Back to Archived Journals » Hypoxia » Volume 2
HIF-mediated innate immune responses: cell signaling and therapeutic implications
Authors Harris A, Thompson AR, Whyte M ![]() , Walmsley S
, Walmsley S
Received 24 January 2014
Accepted for publication 28 February 2014
Published 24 May 2014 Volume 2014:2 Pages 47—58
DOI https://doi.org/10.2147/HP.S50269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Alison J Harris, AA Roger Thompson, Moira KB Whyte, Sarah R Walmsley
Academic Unit of Respiratory Medicine, Department of Infection and Immunity, University of Sheffield, Sheffield, UK
Abstract: Leukocytes recruited to infected, damaged, or inflamed tissues during an immune response must adapt to oxygen levels much lower than those in the circulation. Hypoxia inducible factors (HIFs) are key mediators of cellular responses to hypoxia and, as in other cell types, HIFs are critical for the upregulation of glycolysis, which enables innate immune cells to produce adenosine triphosphate anaerobically. An increasing body of evidence demonstrates that hypoxia also regulates many other innate immunological functions, including cell migration, apoptosis, phagocytosis of pathogens, antigen presentation and production of cytokines, chemokines, and angiogenic and antimicrobial factors. Many of these functions are mediated by HIFs, which are not only stabilized posttranslationally by hypoxia, but also transcriptionally upregulated by inflammatory signals. Here, we review the role of HIFs in the responses of innate immune cells to hypoxia, both in vitro and in vivo, with a particular focus on myeloid cells, on which the majority of studies have so far been carried out.
Keywords: hypoxia, neutrophils, monocytes, macrophages
Introduction
Innate immune cells include both myeloid cells, such as neutrophils and circulating monocytes which differentiate into macrophages within tissues, and innate lymphoid cells (ILCs). These cells have important functions in host defense, including the phagocytosis of pathogens, the release of antimicrobial proteases, and the secretion of cytokines and chemokines that attract and activate other immune cells. Cells of the immune system encounter a range of different oxygen tensions, with lower levels in tissues than in blood, which are further reduced during inflammation. To function under hypoxic conditions, innate immune cells increase anaerobic adenosine triphosphate (ATP) generation by upregulating glycolytic enzymes and glucose transporters.1–5 Hypoxia also, however, has a plethora of additional effects on innate immune responses.
Low oxygen levels act on vascular endothelial cells to upregulate adhesion molecules and chemokines, attracting circulating monocytes and neutrophils.6–11 Neutrophils and monocytes upregulate adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and CD18/CD11b as they reach hypoxic regions, promoting their arrest and transendothelial or transepithelial migration toward the site of the inflammation.2,12 Hypoxia also acts to retain myeloid cells at the site of injury by upregulating factors that inhibit migration.2,3,5,13–16
Hypoxia tends to potentiate inflammatory functions and has a prosurvival effect in neutrophils, monocytes, and eosinophils.4–5,15,17,18 The ability of innate immune cells to survive and function effectively under hypoxic conditions is crucial for host defense in tissues. Immune cell apoptosis is, however, an important means of resolving inflammation; prolonged survival and activation of myeloid cells under hypoxic conditions is likely to play a major role in chronic inflammation, contributing to diseases such as chronic obstructive pulmonary disease, asthma, and rheumatoid arthritis (RA).19–23
Lack of oxygen and a reduction in oxidative phosphorylation, in favor of less efficient glycolysis, might be expected to reduce ATP availability and impair cellular functions; however, there is evidence that cardiac hypoxia inducible factor (HIF) expression can increase ATP availability despite inducing a glycolytic switch.24 This may also be the case in myeloid cells, with hypoxia enhancing many effector functions. Glycolysis is critical for ATP production by myeloid and endothelial cells even under normoxic conditions.25,26 Inhibition of glycolysis reduces endothelial cell proliferation and impairs formation of lamellipodia, causing a defect in directional migration and vessel formation.26 Potentially, therefore, glycolysis (and its upregulation by hypoxia) may promote migration of myeloid cells.
Hypoxia upregulates Fc and complement receptors on macrophages and neutrophils, thereby enhancing phagocytosis.27–32 It also promotes the release of antimicrobial mediators, such as β-defensin by macrophages and elastase by neutrophils.33–35 Monocytes and macrophages upregulate proinflammatory cytokines, such as tumor necrosis factor α (TNFα), interferon γ (IFNγ), interleukin (IL)-1β, and several neutrophil chemoattractants in hypoxia, while downregulating chemokines associated with adaptive immunity.2,3,31,36–38
Macrophages precultured in hypoxia are more effective at inducing ovalbumin-specific CD8 T-cell responses, due to the upregulation of major histocompatibility complex (MHC) class I and costimulatory molecules, but they also produce arginase, which inhibits CD4 T-cell responses.31,39,40
Many effects of hypoxia are mediated by release of HIFs from suppression by the oxygen-dependent hydroxylases. These comprise prolyl hydroxylases (PHDs), which target them for degradation, and the asparaginyl hydroxylase, factor inhibiting HIF, which inhibits HIF transcriptional activity. Many immune-related genes are regulated by HIF, either directly, via binding of HIF to a HIF-responsive element (HRE) in the promoter region, or indirectly, via HIF-mediated induction of other signaling molecules and transcription factors, such as NF-κB. Effects of HIF signaling in innate immune cells in response to hypoxia and other stimuli are summarized in Figure 1.
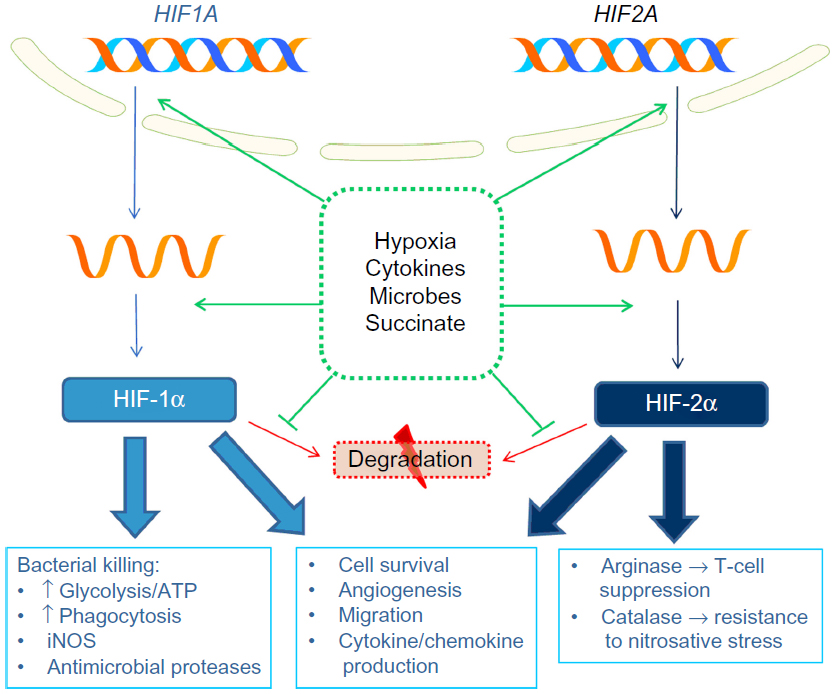 | Figure 1 Role of HIFs in innate immune responses to hypoxia. |
Other mechanisms also play a role in hypoxia signaling; for example, mitochondrial-derived reactive oxygen species stabilize HIFs and also activate other transcription factors, such as activator protein-1 (AP-1), which induces CXC-chemokine ligand (CXCL) 8 in hypoxic macrophages.37,38,41–43 Hypoxia also reduces activity of the Jumonji histone demethylases, resulting in increased methylation of histone H3 residues in the promoter regions of chemokine ligand 2 (CCL2), chemokine (C-C motif) receptor 1 (CCR1), and CCR5, repressing their transcription by macrophages.44
Though research so far has focused predominantly on myeloid cells, there is also some evidence of the effects of hypoxia on other aspects of innate immunity. For instance, mice with chronic hypoxia-induced pulmonary hypertension have reduced natural killer (NK) cell numbers, cytotoxicity, and IFNγ production.45 Hypoxic NK cells downregulate the key cytotoxic mediators granzyme B and perforin and lose their ability to upregulate activating receptors in response to cytokines, thereby reducing their ability to recognize and kill infected or abnormal cells.46,47
In addition to the effects of hypoxia on innate immune responses to infection, hypoxia can also have direct effects on the pathogens themselves, such as modulating the expression of virulence factors,48 but this is beyond the scope of this review.
Roles of HIFs in neutrophil responses to hypoxia
Neutrophils are inherently adapted to low oxygen tensions, with few mitochondria and reliance on glycolysis for much of their energy demand even under normoxic conditions.25
While HIF1A messenger RNA (mRNA) is present in freshly isolated neutrophils, HIF-1α protein is not expressed in unstimulated cells. It can be induced by hypoxic culture, the iron chelator desferrioxamine, or the competitive hydroxylase inhibitor dimethyloxalylglycine (DMOG).5,49 When cultured in hypoxia, neutrophils upregulate the glycolytic HIF-1α target genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and phosphoglycerate kinase (PGK), which may serve to provide additional glycolytic capacity for enhanced effector function.5 Hypoxic induction of Pgk expression is reduced but not abolished in Hif1a−/− neutrophils, suggesting that both HIF-1α and other factors (possibly HIF-2α) are involved in its induction. ATP levels are substantially reduced in Hif1a−/− neutrophils and macrophages, reflecting the central role of HIF-1α in regulation of metabolic activity.50
Expression of the cathelicidin-related antimicrobial peptide (CRAMP) is upregulated by hypoxia and in neutrophils lacking the von Hippel-Lindau protein (VHL) (involved in the proteasomal degradation of HIF), whereas Hif1a−/− neutrophils express lower levels of CRAMP and display reduced ability to breakdown artificial substrates for antimicrobial proteases.51 HIF-1α also promotes extracellular bacterial killing by inducing formation of neutrophil extracellular traps.52 Furthermore, neutrophil hypoxic survival is mediated by HIF, with Hif1a−/− neutrophils displaying a failure of hypoxia-mediated apoptosis suppression.5,49 In wild-type (WT), but not Hif1a−/− neutrophils, hypoxia reverses the decline in nuclear factor (NF)-κB and IκB kinase (IKK)-α levels observed during culture and maintains DNA-binding activity of NF-κB family members, which is essential for hypoxic survival.
HIF-2α is also expressed by human and murine neutrophils and its expression is upregulated by hydroxylase inhibitors, bacteria or lipopolysaccharide (LPS).53 The respiratory burst, phagocytosis, chemotaxis, LPS-induced receptor upregulation and constitutive apoptosis are unaffected by deletion of Hif2a. However, Hif2a−/− inflammatory neutrophils derived from bronchoalveolar lavage (BAL) fluid of LPS-challenged mice express lower levels of the antioxidant enzyme catalase than WT neutrophils and display increased apoptosis in response to nitrosative stress.
While circulating leukocytes that are attracted to a site of inflammation initially upregulate HIF in response to hypoxia, cells exposed to chronic hypoxia may respond differently due to the activation of negative feedback mechanisms. For instance, hypoxia is reported to induce HIF-mediated transcription of PHD2, PHD3, and VHL – all negative regulators of HIF expression.54–57
Although PHDs are generally understood to act upstream of HIFs, promoting their degradation, in neutrophils PHD3 may function downstream of HIF. PHD3 is upregulated during hypoxia; this is significantly impaired in Hif1a−/− neutrophils.54 PHD3 does not, however, appear to regulate HIF-1α expression, since the hypoxic upregulation of HIF target genes is not affected by the deletion of Phd3. Phd3−/− neutrophils fail to upregulate the anti-apoptotic protein B-cell lymphoma extra-large (Bcl-XL), as occurs in hypoxic WT neutrophils, and exhibit reduced survival during hypoxia.
Roles of HIFs in monocyte and macrophage responses to hypoxia
Hypoxic macrophages upregulate HIF-1α and HIF-2α proteins in a phosphatidylinositide 3-kinase (PI3K)/protein kinase B (AKT)-dependent manner.3,58 In contrast to neutrophils, macrophages from Phd3−/− mice exhibit higher expression of HIF-1α and NF-κB than WT, produce more cytokines, and exhibit enhanced chemotactic and phagocytic responses, suggesting that in macrophages, PHD3 is acting upstream of HIF-1α to degrade HIF-1α protein.59
As in neutrophils, HIF-1α has an important role in regulating macrophage metabolism under hypoxic conditions. HIF1A-null or small interfering RNA (siRNA)-treated macrophages have reduced basal expression and hypoxic induction of the glycolytic genes PGK and GLUT1, whereas the loss of VHL mimics hypoxic induction.3,50 Release of lactate, a marker of glycolytic activity and inflammatory activation, is significantly lower in Hif1a−/− macrophages and significantly higher in Vhl−/− cells.50 HIF signaling is, in turn, influenced by metabolism. For instance, the tricarboxylic acid cycle intermediate, succinate, which is generated during aerobic respiration and upregulated in response to LPS stimulation, inhibits PHD activity, resulting in HIF-1α stabilization and Il1b transcription in macrophages.60
Hypoxic upregulation of the adhesion molecule ICAM-1 is inhibited by siRNA targeted against HIF1A or HIF2A, while induction of β-integrins in monocytes is abrogated by mutation of the HRE in the promoter of CD18, the subunit common to the four known β2 integrin heterodimers.3,12 Hif1a−/− macrophages display defects in chemotaxis, in their ability to penetrate the extracellular matrix, and in homotypic adhesion, a process of self-aggregation that amplifies the tissue recruitment of leukocytes.50,61 Hif2a−/− macrophages have reduced expression of the chemokine receptors macrophage colony-stimulating factor receptor and CXC chemokine receptor 4 and the adhesion molecule fibronectin-1, resulting in a 50% reduction in migratory and invasive capacities.62
Phagocytosis and intracellular bactericidal activity are also regulated by HIFs. Hypoxia induces p38 in macrophages and p38 inhibition reverses HIF-1α stabilization and enhancement of phagocytosis.32 HIF-1α induces thrombospondin-1 and the scavenger receptor CD36, which are involved in the recognition of apoptotic cells, therefore hypoxia enhances phagocytosis of both bacteria and apoptotic neutrophils – effects that are abrogated in the presence of HIF1A-directed siRNA.32,63 Killing of intracellular bacteria, such as streptococci and Pseudomonas aeruginosa, is enhanced by iron chelators and in Vhl−/− macrophages, but impaired in Hif1a−/− macrophages, possibly due to HIF-1α-mediated inducible nitric oxide synthase (iNOS) induction.50,51,61
Hypoxia also induces the expression of IFNγ, which promotes phagocytosis of opsonized cells by upregulating surface levels of the CD11b/CD18 complement receptor and the CD16/CD32 Fcγ receptor.31 The Ifng promoter contains a functional HRE and hypoxic induction of IFNγ and the associated upregulation of opsonic receptors are abolished by mutation of this site or by deletion or siRNA knockdown of Hif1a. Hif1a−/− macrophages secrete less TNF-α in response to LPS or group A streptococci (GAS) and fail to upregulate TNF-α when LPS is administered under hypoxic conditions.50,51 HIF-1α also binds to a highly polymorphic region of the promoter encoding solute carrier family member 1 (Slc11a1), a phagocyte-specific solute carrier that induces expression of cytokines, chemokines, and MHC class II molecules.64 Allelic variation affects the ability of HIF-1α to induce SLC11A1 expression and has been associated with susceptibility to various inflammatory diseases. HIF-2α also appears to be involved in cytokine and chemokine transcription, since the hypoxic upregulation of IL1B, IL12, CXCL2, and CXCL6 is impaired in Hif2a−/− macrophages while hypoxic expression of CXCL8 is reduced in macrophages treated with siRNA targeting Hif2A.3,62
In contrast to its many proinflammatory effects, hypoxia also promotes repair mechanisms and macrophage suppression of T-cells in a HIF-dependent manner.65 Induction of the proangiogenic factors vascular endothelial growth factor (VEGF) and adrenomedullin is significantly reduced by siRNA targeting either HIF1A or HIF2A.3 Hif1a−/− macrophages have reduced basal expression and hypoxic induction of VEGF, whereas deletion of VHL induces VEGF expression under normoxic conditions.3,50 Plasminogen activator inhibitor-1 (PAI-1), which promotes thrombosis by inhibiting plasminogen-mediated fibrinolysis, is also activated by HIF-1α.66
HIFs can also act as negative regulators of gene expression. For instance, HIF-1α (but not HIF-2α) binds to the HRE of the histone deacetylase-2 (HDAC2) promoter independently of the HIF-1β subunit and reduces transcription by inhibiting binding of Polymerase II.67 Hypoxia, therefore, reduces mRNA and protein levels of HDAC2, a suppressor of NF-κB-mediated transcription, resulting in increased inflammation and corticosteroid resistance.
Hif1a mRNA has a shorter half-life than Hif2a mRNA and, following intrapulmonary LPS challenge, Hif1a is rapidly upregulated but declines after 24 hours, whereas Hif2a expression is delayed but maintained during the resolution phase.53,68 HIF-1α and HIF-2α bind to the same promoter sequence and the expression of many hypoxia-inducible genes, including VEGF, IL1B, CXCL8, GLUT1, ADM (adrenomedullin), CXCR4, and STAT4, can be impaired by siRNA targeted against either HIF1A or HIF2A.3 Some target genes, however, such as the adenosine A2a receptor (ADORA2A), are only regulated by one HIF and not the other, which is thought to be due to interaction with different coactivators.3,69
Roles of HIF-1α and HIF-2α in macrophage polarization
Although both HIF-1α and HIF-2α are induced by hypoxia, other stimuli such as cytokines also influence their relative expression levels. For instance, Takeda et al68 reported that HIF1-α mRNA and protein are induced during M1 polarization of macrophages (by IFNγ or LPS), whereas HIF-2α is induced during M2 polarization (by IL-4 or IL-13) and is decreased by stimulation with LPS or IFNγ. Hif1a−/− macrophages infiltrating tumor spheroids in vitro develop a more M2-like profile than their WT counterparts, with lower cytotoxicity and decreased production of TNFα, iNOS, and IL-6, suggesting that HIF-1α is important for proinflammatory responses.70 The work of Imtiyaz et al62 indicates that HIF-2α also has important proinflammatory functions, since myeloid-specific Hif2a deletion inhibits in vitro secretion of the proinflammatory cytokines IL-1β, IL-6, and IL-12 in response to a combined IFNγ/LPS stimulus and reduces serum levels of IL-1β, IL-12, TNFα, and IFNγ following in vivo LPS stimulation.
The relative levels of HIF-1α and HIF-2α may play an important role in the regulation of nitric oxide (NO) metabolism by controlling the levels of both iNOS and arginase (which competes with iNOS for the L-arginine substrate required for NO production). Hif2a−/−macrophages exhibit normal hypoxic induction of iNOS (Nos2); however, both the basal expression and hypoxic induction of arginase 1(Arg1) are attenuated.62,68 Conversely, Hif1a deletion dramatically reduces hypoxic iNOS induction, but it has a smaller effect on arginase expression.68 Similar data was obtained by Kobayashi et al,71 who observed an increased iNOS response to LPS in cells with a Vhl/Hif2a double knockout (in which HIF-1α is stabilized while HIF-2α is absent), while Vhl/Hif1a double knockout mice produced more arginase in response to IL-13 stimulation. In accordance with the altered ratio of iNOS/arginase, mice lacking myeloid HIF-1α exhibited reduced NO production 6 hours after LPS treatment, while those lacking HIF-2α exhibited increased NO after 24 hours.68 Takeda et al68 also demonstrated decreased and increased levels of in vitro NO production in IFNγ-stimulated Hif1a−/− and Hif2a−/− thioglycollate-elicited peritoneal macrophages respectively. However, when Imtiyaz et al62 stimulated bone marrow-derived marcophages (BMDMs) with a combination of IFNγ and LPS, no difference was observed in NO production between Hif2a−/− and WT. The discrepancy between the studies could be explained by many factors, including the sourcing of macrophages from different tissues, use of a different stimulus, and the apparent dependency of the Hif2a phenotype on the level of L-arginine availability.68
Mice with a myeloid-specific Phd2 haplodeficiency exhibit greater infiltration of mannose receptor, C type 1 (MRC1)+ proangiogenic/wound-healing M2-like macrophages, which release arteriogenic factors, following femoral artery ligation.72 In comparison to WT macrophages, Phd2+/− macrophages isolated at baseline express higher levels of M2 genes, such as arginase (Arg1), Cxcr4, platelet-derived growth factor beta (Pdgfb), neuropilin 1 (Nrp1), found in inflammatory zone 1 (Fizz1) and transforming growth factor beta (Tgfb) and lower levels of the M1 genes Il12, Nos2, Il1b, Il6, and Tnfa. However, following ligation, WT macrophages from the ischemic musculature upregulate M2 genes, reaching a similar level to that of Phd2+/− macrophages, indicating hypoxia is releasing M2 genes from PHD2-mediated suppression. Given the previous work indicating an association of HIF-1α with iNOS and HIF-2α with arginase expression,68 this data might suggest that PHD2 expression favors an M1 response by preferentially targeting HIF-2α for destruction.
Modulation of HIF signaling by infection and inflammatory stimuli
While hypoxia regulates HIFs largely at the posttranslational level, other factors such as cytokines and infectious agents regulate its transcription, resulting in a synergistic effect when the stimuli are combined.73–78
The HIF1A promoter contains a binding site for NF–κB transcription factors.77 Both the basal transcription and induction of HIF1A (but not HIF2A) in response to stimuli, such as bacteria, TNF-α, or hypoxia, are dependent on NF-κB. Reduced HIF-1α accumulation and target gene induction are observed in IKKβ-deficient macrophages and in cells treated with siRNA targeting members of the NF-κB family.76,77 Hypoxia, in turn, augments NF-κB expression, phosphorylation, and target gene induction in macrophages and neutrophils.3,5,79–81 Hypoxic induction of mRNA encoding IKKα, IKKβ, and p65 is impaired in Hif1a−/− cells, while the increase in p65 phosphorylation is ablated by the deletion of Hif2a.3,5 IKKβ and NF-κB1 are also directly regulated by PHDs and factor inhibiting HIF respectively.80,82 HIF-1α also binds to the Tlr4 promoter and upregulates TLR4 expression, thereby increasing the macrophage response to LPS.83
In addition to activating NF-κB-mediated transcription, LPS induces a mammalian target of rapamycin-dependent increase in HIF-1α translation in neutrophils and promotes succinate-mediated HIF-1α stabilization in macrophages, as detailed previously.52,60 Oxygen consumption by bacteria, resulting in cellular hypoxia, has also been proposed as a mechanism for the upregulation of HIFs during infection.84,85
In vivo models
Immune responses in vivo may be influenced by HIF expression in both leukocytes and tissue cells, and systemic modulation of HIF expression affects both. Myeloid-specific knockouts of HIF signaling components, however, have permitted elucidation of the innate immune cell-intrinsic effects of hypoxia and HIFs.
Administration of the hydroxylase inhibitor DMOG delays inflammatory resolution following tail transection in zebrafish larvae, due to decreased apoptosis of neutrophils and their retention at the site of injury during the resolution phase.86 The effects of DMOG were abrogated by injection with a hif1b morpholino or by expression of a dominant negative form of the zebrafish HIF1A homolog, hif1ab. In contrast, mutation of the proline hydroxylation site targeted by PHDs, to create a systemic or myeloid-specific dominant active form of HIF-1αb, recapitulated the effect of DMOG, indicating a role for HIF-1α in delayed inflammation resolution.
In a murine trauma/hemorrhagic shock model, in which gut ischemia/reperfusion injury leads to both local and systemic inflammatory responses, Hif1a+/− mice were protected against mucosal gut injury and subsequent lung damage, with reduced expression of TNFα, IL-1β, and iNOS in the intestinal mucosa and IL-1β in the lung, further demonstrating a role for HIF-1α in promoting inflammation in vivo.87
Cramer et al50 crossed mice with floxed Hif1a, Vegf, or Vhl with LysM-cre mice to generate strains with myeloid-specific deletions of each of these genes, providing important insights into the role of HIF in innate immunity. In a model of acute skin inflammation, leukocyte infiltration, myeloperoxidase production, and edema were reduced in LysM-cre/Hif1a mice and increased in LysM-cre/Vhl mice. Likewise, in an arthritis model, edema, synovial infiltration, pannus formation, and cartilage destruction were profoundly reduced in LysM-cre/Hif1a mice.
HIF-1α and VEGF upregulation correlates with increased disease severity in a mouse model of allergic rhinitis and inhibition of HIF using 2-methoxyestradiol attenuates VEGF production and inflammation.88 In an OVA-induced murine model of allergic airway disease, inflammatory cell infiltration, levels of Th2 cytokines (IL-4, IL-5, and IL-13), vascular permeability, fibrosis, goblet cell mucus production, OVA-specific IgE, and airway hyperresponsiveness were all significantly reduced by inhibition of HIF-1α or VEGF.89–91 Similar effects were observed in mice with a systemic Hif1b deletion or a myeloid-specific Hif1a deletion.89,92,93
Activation of HIFs may be important for antibacterial responses. Following subcutaneous GAS administration, LysM-cre/Hif1a mice lost more weight than WT mice and developed larger necrotic skin lesions, containing approximately 1,660-fold more bacteria.51 Bacterial counts in blood and spleen were also increased, indicating greater systemic spread of infection. Similarly, HIF-1α-mediated induction of CRAMP in keratinocytes protects against induction of skin lesions by GAS and approximately three-fold more bacteria were found in the skin ulcers of mice with a keratinocyte-specific deletion of Hif1a.94
Conversely, treatment with the HIF agonist mimosine reduced lesion size by 50% following subcutaneous S. aureus challenge.95 However, mice infected intraperitoneally with a mutant strain of S. aureus (SCV) that does not activate HIF-1α survive, whereas those infected with the WT strain have 100% mortality except when given the HIF-1α inhibitor 17-N-allylamino-17-demethoxygeldanamycin (17-DMAG).84 This demonstrates the fine balance between controlling infection and avoiding excessive and deleterious inflammation.
Mice with a myeloid-specific deletion of Hif2a exhibit greater survival, reduced proinflammatory cytokine production and less severe hypothermia and cardiac dysfunction during LPS-induced endotoxemia.62 Deletion of Hif2a also decreases macrophage infiltration in a thioglycollate-induced model of peritonitis and neutrophil infiltration in the 12-O-tetradecanoylphorbol-13-acetate-induced model of acute skin inflammation.
Overexpression of the dominant active zebrafish HIF2a ortholog, hif2aa, did not affect neutrophil recruitment in the tail injury model described previously, but it did delay the resolution of neutrophilic inflammation.53 Conversely, in a neutrophil-mediated LPS-induced model of acute lung injury, mice with a myeloid-specific Hif2a deletion displayed normal recruitment of neutrophils to the lung, but the clearance was more rapid, with reduced neutrophil counts after 48 and 72 hours, increased neutrophil apoptosis, and reduced markers of lung injury.53
Although the majority of evidence supports a role for HIFs predominantly in promoting inflammation, they may have anti-inflammatory roles under certain circumstances. For instance, renal F4/80+ cell (macrophage) accumulation in a unilateral ureteral obstruction-induced kidney injury model is increased in Vhl−/− and LysMcre/Vhl mice and decreased in Hif−/− and LysMcre/Hif mice (in which both Hif1a and Hif2a are deleted), suggesting that HIFs are inhibiting migration toward the site of injury.71
HIF signaling also has a complex role in tumor biology, including effects on the innate immune response, particularly tumor-associated macrophages (TAMs), which preferentially accumulate in hypoxic regions of tumors.96 Roda et al97 demonstrated opposing roles of HIF-1α and HIF-2α in tumor angiogenesis in a malignant melanoma model. In LysM-cre/Hif1a mice, granulocyte-macrophage colony-stimulating factor treatment induced soluble VEGF receptor, but not VEGF, resulting in a greater reduction in tumor growth and vascularization. In contrast, myeloid Hif2a deletion resulted in increased expression of VEGF, but not soluble VEGF receptor, and abrogated the granulocyte-macrophage colony-stimulating factor-induced reduction in tumor growth and angiogenesis observed in control mice. In a murine model of breast cancer, targeted deletion of Hif1a in myeloid cells reduced tumor growth, despite Vegf expression and vascularization remaining unchanged, perhaps due to the release of T-cells from HIF1-α-dependent suppression by macrophages.65 Hypoxia within the tumor microenvironment also confers on myeloid-derived suppressor cells (MDSC) the ability to suppress T-cell proliferation and IFNγ production and promotes their differentiation into macrophages expressing Il10, Il12, Nos2, Arg1, and Il6.96 Stabilization of HIF by desferrioxamine treatment mimics the enhancement in suppressive capacity, whereas Hif1a−/− MDSC are poor suppressors of T-cell proliferation.
The role of hypoxia and HIFs in macrophage polarization in the context of tumor biology remains subject to debate. Laoui et al98 suggest that hypoxia is not an important driver of macrophage polarization, as it induces HIF-1α and HIF-2α simultaneously, therefore promoting the expression of factors associated with both M1 and M2 responses. Tumors of PHD2+/− mice, in which tumor oxygenation is increased, display a similar relative abundance of MHCIIhi and MHCIIlo TAM subsets (corresponding to M1 and M2 phenotypes, respectively) to that seen in WT mice, and the expression of hypoxia-regulated genes associated with both M1 and M2 responses is reduced in TAM infiltrating PHD2 +/− tumors.
Data is also beginning to emerge regarding the role of other components of the HIF signaling pathway, such as PHDs. For example, in an LPS-induced acute lung injury model, where rapid neutrophil influx is followed by resolution, Phd3−/− and WT mice recovered equally well in normoxia. With coexisting hypoxia, however, Phd3−/− mice had lower BAL neutrophil counts and higher neutrophil apoptosis.54 Increased apoptosis and reduced neutrophil numbers were also observed in bowel submucosa following dextran sulfate sodium-induced colitis, a neutrophil-dominated inflammatory model that involves tissue hypoxia. In a fatal model of abdominal sepsis, however, Phd3−/− mice exhibited increased macrophage proinflammatory cytokine production and decreased survival, due to an overwhelming innate immune response.59
HIF-hypoxia signaling in human infectious and inflammatory disease
Hypoxia and HIF activation are features of many human diseases, including infection, cancers, and a variety of inflammatory conditions. Immune cells are an important feature of these conditions, as are the tissue cells, and their relative and context-specific expression of the HIF pathway components may modify clinical outcomes. In skin biopsy samples from patients with a variety of cutaneous infections (including Gram-positive and Gram-negative bacteria, parasites, and viruses), a strong nuclear HIF-1α signal is detected in keratinocytes, dermal capillaries, neutrophils, lymphocytes, and macrophages, which is absent in uninfected biopsies.84 Biopsies from damaged intestinal mucosa of patients with inflammatory bowel disease exhibit increased HIF-1α expression in lamina propria macrophages, compared to those from undamaged sections.63 The level of HIF-1α in damaged mucosa correlates with the expression of the scavenger receptor CD36, suggesting that HIF-1α might promote macrophage phagocytosis of microbes and/or apoptotic cells within the colon.
HIF-1α is also found in central hypoxic regions of human atherosclerotic plaques and colocalizes with Unc5b and netrin-1, proteins that are induced in macrophages by hypoxia and low-density lipoprotein, promoting cell survival and inhibiting migration away from the plaque.15 siRNA knockdown of HIF1A in U937 monocytic cells inhibits foam cell formation and reduces the expression of 57 of the 70 atherosclerotic genes that are induced by oxidized low-density lipoprotein, including PTGS2, IL1B, ICAM1, and VCAM1, suggesting that myeloid HIF-1α may have an important role in atherosclerosis.99
HIF-2α expression is increased in peripheral blood neutrophils of inflammatory arthritis patients compared to those from healthy controls.53 Strong nuclear HIF-2α and cytoplasmic VEGF immunostaining is also found in both neutrophils and neovasculature in lesions of patients with pyoderma gangrenosum, an inflammatory skin condition thought to involve a defect in resolution of neutrophilic inflammation following an inflammatory stimulus.100
Although hypoxia might be expected to activate HIF equally in all cell types present in diseased tissue, this is not always the case. HIF2-α is expressed in neutrophils infiltrating the airway of chronic obstructive pulmonary disease patients, but not in the epithelium.53 Likewise, HIF-2α protein is expressed in TAMs in the majority of uterine cervical cancer specimens, whereas <10% of specimens exhibit tumor cell HIF-2α expression.101 TAMs infiltrating lung tumors also exhibit strong HIF-2α immunostaining in the absence of HIF-2α expression in the tumor cells themselves.102 The frequency of clusters of HIF-2α+ TAM in breast carcinomas correlates with increased density of microvessels and appears to be associated with a reduced overall survival rate.103
In cervical cancer patients, the overall macrophage count in tumor biopsies does not affect the chance of disease-free survival; however, the higher the percentage of tumor-associated macrophages that express HIF2α, the greater the risk of local recurrence and the lower the probability of disease-free survival following radiotherapy.101 In many types of cancer, including breast, ovarian, colon, and pancreatic adenocarcinomas, HIF-1α and HIF-2α are expressed in the tumor cells themselves.102 This might induce factors, such as chemokines, that attract monocytes and MDSC and promote their differentiation into immunosuppressive macrophages that inhibit T-cell responses, as evidenced by murine models. Urothelial carcinomas with higher expression of HIF-1α in tumor cells exhibit higher TAM infiltration and these patients have a reduced disease-free survival rate.104
HIF1A, HIF2A, PHD2 and PHD3 transcripts are increased in peripheral blood neutrophils of RA patients.53,54 HIF-1α is highly expressed in synovial fibroblasts and infiltrating macrophages in the inflamed RA joint, where it enhances TNF-α- and IL-1β-driven activation of the p38 and extracellular signal-regulated kinase signaling pathways, resulting in increased IL-33 expression.105,106 IL-33, in turn, upregulates HIF-1α expression in synovial fibroblasts.105 Synovial fibroblasts derived from osteoarthritis or RA patients subcutaneously engrafted into immunodeficient mice induce significantly greater myeloid cell infiltration and angiogenesis than fibroblasts derived from healthy individuals.107 This effect is dependent on both HIF and VEGF, since it can be reversed by Hif1a siRNA or by pharmacological inhibition of either HIF-1α or VEGF.
Chronically low levels of oxygen may result in the downregulation of HIFs under certain circumstances. For example, pO2 is reduced in the skin of systemic sclerosis patients, yet HIF-1α expression in epidermal keratinocytes is lower than that of controls, perhaps due to increased PHD levels, following induction of negative feedback loops.54,55,57,108
Patients with mutations in components of HIF signaling pathways are beginning to shed light on their role in humans. Neutrophils from erythrocytosis patients with a gain of function mutation in HIF2A express higher levels of the target genes PAI1 and PHD3 and undergo lower rates of apoptosis ex vivo; however, phagocytosis and respiratory burst are unaffected.53 Neutrophils derived from patients who are heterozygous for a mutation in the VHL gene exhibit lower rates of apoptosis and enhanced bacterial phagocytosis under normoxic conditions, resembling the phenotype normally seen in hypoxia, indicating the importance of proteasomal degradation in HIF regulation in human neutrophils.109
Future perspectives and therapeutic potential
Given the large number of inflammatory diseases in which both hypoxia and HIF upregulation occur, and the important role of HIF in determining inflammatory responses, targeting the HIF pathway may be a promising avenue for therapies, although a number of important issues require addressing.
Chronic hypoxia is a feature of many human diseases, yet most research so far has focused on acute responses. One important area for future study will be comparing the effects of acute versus chronic hypoxia and investigating the role of negative feedback mechanisms such as HIF-mediated induction of PHD and VHL transcription in chronic activation of the HIF/hydroxylase pathway.
More detailed studies of the effects of hypoxia on circulating leukocytes compared with resident immune cells derived from different tissues are also warranted. For instance, specialized populations of macrophages exist in tissues, which may well differ from each other and from circulating monocytes in their responses to hypoxic and inflammatory stimuli. Given that certain organs, such as the lungs, have a higher basal level of oxygen than others, macrophages derived from these tissues may exhibit greater responses when exposed to hypoxia, due to lower HIF-mediated transcription of negative regulatory proteins. Furthermore, the study of hypoxic responses and HIF signaling in the innate immune system has largely focused on monocytes, macrophages, and neutrophils, whereas research into other cell types, such as ILCs, eosinophils, and basophils, has been somewhat limited.
Efficacy of HIF-1α inhibition in dampening asthmatic inflammation has recently been demonstrated in vivo, suggesting that inhibition of HIF signaling could be beneficial in chronic inflammatory diseases.89–91 However, given the importance of HIF-1α for antimicrobial responses, care must be taken to ensure that treatments directed at the HIF pathway do not result in susceptibility to infection.
Pharmacological augmentation of HIF-1α activity by mimosine has been proposed as a means of boosting innate immune responses and promoting bacterial clearance during infection, since mimosine was found to enhance myeloid cell bactericidal activity and to inhibit the development of necrotic skin lesions following S. aureus challenge in mice.51,95
Augmentation of either HIF-1α or HIF-2α also enhances angiogenesis and wound healing, which may be beneficial in diabetes or ischemic disease.110,111 However, it is not clear whether this can be done safely in humans without inducing excessive inflammation, which could exacerbate tissue damage or induce shock.
HIF-1α is upregulated more rapidly than HIF-2α and appears to be more important for the acute response to infection, whereas HIF-2α expression dominates during the resolution of inflammation.53,68 Therefore, the targeting of HIF-2α might be useful in dampening chronic inflammation while preserving acute responses. The HIF-2α Per-Arnt-Sim domain contains a cavity that will accommodate small molecule ligands which modulate its ability to form active heterodimers with HIF-1β, and thus provides an avenue for potential pharmacological manipulation.112 However, further investigation into the regulation of HIF-2α and the immune functions it modulates (and how these differ from HIF-1α) is required to determine the likely effects of modifying its expression or activity.
Suppression of downstream targets of HIF might also be a means of delivering therapies that target specific aspects of the hypoxic response while preserving those that are protective. PHD3 has been suggested as a therapeutic target in neutrophils, owing to its specific effects on hypoxic survival in this cell type. Evidence from animal models indicates that the inhibition of PHD3 would preserve neutrophil antimicrobial function, but encourage the resolution of neutrophilic inflammation in hypoxic tissues once the infection is cleared.54 However, it is important to consider the impact of any potential therapy targeting PHD3 on other cell types, as its role in macrophages appears to be different to that in neutrophils.
While modulation of the HIF pathway has had beneficial effects in animal models, further work is required to determine the most appropriate way of manipulating the pathway to promote resolution of infections and to treat inflammatory diseases without causing deleterious side effects. Nonetheless, the crucial role of HIFs in controlling innate immune responses, in response to hypoxia as well as other factors, makes this pathway a promising target for further investigation and potential therapies.
Acknowledgments
SRW is supported by a Wellcome Trust Senior Clinical Fellowship Award (Reference 098516).
Disclosure
The authors report no conflicts of interest in this work.
References
Kawaguchi T, Veech RL, Uyeda K. Regulation of energy metabolism in macrophages during hypoxia. Roles of fructose 2,6-bisphosphate and ribose 1,5-bisphosphate. J Biol Chem. 2001;276(30):28554–28561. | |
Bosco MC, Puppo M, Santangelo C, et al. Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J Immunol. 2006;177(3):1941–1955. | |
Fang HY, Hughes R, Murdoch C, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844–859. | |
Roiniotis J, Dinh H, Masendycz P, et al. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. J Immunol. 2009;182(12):7974–7981. | |
Walmsley SR, Print C, Farahi N, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201(1):105–115. | |
Karakurum M, Shreeniwas R, Chen J, et al. Hypoxic induction of interleukin-8 gene expression in human endothelial cells. J Clin Invest. 1994;93(4):1564–1570. | |
Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol. 2006;177(10):7211–7224. | |
Beck-Schimmer B, Schimmer RC, Madjdpour C, Bonvini JM, Pasch T, Ward PA. Hypoxia mediates increased neutrophil and macrophage adhesiveness to alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;25(6):780–787. | |
Colgan SP, Dzus AL, Parkos CA. Epithelial exposure to hypoxia modulates neutrophil transepithelial migration. J Exp Med. 1996;184(3):1003–1015. | |
Meyer S, Z’graggen BR, Blumenthal S, et al. Hypoxia attenuates effector-target cell interaction in the airway and pulmonary vascular compartment. Clin Exp Immunol. 2007;150(2):358–367. | |
Zünd G, Uezono S, Stahl GL, et al. Hypoxia enhances induction of endothelial ICAM-1: role for metabolic acidosis and proteasomes. Am J Physiol. 1997;273(5 Pt 1):C1571–C1580. | |
Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101(28):10440–10445. | |
Bosco MC, Reffo G, Puppo M, Varesio L. Hypoxia inhibits the expression of the CCR5 chemokine receptor in macrophages. Cell Immunol. 2004;228(1):1–7. | |
Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation – a potential mechanism. Eur J Immunol. 2001;31(2):480–489. | |
Ramkhelawon B, Yang Y, van Gils JM, et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: mechanism for macrophage retention and survival. Arterioscler Thromb Vasc Biol. 2013;33(6):1180–1188. | |
White JR, Harris RA, Lee SR, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;83(1):1–8. | |
Hannah S, Mecklenburgh K, Rahman I, et al. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995;372(2–3):233–237. | |
Nissim Ben Efraim AH, Eliashar R, Levi-Schaffer F. Hypoxia modulates human eosinophil function. Clin Mol Allergy. 2010;8:10. | |
Bianchi SM, Dockrell DH, Renshaw SA, Sabroe I, Whyte MK. Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin Sci (Lond). 2006;110(3):293–304. | |
Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ. Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leukoc Biol. 2006;80(3):521–528. | |
Haanen C, Vermes I. Apoptosis and inflammation. Mediators Inflamm. 1995;4(1):5–15. | |
Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999; 160(5 Pt 2):S5–S11. | |
Rossi AG, Sawatzky DA, Walker A, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12(9):1056–1064. | |
Bekeredjian R, Walton CB, MacCannell KA, et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS ONE. 2010;5(7):e11693. | |
Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. 1982;70(3):550–557. | |
De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–663. | |
Knowles R, Keeping H, Graeber T, et al. Cytokine control of PMN phagocytosis: regulatory effects of hypoxemia and hypoxemia-reoxygenation. Am J Physiol. 1997;272(4 Pt 1):C1352–C1364. | |
Simms HH, D’Amico R. Regulation of whole blood polymorphonuclear leukocyte phagocytosis following hypoxemia and hypoxemia/reoxygenation. Shock. 1994;1(1):10-18. | |
Wang JS, Chiu YT. Systemic hypoxia enhances exercise-mediated bactericidal and subsequent apoptotic responses in human neutrophils. J Appl Physiol (1985). 2009;107(4):1213–1222. | |
Wang JS, Liu HC. Systemic hypoxia enhances bactericidal activities of human polymorphonuclear leuocytes. Clin Sci (Lond). 2009;116(11):805–817. | |
Acosta-Iborra B, Elorza A, Olazabal IM, et al. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. J Immunol. 2009;182(5):3155–3164. | |
Anand RJ, Gribar SC, Li J, et al. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J Leukoc Biol. 2007;82(5):1257–1265. | |
McGovern NN, Cowburn AS, Porter L, et al. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol. 2011;186(1):453–463. | |
Tamura DY, Moore EE, Partrick DA, Johnson JL, Offner PJ, Silliman CC. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock. 2002;17(4):269–273. | |
Nickel D, Busch M, Mayer D, Hagemann B, Knoll V, Stenger S. Hypoxia triggers the expression of human β defensin 2 and antimicrobial activity against Mycobacterium tuberculosis in human macrophages. J Immunol. 2012;188(8):4001–4007. | |
Bosco MC, Puppo M, Pastorino S, et al. Hypoxia selectively inhibits monocyte chemoattractant protein-1 production by macrophages. J Immunol. 2004;172(3):1681–1690. | |
Hirani N, Antonicelli F, Strieter RM, et al. The regulation of interleukin-8 by hypoxia in human macrophages – a potential role in the pathogenesis of the acute respiratory distress syndrome (ARDS). Mol Med. 2001;7(10):685–697. | |
Rydberg EK, Salomonsson L, Hultén LM, et al. Hypoxia increases 25-hydroxycholesterol-induced interleukin-8 protein secretion in human macrophages. Atherosclerosis. 2003;170(2):245–252. | |
Albina JE, Mahoney EJ, Daley JM, Wesche DE, Morris SM Jr, Reichner JS. Macrophage arginase regulation by CCAAT/enhancer-binding protein beta. Shock. 2005;23(2):168–172. | |
Pesce JT, Ramalingam TR, Mentink-Kane MM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5(4):e1000371. | |
Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275(33):25130–25138. | |
Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401–408. | |
Pan Y, Mansfield KD, Bertozzi CC, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27(3):912–925. | |
Tausendschön M, Dehne N, Brüne B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine. 2011;53(2):256–262. | |
Ormiston ML, Chang C, Long LL, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation. 2012;126(9):1099–1109. | |
Balsamo M, Manzini C, Pietra G, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013; 43(10):2756–2764. | |
Sarkar S, Germeraad WT, Rouschop KM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS One. 2013;8(5):e64835. | |
Marteyn B, West NP, Browning DF, et al. Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010;465(7295):355–358. | |
Mecklenburgh KI, Walmsley SR, Cowburn AS, et al. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002;100(8):3008–3016. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Peyssonnaux C, Datta V, Cramer T, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115(7):1806–1815. | |
McInturff AM, Cody MJ, Elliott EA, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood. 2012;120(15):3118–3125. | |
Thompson AA, Elks PM, Marriott HM, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood. 2014;123(3):366–376. | |
Walmsley SR, Chilvers ER, Thompson AA, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121(3):1053–1063. | |
Henze AT, Riedel J, Diem T, et al. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 2010;70(1):357–366. | |
Karhausen J, Kong T, Narravula S, Colgan SP. Induction of the von Hippel-Lindau tumor suppressor gene by late hypoxia limits HIF-1 expression. J Cell Biochem. 2005;95(6):1264–1275. | |
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | |
Kim SY, Jeong E, Joung SM, Lee JY. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochem Biophys Res Commun. 2012;419(3):466–471. | |
Kiss J, Mollenhauer M, Walmsley SR, et al. Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol. 2012;189(4):1955–1965. | |
Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. | |
Mahabeleshwar GH, Qureshi MA, Takami Y, Sharma N, Lingrel JB, Jain MK. A myeloid hypoxia-inducible factor 1α- Krüppel-like factor 2 pathway regulates gram-positive endotoxin-mediated sepsis. J Biol Chem. 2012;287(2):1448–1457. | |
Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120(8):2699–2714. | |
Ortiz-Masià D, Díez I, Calatayud S, et al. Induction of CD36 and thrombospondin-1 in macrophages by hypoxia-inducible factor 1 and its relevance in the inflammatory process. PLoS One. 2012;7(10):e48535. | |
Bayele HK, Peyssonnaux C, Giatromanolaki A, et al. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood. 2007;110(8):3039–3048. | |
Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465–7475. | |
Liao H, Hyman MC, Lawrence DA, Pinsky DJ. Molecular regulation of the PAI-1 gene by hypoxia: contributions of Egr-1, HIF-1 alpha, and C/EBPalpha. FASEB J. 2007;21(3):935–949. | |
Charron CE, Chou PC, Coutts DJ, et al. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem. 2009;284(52):36047–36054. | |
Takeda N, O’Dea EL, Doedens A, et al. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. | |
Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18(11):4528–4542. | |
Werno C, Menrad H, Weigert A, et al. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. 2010;31(10):1863–1872. | |
Kobayashi H, Gilbert V, Liu Q, et al. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol. 2012;188(10):5106–5115. | |
Takeda Y, Costa S, Delamarre E, et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479(7371):122–126. | |
Blouin CC, Pagé EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood. 2004;103(3):1124–1130. | |
Frede S, Freitag P, Otto T, Heilmaier C, Fandrey J. The proinflammatory cytokine interleukin 1beta and hypoxia cooperatively induce the expression of adrenomedullin in ovarian carcinoma cells through hypoxia inducible factor 1 activation. Cancer Res. 2005;65(11):4690–4697. | |
Mi Z, Rapisarda A, Taylor L, et al. Synergystic induction of HIF-1alpha transcriptional activity by hypoxia and lipopolysaccharide in macrophages. Cell Cycle. 2008;7(2):232–241. | |
Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. | |
van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412(3):477–484. | |
Westra J, Brouwer E, Bos R, et al. Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann N Y Acad Sci. 2007;1108:340–348. | |
Lahat N, Rahat MA, Ballan M, Weiss-Cerem L, Engelmayer M, Bitterman H. Hypoxia reduces CD80 expression on monocytes but enhances their LPS-stimulated TNF-alpha secretion. J Leukoc Biol. 2003;74(2):197–205. | |
Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. | |
Leeper-Woodford SK, Detmer K. Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-kappaB expression. Am J Physiol. 1999;276(6 Pt 1):L909–L916. | |
Cockman ME, Lancaster DE, Stolze IP, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci U S A. 2006;103(40):14767–14772. | |
Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS, Lee JY. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology. 2010;129(4):516–524. | |
Werth N, Beerlage C, Rosenberger C, et al. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One. 2010;5(7):e11576. | |
Kempf VA, Lebiedziejewski M, Alitalo K, et al. Activation of hypoxia-inducible factor-1 in bacillary angiomatosis: evidence for a role of hypoxia-inducible factor-1 in bacterial infections. Circulation. 2005;111(8):1054–1062. | |
Elks PM, van Eeden FJ, Dixon G, et al. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. 2011;118(3):712–722. | |
Feinman R, Deitch EA, Watkins AC, et al. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G833–G843. | |
Zhou H, Chen X, Zhang WM, Zhu LP, Cheng L. HIF-1α inhibition reduces nasal inflammation in a murine allergic rhinitis model. PLoS One. 2012;7(11):e48618. | |
Crotty Alexander L, Akong-Moore K, Feldstein S, et al. Myeloid cell HIF-1α regulates asthma airway resistance and eosinophil function. J Mol Med (Berl). 2013;91(5):637–644. | |
Huerta-Yepez S, Baay-Guzman GJ, Garcia-Zepeda R, et al. 2-Methoxyestradiol (2-ME) reduces the airway inflammation and remodeling in an experimental mouse model. Clin Immunol. 2008;129(2):313–324. | |
Kim SR, Lee KS, Park HS, et al. HIF-1α inhibition ameliorates an allergic airway disease via VEGF suppression in bronchial epithelium. Eur J Immunol. 2010;40(10):2858–2869. | |
Huerta-Yepez S, Baay-Guzman GJ, Bebenek IG, et al. Hypoxia Inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy. 2011;66(7):909–918. | |
Baay-Guzman GJ, Bebenek IG, Zeidler M, et al. HIF-1 expression is associated with CCL2 chemokine expression in airway inflammatory cells: implications in allergic airway inflammation. Respir Res. 2012;13:60. | |
Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS. Critical role of HIF-1alpha in keratinocyte defense against bacterial infection. J Invest Dermatol. 2008;128(8):1964–1968. | |
Zinkernagel AS, Peyssonnaux C, Johnson RS, Nizet V. Pharmacologic augmentation of hypoxia-inducible factor-1alpha with mimosine boosts the bactericidal capacity of phagocytes. J Infect Dis. 2008;197(2):214–217. | |
Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. | |
Roda JM, Sumner LA, Evans R, Phillips GS, Marsh CB, Eubank TD. Hypoxia-inducible factor-2α regulates GM-CSF-derived soluble vascular endothelial growth factor receptor 1 production from macrophages and inhibits tumor growth and angiogenesis. J Immunol. 2011;187(4):1970–1976. | |
Laoui D, Van Overmeire E, Di Conza G, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. | |
Jiang G, Li T, Qiu Y, Rui Y, Chen W, Lou Y. RNA interference for HIF-1alpha inhibits foam cells formation in vitro. Eur J Pharmacol. 2007;562(3):183–190. | |
Chua RA, Mackelfresh J, Cohen C, Varma V, Fried L, Arbiser JL. Disseminated pyoderma gangrenosum: role for vascular endothelial growth factor and hypoxia inducible factor-2. J Am Acad Dermatol. 2009;61(4):730–732. | |
Kawanaka T, Kubo A, Ikushima H, Sano T, Takegawa Y, Nishitani H. Prognostic significance of HIF-2alpha expression on tumor infiltrating macrophages in patients with uterine cervical cancer undergoing radiotherapy. J Med Invest. 2008;55(1–2):78–86. | |
Talks KL, Turley H, Gatter KC, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157(2):411–421. | |
Leek RD, Talks KL, Pezzella F, et al. Relation of hypoxia-inducible factor-2 alpha (HIF-2 alpha) expression in tumor-infiltrative macrophages to tumor angiogenesis and the oxidative thymidine phosphorylase pathway in Human breast cancer. Cancer Res. 2002;62(5):1326–1329. | |
Chai CY, Chen WT, Hung WC, et al. Hypoxia-inducible factor-1alpha expression correlates with focal macrophage infiltration, angiogenesis and unfavourable prognosis in urothelial carcinoma. J Clin Pathol. 2008;61(5):658–664. | |
Hu F, Shi L, Mu R, et al. Hypoxia-inducible factor-1α and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLoS One. 2013;8(8):e72650. | |
Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001;44(7):1540–1544. | |
del Rey MJ, Izquierdo E, Caja S, et al. Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheum. 2009;60(10):2926–2934. | |
Distler O, Distler JH, Scheid A, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95(1):109–116. | |
Walmsley SR, Cowburn AS, Clatworthy MR, et al. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108(9):3176–3178. | |
Mace KA, Yu DH, Paydar KZ, Boudreau N, Young DM. Sustained expression of Hif-1alpha in the diabetic environment promotes angiogenesis and cutaneous wound repair. Wound Repair Regen. 2007;15(5):636–645. | |
Chen L, Uchida K, Endler A, Shibasaki F. Mammalian tumor suppressor Int6 specifically targets hypoxia inducible factor 2alpha for degradation by hypoxia- and pVHL-independent regulation. J Biol Chem. 2007;282(17):12707–12716. | |
Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci U S A. 2009;106(2):450–455. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.