle V, Langlois MP, Lapierre P, Charpentier T, Poliquin L, Lamarre A. The strength of the T cell response against a surrogate tumor antigen induced by oncolytic VSV therapy does not correlate with tumor control. Mol Ther. 2014;22(6):1198–1210. | |
Bridle BW, Stephenson KB, Boudreau JE, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18(8): 1430–1439. | |
Kottke T, Errington F, Pulido J, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17(7):854–859. | |
Fournier P, Arnold A, Wilden H, Schirrmacher V. Newcastle disease virus induces pro-inflammatory conditions and type I interferon for counter-acting Treg activity. Int J Oncol. 2012;40(3):840–850. | |
de Vries CR, Monken CE, Lattime EC. The addition of recombinant vaccinia HER2/neu to oncolytic vaccinia-GMCSF given into the tumor microenvironment overcomes MDSC-mediated immune escape and systemic anergy. Cancer Gene Ther. 2015;22(3):154–162. | |
Cerullo V, Diaconu I, Romano V, et al. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther. 2012;20(11):2076–2086. | |
Panda D, Das A, Dinh PX, et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc Natl Acad Sci U S A. 2011;108(47): 19036–19041. | |
Mercer J, Snijder B, Sacher R, et al. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012;2(4):1036–1047. | |
Lee AS, Burdeinick-Kerr R, Whelan SP. A genome-wide small interfering RNA screen identifies host factors required for vesicular stomatitis virus infection. J Virol. 2014;88(15):8355–8360. | |
Samanta D, Datta PK. Alterations in the Smad pathway in human cancers. Front Biosci (Landmark Ed). 2012;17:1281–1293. | |
Xie W, Rimm DL, Lin Y, Shih WJ, Reiss M. Loss of Smad signaling in human colorectal cancer is associated with advanced disease and poor prognosis. Cancer J. 2003;9(4):302–312. | |
Sjoblom T, Yakymovych I, Heldin CH, Ostman A, Souchelnytskyi S. Smad2 suppresses the growth of Mv1Lu cells subcutaneously inoculated in mice. Eur J Cancer. 2004;40(2):267–274. | |
Beard PM, Griffiths SJ, Gonzalez O, et al. A loss of function analysis of host factors influencing vaccinia virus replication by RNA interference. PLoS One. 2014;9(6):e98431. | |
Haga IR, Pechenick Jowers T, Griffiths SJ, Haas J, Beard PM. TRAF2 facilitates vaccinia virus replication by promoting rapid virus entry. J Virol. 2014;88(7):3664–3677. | |
Sivan G, Martin SE, Myers TG, et al. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc Natl Acad Sci U S A. 2013;110(9):3519–3524. | |
Varble A, Benitez AA, Schmid S, et al. An in vivo RNAi screening approach to identify host determinants of virus replication. Cell Host Microbe. 2013;14(3):346–356. | |
Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. | |
Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23(7):837–848. | |
Passer BJ, Cheema T, Zhou B, et al. Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res. 2010;70(10):3890–3895. | |
Peng T, Hunter JR, Nelson JW. The novel protein kinase of the RR1 subunit of herpes simplex virus has autophosphorylation and transphosphorylation activity that differs in its ATP requirements for HSV-1 and HSV-2. Virology. 1996;216(1):184–196. | |
Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21(1):39–49. | |
Mahoney DJ, Lefebvre C, Allan K, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell. 2011;20(4): 443–456. | |
Panaretakis T, Laane E, Pokrovskaja K, et al. Doxorubicin requires the sequential activation of caspase-2, protein kinase Cdelta, and c-Jun NH2-terminal kinase to induce apoptosis. Mol Biol Cell. 2005;16(8): 3821–3831. | |
Workenhe ST, Ketela T, Moffat J, Cuddington BP, Mossman KL. Genome-wide lentiviral shRNA screen identified serine-arginine-rich splicing factor 2 as a determinant of oncolytic virus activity in breast cancer cells. Oncogene 2015; doi:10.1038/onc.2015.303. | |
Mossman KL, Saffran HA, Smiley JR. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000;74(4): 2052–2056. | |
Mo S, Ji X, Fu XD. Unique role of SRSF2 in transcription activation and diverse functions of the SR and hnRNP proteins in gene expression regulation. Transcription. 2013;4(5):251–259. | |
Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14(3):185–193. | |
Stickeler E, Kittrell F, Medina D, Berget SM. Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene. 1999;18(24):3574–3582. | |
Labourier E, Rossi F, Gallouzi IE, Allemand E, Divita G, Tazi J. Interaction between the N-terminal domain of human DNA topoisomerase I and the arginine-serine domain of its substrate determines phosphorylation of SF2/ASF splicing factor. Nucleic Acids Res. 1998;26(12):2955–2962. | |
Teferi WM, Dodd K, Maranchuk R, Favis N, Evans DH. A whole-genome RNA interference screen for human cell factors affecting myxoma virus replication. J Virol. 2013;87(8):4623–4641. | |
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | |
Diallo JS, Le Boeuf F, Lai F, et al. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol Ther. 2010;18(6):1123–1129. | |
Stojdl DF, Lichty BD, tenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4(4):263–275. | |
Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L-C7L- mutant. MBio. 2015;6(4):e01122. | |
Liu J, McFadden G. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol. 2015;89(3):1925–1931. | |
Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–597. | |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | |
Lang SH, Frame FM, Collins AT. Prostate cancer stem cells. J Pathol. 2009;217(2):299–306. | |
Singh SK, Venugopal C, Hallett R, et al. Pyrvinium targets CD133 in human glioblastoma brain tumor-initiating cells. Clin Cancer Res. 2015;21(23):5324–5337. | |
Moncharmont C, Levy A, Gilormini M, et al. Targeting a cornerstone of radiation resistance: cancer stem cell. Cancer Lett. 2012;322(2): 139–147. | |
Pont LM, Balvers RK, Kloezeman JJ, et al. In vitro screening of clinical drugs identifies sensitizers of oncolytic viral therapy in glioblastoma stem-like cells. Gene Ther. 2015;22(12):947–959. | |
McKenzie BA, Zemp FJ, Pisklakova A, et al. In vitro screen of a small molecule inhibitor drug library identifies multiple compounds that synergize with oncolytic myxoma virus against human brain tumor-initiating cells. Neuro Oncol. 2015;17(8):1086–1094. | |
Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. | |
Lun XQ, Jang JH, Tang N, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15(8):2777–2788. | |
Pol J, Vacchelli E, Aranda F, et al. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology. 2015;4(4):e1008866. | |
Workenhe ST, Pol JG, Lichty BD, Cummings DT, Mossman KL. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol Res. 2013;1(5):309–319. | |
Cerullo V, Diaconu I, Kangasniemi L, et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther. 2011;19(9):1737–1746. | |
McGuire MJ, Johnston SA, Sykes KF. Novel immune-modulator identified by a rapid, functional screen of the parapoxvirus ovis (Orf virus) genome. Proteome Sci. 2012;10(1):4. | |
Bridle BW, Clouthier D, Zhang L, et al. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8 T-cell responses to anticancer vaccines. Oncoimmunology. 2013;2(8):e26013. | |
Pulido J, Kottke T, Thompson J, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30(4):337–343. | |
Hegde M, Corder A, Chow KK, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087–2101. | |
Spranger S, Gajewski T. Rational combinations of immunotherapeutics that target discrete pathways. J Immunother Cancer. 2013;1:16. | |
Alonso-Camino V, Rajani K, Kottke T, et al. The profile of tumor antigens which can be targeted by immunotherapy depends upon the tumor’s anatomical site. Mol Ther. 2014;22(11):1936–1948. | |
Lefkowitz EJ, Wang C, Upton C. Poxviruses: past, present and future. Virus Research 2006;117:105–118. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.