Back to Journals » International Journal of General Medicine » Volume 16
Heterozygous Pathogenic and Likely Pathogenic Symptomatic HTRA1 Variant Carriers in Cerebral Small Vessel Disease
Authors Xu SY ![]() , Li HJ, Li S, Ren QQ, Liang JL, Li CX
, Li HJ, Li S, Ren QQ, Liang JL, Li CX ![]()
Received 14 January 2023
Accepted for publication 23 March 2023
Published 29 March 2023 Volume 2023:16 Pages 1149—1162
DOI https://doi.org/10.2147/IJGM.S404813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Sui-Yi Xu,1,* Hui-Juan Li,2,* Shun Li,2 Qian-Qian Ren,1 Jian-Lin Liang,1 Chang-Xin Li1
1Department of Neurology, Headache Center, The First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2Center for Medical Genetics & Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chang-Xin Li, Department of Neurology, The First Hospital of Shanxi Medical University, Jiefangnan 85 Road, Taiyuan, Shanxi Province, 030001, People’s Republic of China, Tel +86 15103513579, Email [email protected]
Abstract: High temperature requirement serine peptidase A1 (HTRA1) related cerebral small vessel disease (CSVD) includes both symptomatic heterozygous HTRA1 variant carrier and cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) patients. Presently, most reported symptomatic heterozygous HTRA1 variant carrier cases are sporadic family reports with a lack of specific characteristics. Additionally, the molecular mechanism of heterozygous HTRA1 gene variants is unclear. We conducted this review to collect symptomatic carriers of heterozygous HTRA1 gene variants reported as of 2022, analyzed all pathogenicity according to American College of Medical Genetics and Genomics (ACMG) variant classification, and summarized the cases with pathogenic and likely pathogenic HTRA1 variants gender characteristics, age of onset, geographical distribution, initial symptoms, clinical manifestations, imaging signs, HTRA1 gene variant information and to speculate its underlying pathogenic mechanisms. In this review, we summarized the following characteristics of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers: to date, the majority of reported symptomatic HTRA1 carriers are in European and Asian countries, particularly in China which was found to have the highest number of reported cases. The age of first onset is mostly concentrated in the fourth and fifth decades. The heterozygous HTRA1 gene variants were mostly missense variants. The two variant sites, 166– 182 aa and 274– 302 aa, were the most concentrated. Clinicians need to pay attention to de novo data and functional data, which may affect the pathogenicity analysis. The decrease in HtrA1 protease activity is currently the most important explanation for the genetic pathogenesis.
Keywords: stroke, HTRA1 gene, ACMG criteria, heterozygous variant, symptomatic carrier
Introduction
Cerebral small vessel disease (CSVD) is an age-related cerebrovascular disease. A variety of factors may affect the small arteries, arterioles, capillaries, and venules in the brain, leading to clinical, imaging, and pathological syndromes of CSVD.1 The clinical symptoms associated with CSVD are complex and include cognitive impairment, stroke, spine disorders, gait disorders, alopecia, psychiatric disorders, migraine, epilepsy, and more.2,3 The consequences of CSVD on the brain lesions were easily captured in magnetic resonance imaging (MRI). White matter hyperintensities (WMHs), lacunar infarctions (LIs), and cerebral microbleeds (CMBs) were the most common imaging manifestations.2 With improvements in the understanding of genetic precision medicine, a number of disease-causing genes, including NOTCH3 (OMIM 600276),4 HTRA1 (OMIM 602194),5,6 TREX1 (OMIM 606609),7 COL4A1 (OMIM 120130),8 COL4A2 (OMIM 120090),9 CTSA (OMIM 613111),10 and GLA (OMIM 300644),11 have been discovered; among these, heterozygous HTRA1 gene variant is related to autosomal dominant cerebral arteriopathy with subcortical infarcts and leukoencephalopathy type 2 (CADASIL2).6 CADASIL caused by NOTCH3 gene mutation, the most common dominantly inherited monogenic cerebrovascular disease, has typical abnormal temporal pole signals.12 Additionally, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), also known as the Maeda syndrome, which pathogenic gene is the same as CADASIL2, and has more specific manifestations, such as alopecia and spine disorders.13
Presently, most reported symptomatic carriers of heterozygous HTRA1 gene variants are sporadic family reports with a lack of specific characteristics. Moreover, the molecular mechanism of heterozygous HTRA1 gene variants is unclear. Thus, in this review, we used PubMed, Scopus, Web of Science, and Google Scholar to collect symptomatic carriers of heterozygous HTRA1 gene variants with complete index cases information reported as of 2022, analyzed all pathogenicity according to American College of Medical Genetics and Genomics (ACMG) variant classification,14 excluded all variants of uncertain significance, and summarized the cases with pathogenic and likely pathogenic HTRA1 variants gender characteristics, age of onset, geographical distribution, initial symptoms, clinical manifestations, imaging signs, HTRA1 gene variant information and to speculate its underlying pathogenic mechanisms.
HTRA1 Gene
The HTRA1 gene (OMIM 602194) is located on the autosome 10q26.13, and its most commonly used transcript code is NM_002775.5. The HTRA1 gene includes nine exons and a 1443 bp coding domain sequence, and it encodes the HtrA1 protease comprising 480 amino acids (aa).15 HtrA1 protease can be widely expressed in normal human tissues, including four domains such as the insulin-like growth factor binding protein (33–98 aa), Kazal-type serine protease inhibitor (99–157 aa), trypsin-like serine protease (204–364 aa), and PDZ domains (365–467 aa).16 Its linker region (158–203 aa), Loop D (283–291 aa), and Loop 3 (301–314 aa) are the key structures for activation of enzyme activity.17
In CSVDs, homozygous and heterozygous variants in the HTRA1 gene cause different disease phenotypes.17,18 Homozygous or compound heterozygous HTRA1 gene mutations are related to CARASIL19 while heterozygous HTRA1 gene variants are related to CADASIL2;6 however, the pathogenic mechanisms of both diseases are unclear till date.20
Clinical Manifestations of Symptomatic HTRA1 Variant Carriers
In 2015, Verdura6 used next generation sequencing to screen all candidate pathogenic genes carried by probands with familial cerebrovascular disease of unknown etiology. After verification by Standard PCR amplification and Sanger sequencing, it was first proposed that heterozygous HTRA1 gene variants were associated with autosomal dominant cerebrovascular disease. In 2016, the Online Mendelian Inheritance in Man (OMIM) database officially named the CSVD caused due to heterozygous HTRA1 gene variants as CADASIL2 (https://www.omim.org/). However, the correlation between the genotype of the heterozygous HTRA1 variant and the clinical phenotype has not been confirmed,21 and not all carriers of the heterozygous HTRA1 gene variant will have the corresponding clinical manifestations. The penetrance appears to be low.17 Therefore, the naming of CADASIL2 is still controversial. Some scholars17 believe that it is more suitable to use “symptomatic HTRA1 variant carriers” to describe this type of disease.
This study reviews a total of 76 symptomatic HTRA1 variant carriers’ family probands with relatively complete data from PubMed, Scopus, Web of Science, and Google Scholar databases as of 2022. We analyzed all pathogenicity according to ACMG variant classification,14 and included 55 pathogenic and likely pathogenic variants (Table 1). The gender, age of first onset, nationality, initial symptoms, clinical manifestations, imaging signs, accompanying diseases, HTRA1 gene variant information were analyzed by 55 pathogenic and likely pathogenic symptomatic carriers to summarize the characteristics of this type of disease.
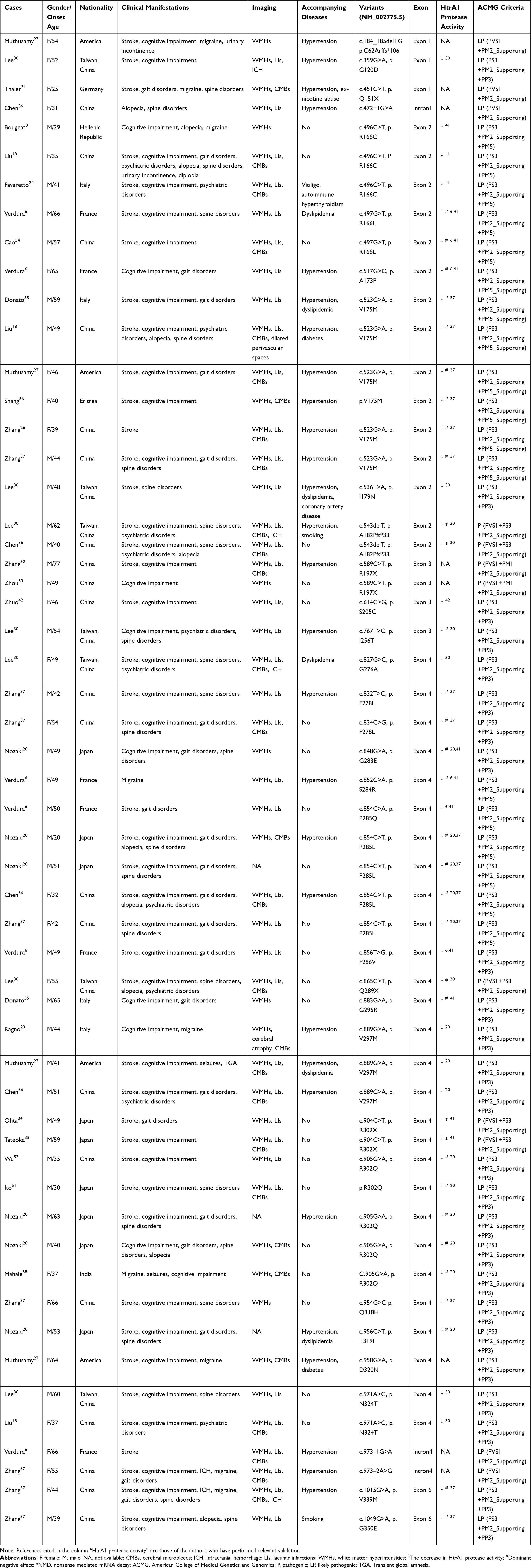 |
Table 1 Worldwide Distribution of P and LP Symptomatic HTRA1 Variant Carriers |
Among 55 probands, no consanguinity of the proband’s parents were found, and males accounted for about 56.36%. These indicate that a male sex advantage may exist, and similar to CARASIL.22 Most probands are from Asian and European countries, especially from China (50.91%), followed by Japan, France, and Italy. In recent years, related cases have also been reported in African and American countries (Figure 1).
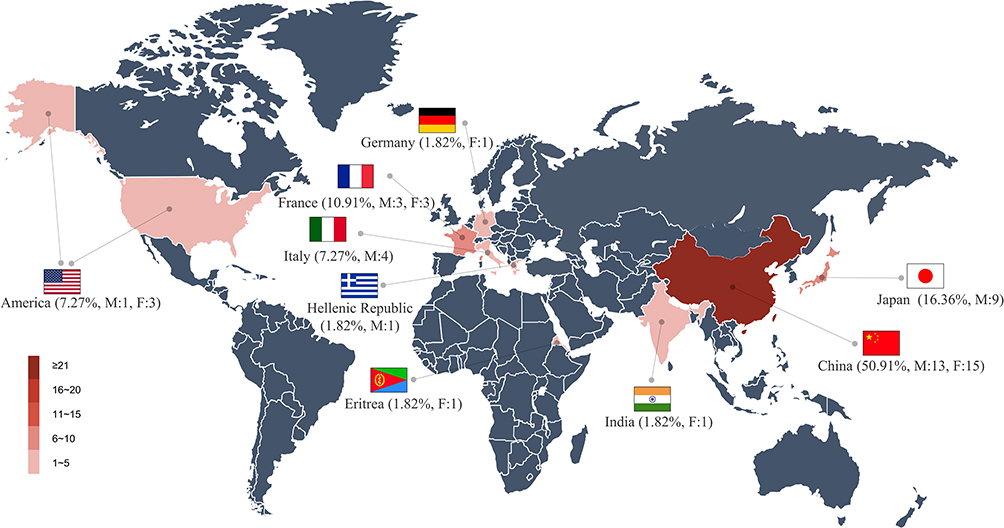 |
Figure 1 Geographical and gender distribution of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers. Symptomatic carrier is prevalent in Asian and European countries, especially in China (50.91%). Symptomatic carrier has gender differences, and it mostly affects males. The color depth represents the number of cases. Abbreviations: F, female; M, male. |
The age of onset of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers was later than that of CARASIL. The former onsets mostly during the fourth to fifth decade while the latter onsets mostly during the second to third decade.13 The initial symptoms of stroke (45%) the most common, followed by the cognitive impairment (Figure 2). The clinical manifestations of symptomatic HTRA1 variant carriers were mainly progressive cognitive impairment. Some patients had gait and psychiatric disorders. The symptoms of symptomatic carriers outside the nervous system are less common than those of CARASIL. Patients with spine disorders and alopecia accounted for 47.27% and 20% of the total number of patients, respectively. Ragno23 has reported that symptomatic carriers may have cutaneous sensory and autonomic small-fiber neuropathy.
 |
Figure 2 The frequency of initial symptoms, clinical manifestations, and imaging manifestations in pathogenic and likely pathogenic symptomatic HTRA1 variant carriers. Abbreviations: TGA, Transient global amnesia; WMHs, white matter hyperintensities; LIs, lacunar infarctions; CMBs, cerebral microbleeds; ICH, intracranial hemorrhage. |
Similar to common hereditary CSVDs such as CADASIL and CARASIL, some pathogenic and likely pathogenic symptomatic HTRA1 variant carriers do not have traditional vascular risk factors (Table 1). Some patients have hypertension (50.91%) and dyslipidemia (10.91%), and one patient have vitiligo, autoimmune hyperthyroidism.24 Consensus recommendations of the European Academy of Neurology25 suggest that even if patients are accompanied by traditional cerebrovascular disease risk factors, the diagnosis of monogenic CSVD should be considered. Ordinal logistic regression analysis was performed to reveal risk factors for the clinical phenotype.26 Patients with vascular risk factors presented with more severe clinical symptoms.
Clinicoradiographic characteristics of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers may overlap with sporadic CSVD.27 Presently, there are no specific imaging signs for symptomatic carriers. White matter hyperintensities (WMHs, 100%), lacunar infarctions (LIs, 75%), and cerebral microbleeds (CMBs, 55.77%) were the most common imaging manifestations (Figure 2). Rare genetic variants of the HTRA1 gene and HtrA1 protease play an important role in the burden of WMH in the general population.28 Loss-of-function variants in HTRA1 gene associated with an increased WMH volume. Domain-specific burden tests revealed that the association with WMH volume was restricted to variants in the protease domain. The WMH volume was brought forward by 11 years in carriers of a rare protease domain variant. With the development of brain imaging genomics,29 quantitative analysis of the location and number of WMHs, LIs, and CMBs; brain volume; cerebral blood flow; and tissue metabolism capacity in symptomatic carriers has become feasible.
Most pathogenic and likely pathogenic heterozygous HTRA1 gene variants of symptomatic carriers were missense variants, and some of them were nonsense variants,30–35 frameshift variants,27,30,36 and splice site variants.6,36,37 The variant sites of the heterozygous HTRA1 gene were mostly located in exon 4 (50.91%), and the trypsin-like serine protease domain was the most common domain in HtrA1 protease (61.82%). Moreover, we found two variant site aggregation regions (166–182 aa and 276–302 aa). The former was located in the linker region of the trypsin-like serine protease domain while the latter was located in Loop D and Loop 3, which activate the serine protease activity (Figure 3). The severity of the clinical manifestations of symptomatic carriers was related to the location of the HTRA1 gene variant. When the variant was located in the loop 3/loop D domain or exon 4, the patient had a more severe clinical phenotype.26
 |
Figure 3 Variant sites of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers. In NP_002766.1, Domains 1–4 are insulin-like growth factor binding protein, Kazal-type serine protease inhibitor, trypsin-like serine protease, and PDZ domains, respectively. The corner numbers indicate the number of probands at the particular site. According to ACMG guidelines, red represents a pathogenicity rating of pathogenic, and blue represents a pathogenicity rating of likely pathogenic. |
According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines,14 we conducted pathogenicity analysis on the collected proband data (Table 1). The variants of uncertain significance (VUS) were presented in Supplement Table 1. Not all rare heterozygous HTRA1 missense variants are pathogenic or likely pathogenic,25 and heterozygous HTRA1 nonsense or frameshift variants are pathogenic or likely pathogenic for the moment.38 Among them, 72.37% of the variants were pathogenic or likely pathogenic, and 27.63% of the variants of uncertain significance. Among probands rated as variants of indeterminate significance, the judgment of pathogenicity was often affected by lack of evidence such as de novo data (with or without paternity and maternity confirmed) —PS2/PM6, and functional data —PS3/PM1/PP2. When considering a patient’s diagnosis of heterozygous HTRA1 gene variants of symptomatic carriers or other hereditary diseases, clinicians need to pay attention to de novo data and functional data, which may affect the pathogenicity analysis.
Possible Pathogenic Mechanism
The molecular mechanism of heterozygous HTRA1 gene variant is not completely clear,21 and the molecular pathogenesis caused by variants at different sites is different.39 Presently, it is mainly focused on preliminary studies on the effect of HtrA1 protease activity and downstream TGF-β1 signaling pathway (Figure 4). The HtrA1 protease is composed of trimers, and each adjacent HtrA1 subunit activates each other through the linker region, the sensor domains of Loop 3 and Loop D.40 Variant sites p.R166C,41 p.R166L,41 p.A173P,41 p.G283E,20 p.G295R,41 and p.T319I20 hinder the formation of stable trimers by HtrA1, thus, affecting the activation cascade. It has been suggested that if the variant is located in the key structures of HtrA1 protease, such as the linker region, Loop 3, or Loop D, the communication between HtrA1 subunits can be affected.17 It also has a dominant negative effect, which reduces the activity of HtrA1. For example, variants in p.P285L and p.R302Q cause damage to the HtrA1 enzyme activation cascade.20 However, the heterozygous HTRA1 gene variant site located outside the linker region and the Loop 3/Loop D domain require a careful assessment of pathogenicity.
 |
Figure 4 Possible pathogenic mechanism of heterozygous HTRA1 gene variant. The decrease in HtrA1 protease activity has three pathogenic mechanisms (1–3). 1: variant HtrA1 enzyme is unable to form stable trimers, and it has a dominant negative effect to inhibit wild HtrA1 activity; 2: variant HtrA1 enzyme affects the communication between its subunits, and it has a dominant negative effect on inhibiting wild HtrA1 activity; 3: variant HtrA1 enzyme shows a decrease in its own activity, and it has no dominant negative effect on wild HtrA1. Abbreviations: CTGF, connective tissue growth factor; PAI-1, plasminogen activator inhibitor-1. |
Moreover, mutant HtrA1 inhibited the activity of wild HtrA1, and it had a dominant negative effect, which was defined as the mutated allele being a loss of function mutant that interfered with the normal function of the remaining wild-type allele, leading to a further decrease in enzyme activity. As for heterozygous HTRA1 gene variants, missense variants in p.R166L,6,41 p.A173P,6,41 p.V175M,37 p.I256T,30 p.F278L,37 p.G283E,20,41 p.S284R,6,41 p.P285L,20,37 p.G295R,41 p. R302Q,20 p.Q318H,37 p.T319I,20 p.V339M37 and p.G350E37 have dominant negative effects. However, missense variants in p.G120D,30 p.R166C,41 p.I179N,30 p.S205C,42 p.G276A,30 p.P285Q,6,41 p.F286V,6,41 p.V297M20 and p.N324T30 do not show a dominant negative effect. Nonsense variant site p.Q289X may be submitted to nonsense mediated mRNA decay (NMD).30 Frame shift variant site p.A182Pfs*33 caused premature termination codons (PTC), and the pathogenesis may be submitted to NMD.30 Nonsense variant site p.R302X was also related to NMD.41 In summary, the decrease in HtrA1 protease activity is currently the most important explanation for the genetic pathogenic mechanism of symptomatic heterozygous HTRA1 variant carriers.
HtrA1 protease is involved in the transduction of TGF-β1 signaling pathway, which can inhibit the expression of TGF-β1.43 A recent study demonstrated that in HTRA1-/- mouse model, the decreased HtrA1 enzymatic activity can lead to the accumulation of matrisome proteins such as latent TGF-β binding protein 4 (LTBP-4) and hub protein fibronectin (FN), which are substrates of HtrA1. Candesartan treatment alleviated matrisome protein accumulation, and may be a potential treatment for CARASIL.44 The p.S205C variant site can upregulate the expression of TGF-β1, Smad2/3, Smad4, phosphorylated Smad2/3, and Smad4.42 The abnormal signal transduction of the TGF-β1/Smads pathway may be a potential molecular pathogenic mechanism, suggesting that TGF-β1 antagonists can be used for the treatment of symptomatic heterozygous HTRA1 variant carriers.42 In addition to the TGF-β1/Smad signaling pathway, a high expression of CTGF and PAI-1 genes was observed in patients with p.E42fs and p.A321T variants.39 It is suggested that the TGF-β1/non-Smad pathway may also be involved in the molecular pathogenesis of symptomatic heterozygous HTRA1 variant carriers. Increased expression of TGF-β signaling pathway is a common cause of increased Fn1 mRNA expression and vascular intimal thickening.45 However, a recent study analyzed various signaling pathways regulated by TGF-β and found that TGF-β is deposited in HTRA1-/- mice, but Fn1 mRNA expression has not been significantly increased or decreased, suggesting that TGF-β signaling pathway may be less effective in CARASIL.44
Interestingly, TGF-β signaling was found to be associated with some clinical manifestations of HTRA1-related CSVD. Bone morphogenetic protein (BMP), a member of the TGF-β family, is involved in the maintenance of hair follicle stem cells.46,47 When the secretion of BMP is increased or its function is hyperactive, it will inhibit the activity of hair follicle stem cells, thereby promoting alopecia. TGF-β may also promote the production of IL-17 by cooperating with IL-6, aggravate the inflammatory response, stimulate the increase of NO secretion, and eventually lead to migraine.48,49 TGF-β receptors in astrocytes can be activated by binding to serum albumin, increasing the level of downstream p-Smad1/5/8, and participating in the occurrence of epilepsy.50
Pathology and Pathophysiology
The pathology and pathophysiology of CSVD related to heterozygote HTRA1 variant (symptomatic carrier) is overlapped by that to homozygote HTRA1 variant (CARASIL). Autopsy study of a heterozygous HTRA1 variant case (p.G283E) revealed cerebral small arteries showed intimal proliferation, splitting of the internal elastic lamina, hyaline degeneration of media, and diffuse and focally intensive myelin pallor in the white matter.20 Another autopsy study of a patient with heterozygous HTRA1 variant (p.R302Q) found that internal elastic lamina splitting, myointimal cell proliferation, smooth muscle cell (SMC) loss, intima and adventitia fibrosis, lipohyalinosis, lumen narrow or occlusive in arterioles smaller than 100 μm.51 100–500 μm arteries revealed lumen distorted dilation, aneurysm-like structures, and tunica media TGF-β1 high expression.51 Pathological observation of frontal white matter suggested focal myelin pallor with preserved U-fibers.51 Cerebrovascular pathophysiological regulations were secondary to the vascular structure pathological changes. Disruption of the autoregulatory mechanism of cerebral blood flow may ultimately lead to white matter ischemia.13,52 Animal study also confirmed that cerebral blood flow decreased in HTRA1 -/- mice.44
Conclusion and Perspectives
Symptomatic HTRA1 variant carriers is a monogenic CSVD, and the penetrance appears to be low. According to ACMG standards and guidelines, There are mostly pathogenic or likely pathogenic variants, and a little variants of uncertain significance. Through a literature review, we summarized the following characteristics of pathogenic and likely pathogenic symptomatic HTRA1 variant carriers: (1) To date, the majority of reported symptomatic HTRA1 carriers are in European and Asian countries, particularly in China which was found to have the highest number of reported cases. (2) The age of first onset was mostly concentrated in the fourth and fifth decades. The clinical manifestations and imaging signs were similar to those of sporadic CSVDs, and there was no specificity. Diagnosis mainly depends on genetic testing. (3) The heterozygous HTRA1 variants were mostly missense variants. The variant sites were mostly concentrated in Exon 4, which is located in the trypsin-like serine protease domain of HtrA1 protease. The two variant sites, 166–182 aa and 274–302 aa, were the most concentrated. (4) The molecular pathogenesis caused by variants at different sites is different, and the decrease in HtrA1 protease activity is currently the most important explanation for the genetic pathogenesis.
However, the correlation between the genotype and clinical phenotype still needs to be further confirmed. In the future, imaging genomic methods can be used to find characteristic images of the disease and to differentiate it from other common monogenic CSVDs, such as CADASIL and CARASIL. Additionally, studies should focus on the genetic testing of patients with sporadic CSVDs to further enrich the spectrum of heterozygous variants in the HTRA1 gene. The pathogenic mechanism of symptomatic carrier has not been fully clarified yet, and the study of HtrA1 protease activity is currently a hot topic. The downstream TGF-β1 signaling pathway transduction may be taken into consideration for future study.
Funding
This study was supported by grants from Shanxi Provincial Health and Family Planning Commission (No. 2017033, No. 2020080); Doctoral Fund of the First Hospital of Shanxi Medical University (No. YB161706, No. BS03201631, No. SD2215); Shanxi Applied Basic Research Program (No. 201801D221426, No. 20210302124404).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Li T, Huang Y, Cai W, et al. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020;11(10):932. doi:10.1038/s41419-020-03137-x
2. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689–701. doi:10.1016/S1474-4422(10)70104-6
3. Haffner C, Malik R, Dichgans M. Genetic factors in cerebral small vessel disease and their impact on stroke and dementia. J Cereb Blood Flow Metab. 2016;36(1):158–171. doi:10.1038/jcbfm.2015.71
4. Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–710. doi:10.1038/383707a0
5. Hara K, Shiga A, Fukutake T, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360(17):1729–1739.
6. Verdura E, Herve D, Scharrer E, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain. 2015;138(Pt 8):2347–2358. doi:10.1093/brain/awv155
7. Richards A, van den Maagdenberg AM, Jen JC, et al. C-terminal truncations in human 3’-5’ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39(9):1068–1070. doi:10.1038/ng2082
8. Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308(5725):1167–1171. doi:10.1126/science.1109418
9. Yoneda Y, Haginoya K, Arai H, et al. De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly. Am J Hum Genet. 2012;90(1):86–90. doi:10.1016/j.ajhg.2011.11.016
10. Bugiani M, Kevelam SH, Bakels HS, et al. Cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology. 2016;87(17):1777–1786. doi:10.1212/WNL.0000000000003251
11. Bernstein HS, Bishop DF, Astrin KH, et al. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J Clin Invest. 1989;83(4):1390–1399. doi:10.1172/JCI114027
12. Mizuno T, Mizuta I, Watanabe-Hosomi A, Mukai M, Koizumi T. Clinical and genetic aspects of CADASIL. Front Aging Neurosci. 2020;12:91. doi:10.3389/fnagi.2020.00091
13. Uemura M, Nozaki H, Onodera O. Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL). Brain Nerve. 2017;69(1):25–33. doi:10.11477/mf.1416200631
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
15. De Luca A, De Falco M, Severino A, et al. Distribution of the serine protease HtrA1 in normal human tissues. J Histochem Cytochem. 2003;51(10):1279–1284. doi:10.1177/002215540305101004
16. Eigenbrot C, Ultsch M, Lipari MT, et al. Structural and functional analysis of HtrA1 and its subdomains. Structure. 2012;20(6):1040–1050. doi:10.1016/j.str.2012.03.021
17. Uemura M, Nozaki H, Kato T, et al. HTRA1-related cerebral small vessel disease: a review of the literature. Front Neurol. 2020;11:545. doi:10.3389/fneur.2020.00545
18. Liu J, Zhu Y, Zhou L, et al. HTRA1-related autosomal dominant cerebral small vessel disease. Chin Med J. 2020;134(2):178–184. doi:10.1097/CM9.0000000000001176
19. Yu Z, Cao S, Wu A, et al. Genetically confirmed CARASIL: case report with novel HTRA1 mutation and literature review. World Neurosurg. 2020;143:121–128. doi:10.1016/j.wneu.2020.05.128
20. Nozaki H, Kato T, Nihonmatsu M, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology. 2016;86(21):1964–1974. doi:10.1212/WNL.0000000000002694
21. Bougea A. Do heterozygous HTRA1 mutations carriers form a distinct clinical entity? CNS Neurosci Ther. 2018;24(12):1299–1300. doi:10.1111/cns.13047
22. Xie F, Zhang LS. A Chinese CARASIL patient caused by novel compound heterozygous mutations in HTRA1. J Stroke Cerebrovasc Dis. 2018;27(10):2840–2842. doi:10.1016/j.jstrokecerebrovasdis.2018.06.017
23. Ragno M, Pianese L, Caulo M, et al. Cutaneous sensory and autonomic small fiber neuropathy in HTRA1-related cerebral small vessel disease. J Neuropathol Exp Neurol. 2020;80:713.
24. Favaretto S, Margoni M, Salviati L, Pianese L, Manara R, Baracchini C. A new Italian family with HTRA1 mutation associated with autosomal-dominant variant of CARASIL: are we pointing towards a disease spectrum? J Neurol Sci. 2019;396:108–111. doi:10.1016/j.jns.2018.11.008
25. Mancuso M, Arnold M, Bersano A, et al. Monogenic cerebral small-vessel diseases: diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur J Neurol. 2020;27(6):909–927. doi:10.1111/ene.14183
26. Zhang H, Qin X, Shi Y, et al. Genotype-phenotype correlations of heterozygous HTRA1-related cerebral small vessel disease: case report and systematic review. Neurogenetics. 2021;22(3):187–194. doi:10.1007/s10048-021-00646-5
27. Muthusamy K, Ferrer A, Klee EW, Wierenga KJ, Gavrilova RH. Clinicoradiographic and genetic features of cerebral small vessel disease indicate variability in mode of inheritance for monoallelic HTRA1 variants. Mol Genet Genomic Med. 2021;9(10):e1799. doi:10.1002/mgg3.1799
28. Malik R, Beaufort N, Frerich S, et al. Whole-exome sequencing reveals a role of HTRA1 and EGFL8 in brain white matter hyperintensities. Brain. 2021;144(9):2670–2682. doi:10.1093/brain/awab253
29. Shen L, Thompson PM. Brain imaging genomics: integrated analysis and machine learning. Proc IEEE Inst Electr Electron Eng. 2020;108(1):125–162. doi:10.1109/JPROC.2019.2947272
30. Lee YC, Chung CP, Chao NC, et al. Characterization of heterozygous HTRA1 mutations in Taiwanese patients with cerebral small vessel disease. Stroke. 2018;49(7):1593–1601. doi:10.1161/STROKEAHA.118.021283
31. Thaler FS, Catak C, Einhäupl M, et al. Cerebral small vessel disease caused by a novel heterozygous mutation in HTRA1. J Neurol Sci. 2018;388:19–21. doi:10.1016/j.jns.2018.02.043
32. Zhang WY, Xie F, Lu PL. Two novel heterozygous HTRA1 mutations in two pedigrees with cerebral small vessel disease families. Neurol Sci. 2018;39(3):497–501. doi:10.1007/s10072-017-3231-z
33. Zhou H, Jiao B, Ouyang Z, Wu Q, Shen L, Fang L. Report of two pedigrees with heterozygous HTRA1 variants-related cerebral small vessel disease and literature review. Mol Genet Genomic Med. 2022;10(10):e2032. doi:10.1002/mgg3.2032
34. Ohta K, Ozawa T, Fujinaka H, Goto K, Nakajima T. Cerebral small vessel disease related to a heterozygous nonsense mutation in HTRA1. Intern Med. 2020;59(10):1309–1313. doi:10.2169/internalmedicine.4041-19
35. Tateoka T, Onda H, Hirota K, et al. Unusual case of cerebral small vessel disease with a heterozygous nonsense mutation in HTRA1. J Neurol Sci. 2016;362:144–146. doi:10.1016/j.jns.2016.01.037
36. Chen MJ, Zhang Y, Luo WJ, et al. Identified novel heterozygous HTRA1 pathogenic variants in Chinese patients with HTRA1-associated dominant cerebral small vessel disease. Front Genet. 2022;13:909131. doi:10.3389/fgene.2022.909131
37. Zhang C, Zheng H, Li X, et al. Novel mutations in HTRA1-related cerebral small vessel disease and comparison with CADASIL. Ann Clin Transl Neurol. 2022;9(10):1586–1595. doi:10.1056/NEJMoa0801560
38. Coste T, Hervé D, Neau J, et al. Heterozygous HTRA1 nonsense or frameshift mutations are pathogenic. Brain. 2021;144(9):2616–2624. doi:10.1093/brain/awab271
39. Fasano A, Formichi P, Taglia I, et al. HTRA1 expression profile and activity on TGF-β signaling in HTRA1 mutation carriers. J Cell Physiol. 2020;235(10):7120–7127. doi:10.1002/jcp.29609
40. Truebestein L, Tennstaedt A, Mönig T, et al. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat Struct Mol Biol. 2011;18(3):386–388.
41. Uemura M, Nozaki H, Koyama A, et al. HTRA1 mutations identified in symptomatic carriers have the property of interfering the trimer-dependent activation cascade. Front Neurol. 2019;10:693. doi:10.3389/fneur.2019.00693
42. Zhuo ZL, Cong L, Zhang J, Zhao XT. A novel heterozygous HTRA1 mutation is associated with autosomal dominant hereditary cerebral small vessel disease. Mol Genet Genomic Med. 2020;8(6):e1111. doi:10.1002/mgg3.1111
43. Jin SF, Wang YX, Xu N, et al. High temperature requirement factor A1 (HTRA1) regulates the activation of latent TGF-β1 in keloid fibroblasts. Cell Mol Biol. 2018;64(1):107–110. doi:10.14715/cmb/2018.64.2.19
44. Kato T, Manabe RI, Igarashi H, et al. Candesartan prevents arteriopathy progression in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy model. J Clin Invest. 2021;131:22. doi:10.1172/JCI140555
45. Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, et al. TGF-β signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi:10.1016/j.cardiores.2007.02.008
46. Onodera O. TGF-β family signaling contributes to human cerebral small vessel disease. Rinsho shinkeigaku. 2011;51(11):943–944. doi:10.5692/clinicalneurol.51.943
47. Plikus MV, Mayer JA, Damon D, et al. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature. 2008;451(7176):340. doi:10.1038/nature06457
48. Ghorbani Z, Rafiee P, Haghighi S, Jahromi SR, Togha M. The effects of vitamin D3 supplementation on TGF-β and IL-17 serum levels in migraineurs: post hoc analysis of a randomized clinical trial. J Pharm Health Care Sci. 2021;7(1):9. doi:10.1186/s40780-021-00192-0
49. Joshi N, Minz RW, Anand S, Parmar NV, Kanwar AJ. Vitamin D deficiency and lower TGF-β/IL-17 ratio in a North Indian cohort of pemphigus vulgaris. BMC Res Notes. 2014;7(1):536. doi:10.1186/1756-0500-7-536
50. Bar-Klein G, Cacheaux LP, Kamintsky L, et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014;75(6):864–875. doi:10.1002/ana.24147
51. Ito J, Nozaki H, Toyoshima Y, et al. Histopathologic features of an autopsied patient with cerebral small vessel disease and a heterozygous HTRA1 mutation. Neuropathology. 2018;38(4):428–432. doi:10.1111/neup.12473
52. Caplan L, Gomes J. Binswanger disease--an update. J Neurol Sci. 2010;299:9–10. doi:10.1016/j.jns.2010.08.041
53. Bougea A, Velonakis G, Spantideas N, et al. The first Greek case of heterozygous cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy: an atypical clinico-radiological presentation. Neuroradiol J. 2017;30(6):583–585. doi:10.1177/1971400917700168
54. Cao H, Liu J, Tian W, et al. A novel heterozygous HTRA1 mutation in an Asian family with CADASIL-like disease. J Clin Lab Anal. 2022;36(2):e24174. doi:10.1002/jcla.24174
55. Di Donato I, Bianchi S, Gallus GN, et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci Ther. 2017;23(9):759–765. doi:10.1111/cns.12722
56. Shang T, Pinho M, Ray D, Khera A. Two unique mutations in HTRA1-related cerebral small vessel disease in North America And Africa and literature review. J Stroke Cerebrovasc Dis. 2021;30(11):106029. doi:10.1016/j.jstrokecerebrovasdis.2021.106029
57. Wu X, Li C, Mao J, Li L, Liu Y, Hou Y. Heterozygous HTRA1 missense mutation in CADASIL-like family disease. Braz J Med Biol Res. 2018;51(5):e6632. doi:10.1590/1414-431x20176632
58. Mahale R, Agarwal A, Gautam J, et al. HTRA1 autosomal dominant cerebral small vessel disease in gene mutation. Ann Indian Acad Neurol. 2021;24(2):297–299. doi:10.4103/aian.AIAN_381_20
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.